Mechanistic Insights into Neurotoxicity Induced by Anesthetics in the Developing Brain

{kind=link}
Abstract
:1. Introduction
2. Anesthetic Exposure and Timing
2.1. Exposure Concentration and Duration
2.2. Age Dependency of Apoptotic Neurodegeneration
2.3. Nonhuman Primate Studies
2.4. Clinical Evidence and Implication
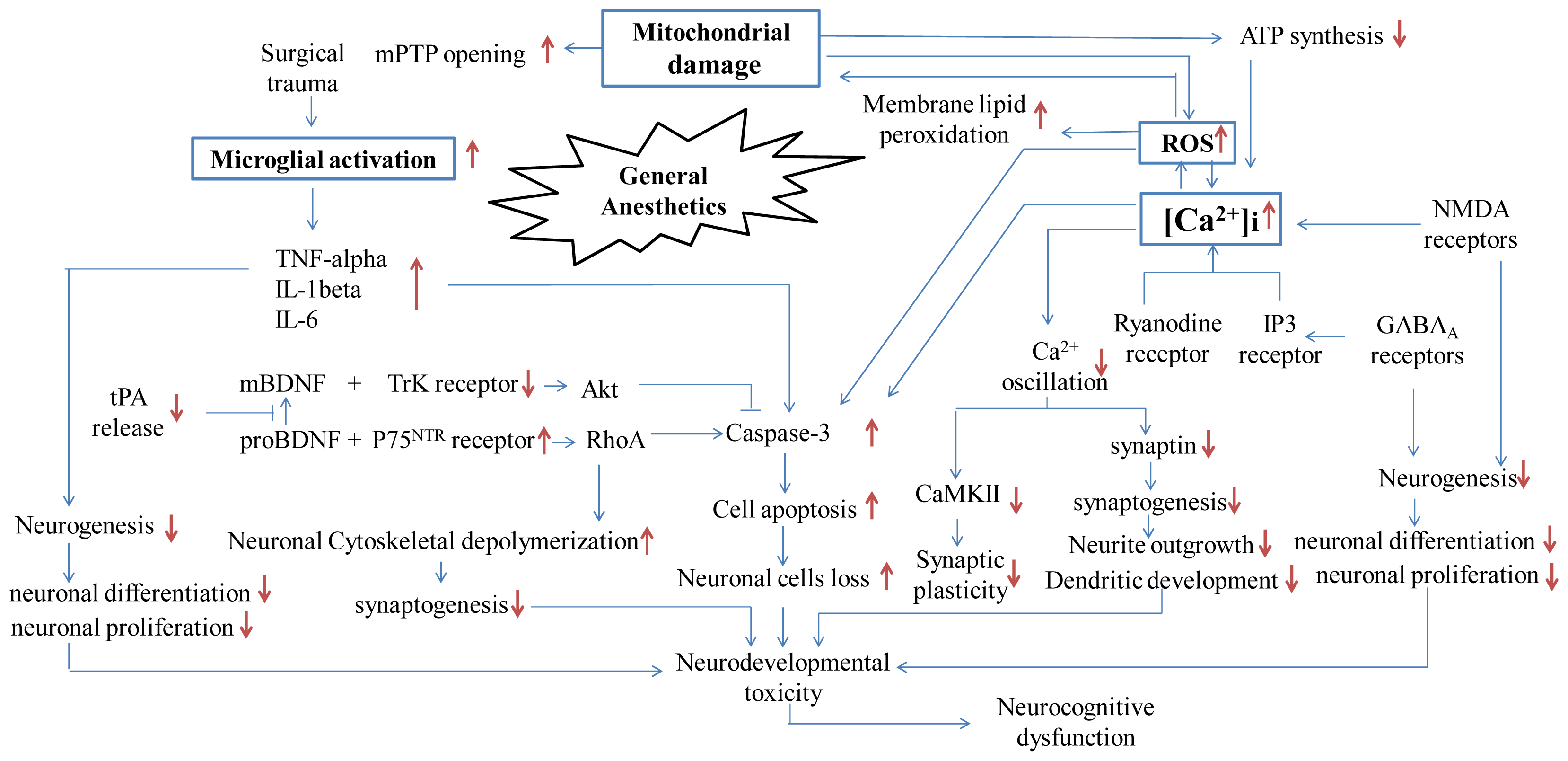
3. Molecular Mechanisms: Neuroapoptosis In Vitro and In Vivo
3.1. NMDA Receptors and GABAA Receptors
3.2. Mitochondrial Perturbations
3.3. Dysregulation of Intracellular Ca2+ Homeostasis
3.4. Neuroinflammatory Pathway
3.5. The BDNF Pathway
3.6. Other Signal Transduction Pathways
4. Cellular Processes in Neurodevelopment
4.1. Neurogenesis
4.2. Dendritic Development
4.3. Neurite Outgrowth
4.4. Glial Development
5. Neurobehavior Mechanisms
6. Neuroprotection against Anesthetic-Induced Neurodegeneration
6.1. Erythropoietin (EPO)
6.2. Brain Preconditioning with Anesthetics
6.3. Vitamins
6.4. Alpha2(α2) Adrenoceptor Agonist
6.5. Lithium
6.6. Activity-Dependent Neuroprotective Protein (ADNP)
6.7. Other Neuroprotectants
7. Summary
Acknowledgement
References
- Jevtovic-Todorovic, V.; Wozniak, D.W.; Benshoff, N.; Olney, J.W. Commonly used anesthesia protocol causes neuronal suicide in the immature rat brain. Soc. Neurosci. Abstr 2001, 27, 2050. [Google Scholar]
- Sun, L. Early childhood general anaesthesia exposure and neurocognitive development. Br. J. Anaesth 2010, 105, i61–i68. [Google Scholar]
- Hansen, T.G.; Pedersen, J.K.; Henneberg, S.W.; Pedersen, D.A.; Murray, J.C.; Morton, N.S.; Christensen, K. Academic performance in adolescence after inguinal hernia repair in infancy: A nationwide cohort study. Anesthesiology 2011, 114, 1076–1085. [Google Scholar]
- DiMaggio, C.; Sun, L.S.; Li, G. Early childhood exposure to anesthesia and risk of developmental and behavioral disorders in a sibling birth cohort. Anesth. Analg 2011, 113, 1143–1151. [Google Scholar]
- Wilder, R.T.; Flick, R.P.; Sprung, J.; Katusic, S.K.; Barbaresi, W.J.; Mickelson, C.; Gleich, S.J.; Schroeder, D.R.; Weaver, A.L.; Warner, D.O. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology 2009, 110, 796–804. [Google Scholar]
- Sprung, J.; Flick, R.P.; Wilder, R.T.; Katusic, S.K.; Pike, T.L.; Dingli, M.; Gleich, S.J.; Schroeder, D.R.; Barbaresi, W.J.; Hanson, A.C. Anesthesia for cesarean delivery and learning disabilities in a population-based birth cohort. Anesthesiology 2009, 111, 302–310. [Google Scholar]
- Kalkman, C.J.; Peelen, L.; Moons, K.G.; Veenhuizen, M.; Bruens, M.; Sinnema, G.; de Jong, T.P. Behavior and development in children and age at the time of first anesthetic exposure. Anesthesiology 2009, 110, 805–812. [Google Scholar]
- DiMaggio, C.; Sun, L.S.; Kakavouli, A.; Byrne, M.W.; Li, G. A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. J. Neurosurg. Anesth 2009, 21, 286–291. [Google Scholar]
- Bartels, M.; Althoff, R.R.; Boomsma, D.I. Anesthesia and cognitive performance in children: No evidence for a causal relationship. Twin Res. Hum. Genet 2009, 12, 246–253. [Google Scholar]
- Sun, L.; Li, G.; Dimaggio, C.; Byrne, M.; Rauh, V.; Brooks-Gunn, J.; Kakavouli, A.; Wood, A. Coinvestigators of the pediatric anesthesia neurodevelopment assessment (PANDA) research network: Anesthesia and neurodevelopment in children: Time for an answer. Anesthesiology 2008, 109, 757–761. [Google Scholar]
- Davidson, A.J.; McCann, M.E.; Morton, N.S.; Myles, P.S. Anesthesia and outcome after neonatal surgery: The role for randomized trials. Anesthesiology 2008, 109, 941–944. [Google Scholar]
- Kabra, N.S.; Schmidt, B.; Roberts, R.S.; Doyle, L.W.; Papile, L.; Fanaroff, A. Neurosensory impairment after surgical closure of patent ductus arteriosus in extremely low birth weight infants: Results from the Trial of Indomethacin Prophylaxis in Preterms. J. Pediatr 2007, 150, 229–234. [Google Scholar]
- Ludman, L.; Spitz, L.; Wade, A. Educational attainments in early adolescence of infants who required major neonatal surgery. J. Pediatr. Surg 2001, 36, 858–862. [Google Scholar]
- Chacko, J.; Ford, W.D.A.; Haslam, R. Growth and neurodevelopmental outcome in extremely-low-birth-weight infants after laparotomy. Pediatr. Surg. Int 1999, 15, 496–499. [Google Scholar]
- Rizzi, S.; Carter, L.B.; Ori, C.; Jevtovic-Todorovic, V. Clinical anesthesia causes permanent damage to the fetal guinea pig brain. Brain Pathol 2008, 18, 198–210. [Google Scholar]
- Paule, M.; Li, M.; Allen, R.; Liu, F.; Zou, X.; Hotchkiss, C.; Hanig, J.; Patterson, T.; Slikker, W. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol. Teratol 2011, 33, 220–230. [Google Scholar]
- Zou, X.; Liu, F.; Zhang, X.; Patterson, T.A.; Callicott, R.; Liu, S.; Hanig, J.P.; Paule, M.G.; Slikker, W., Jr; Wang, C. Inhalation anesthetic-induced neuronal damage in the developing rhesus monkey. Neurotoxicol. Teratol. 2011, 33, 592–597. [Google Scholar]
- Jevtovic-Todorovic, V.; Hartman, R.E.; Izumi, Y.; Benshoff, N.D.; Dikranian, K.; Zorumski, C.F.; Olney, J.W.; Wozniak, D.F. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J. Neurosci 2003, 23, 876–882. [Google Scholar]
- Stratmann, G.; Bell, J.; Alvi, R.S.; Ku, B.; Saw, L.; Encarnacion, A.; Magnusson, K.; Bickler, P.; Liu, J. Neonatal isoflurane anesthesia causes a permanent neurocognitive deficit in rats. J. Neurosurg. Anesth 2006, 18. [Google Scholar] [CrossRef]
- Loepke, A.W.; Istaphanous, G.K.; McAuliffe, J.J., III; Miles, L.; Hughes, E.A.; McCann, J.C.; Harlow, K.E.; Kurth, C.D.; Williams, M.T.; Vorhees, C.V.; et al. The effects of neonatal isoflurane exposure in mice on brain cell viability, adult behavior, learning, and memory. Anesth. Analg 2009, 108, 90–104. [Google Scholar]
- Satomoto, M.; Satoh, Y.; Terui, K.; Miyao, H.; Takishima, K.; Ito, M.; Imaki, J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology 2009, 110, 628–637. [Google Scholar]
- Wang, Y.; Cheng, Y.; Liu, G.; Tian, X.; Tu, X.; Wang, J. Chronic exposure of gestation rat to sevoflurane impairs offspring brain development. Neurol. Sci 2012, 33, 535–544. [Google Scholar]
- Zhang, B.; Dong, Y.; Zhang, G.; Moir, R.D.; Xia, W.; Yue, Y.; Tian, M.; Culley, D.J.; Crosby, G.; Tanzi, R.E.; et al. The inhalation anesthetic desflurane induces caspase activation and increases amyloid beta-protein levels under hypoxic conditions. J. Biol. Chem 2008, 283, 11866–11875. [Google Scholar]
- Young, C.; Tenkova, T.; Wang, H.; Qin, Y.; Labruyere, J.; Jevtovic-Todorovic, V.; Olney, J.W. A single sedating dose of ketamine causes neuronal apoptosis in developing mouse brain. Soc. Neurosci. Abstr. Viewer Itiner. Plan 2003, 2003, 748. [Google Scholar]
- Scallet, A.; Schmued, L.; Slikker, W.; Grunberg, N.; Faustino, P.; Davis, H.; Lester, D.; Pine, P.; Sistare, F.; Hanig, J. Developmental neurotoxicity of ketamine: Morphometric confirmation, exposure parameters, and multiple fluorescent labeling of apoptotic neurons. Toxicol. Sci 2004, 81, 364–370. [Google Scholar]
- Viberg, H.; Ponten, E.; Eriksson, P.; Gordh, T.; Fredriksson, A. Neonatal ketamine exposure results in changes in biochemical substrates of neuronal growth and synaptogenesis, and alters adult behavior irreversibly. Toxicology 2008, 249, 153–159. [Google Scholar]
- Cattano, D.; Young, C.; Straiko, M.M.W.; Olney, J.W. Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth. Analg 2008, 106, 1712–1714. [Google Scholar]
- Fredriksson, A.; Ponten, E.; Gordh, T.; Eriksson, P. Neonatal exposure to a combination of N-methyl-d-aspartate and gamma-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology 2007, 107, 427–436. [Google Scholar]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Hörster, F.; Tenkova, T. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar]
- Zhao, Y.; Xiang, Q.; Shi, Q.; Li, S.; Tan, L.; Wang, J.; Jin, X.; Luo, A. GABAergic excitotoxicity injury of the immature hippocampal pyramidal neurons’ exposure to isoflurane. Anesth. Analg 2011, 113, 1152–1160. [Google Scholar]
- Ozer, M.; Baris, S.; Karakaya, D.; Kocamanoglu, S.; Tur, A. Behavioural effects of chronic exposure to sub-anesthetic concentrations of halothane, sevoflurane and desflurane in rats. Can. J. Anesth 2006, 53, 653–658. [Google Scholar]
- Kodama, M.; Satoh, Y.; Otsubo, Y.; Araki, Y.; Yonamine, R.; Masui, K.; Kazama, T. Neonatal desflurane exposure induces more robust neuroapoptosis than do isoflurane and sevoflurane and impairs working memory. Anesthesiology 2011, 115, 979–991. [Google Scholar]
- Istaphanous, G.K.; Howard, J.; Nan, X.; Hughes, E.A.; McCann, J.C.; McAuliffe, J.J.; Danzer, S.C.; Loepke, A.W. Comparison of the neuroapoptotic properties of equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in neonatal mice. Anesthesiology 2011, 114, 578–587. [Google Scholar]
- Zhang, Y.; Xu, Z.; Wang, H.; Dong, Y.; Shi, H.N.; Culley, D.J.; Crosby, G.; Marcantonio, E.R.; Tanzi, R.E.; Xie, Z. Anesthetics isoflurane and desflurane differently affect mitochondrial function, learning, and memory. Ann. Neurol 2012. [Google Scholar] [CrossRef]
- Stratmann, G.; Sall, J.W.; May, L.D.V.; Bell, J.S.; Magnusson, K.R.; Rau, V.; Visrodia, K.H.; Alvi, R.S.; Ku, B.; Lee, M.T.; et al. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology 2009, 110, 834–848. [Google Scholar]
- Zhu, C.; Gao, J.; Karlsson, N.; Li, Q.; Zhang, Y.; Huang, Z.; Li, H.; Kuhn, H.G.; Blomgren, K. Isoflurane anesthesia induced persistent, progressive memory impairment, caused a loss of neural stem cells, and reduced neurogenesis in young, but not adult, rodents. J. Cereb. Blood Flow Metab 2010, 30, 1017–1030. [Google Scholar]
- Gutierrez, S.; Carnes, A.; Finucane, B.; Musci, G.; Oelsner, W.; Hicks, L.; Russell, G.; Liu, C.; Turner, C. Is age-dependent, ketamine-induced apoptosis in the rat somatosensory cortex influenced by temperature? Neuroscience 2010, 168, 253–262. [Google Scholar]
- Brambrink, A.M.; Evers, A.S.; Avidan, M.S.; Farber, N.B.; Smith, D.J.; Zhang, X.; Dissen, G.A.; Creeley, C.E.; Olney, J.W. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology 2010, 112, 834–841. [Google Scholar]
- Slikker, W., Jr; Zou, X.; Hotchkiss, C.E.; Divine, R.L.; Sadovova, N.; Twaddle, N.C.; Doerge, D.R.; Scallet, A.C.; Patterson, T.A.; Hanig, J.P.; et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol. Sci. 2007, 98, 145–158. [Google Scholar]
- Zou, X.; Patterson, T.A.; Divine, R.L.; Sadovova, N.; Zhang, X.; Hanig, J.P.; Paule, M.G.; Slikker, W., Jr; Wang, C. Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int. J. Dev. Neurosci. 2009, 27, 727–731. [Google Scholar]
- Brambrink, A.M.; Evers, A.S.; Avidan, M.S.; Farber, N.B.; Smith, D.J.; Martin, L.D.; Dissen, G.A.; Creeley, C.E.; Olney, J.W. Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology 2012, 116, 372–384. [Google Scholar]
- Flick, R.P.; Katusic, S.K.; Colligan, R.C.; Wilder, R.T.; Voigt, R.G.; Olson, M.D.; Sprung, J.; Weaver, A.L.; Schroeder, D.R.; Warner, D.O. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics 2011, 128, e1053–e1061. [Google Scholar]
- Miller, M.W. Effects of alcohol on the generation and migration of cerebral cortical neurons. Science 1986, 233, 1308–1311. [Google Scholar]
- Miller, M.W. Mechanisms of ethanol induced neuronal death during development: From the molecule to behavior. Alcohol. Clin. Exp. Res 1996, 20, 128a–132a. [Google Scholar]
- Ullah, I.; Ullah, N.; Naseer, M.I.; Lee, H.Y.; Kim, M.O. Neuroprotection with metformin and thymoquinone against ethanol-induced apoptotic neurodegeneration in prenatal rat cortical neurons. BMC Neurosci 2012, 13. [Google Scholar] [CrossRef]
- Qin, L.; Crews, F.T. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflammation 2012, 9. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Siler-Marsiglio, K. Ethanol influences on bax translocation, mitochondrial membrane potential, and reactive oxygen species generation are modulated by vitamin e and brain-derived neurotrophic factor. Alcohol. Clin. Exp. Res 2011, 35, 1122–1133. [Google Scholar]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vöckler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar]
- Bhutta, A.T.; Schmitz, M.L.; Swearingen, C.; James, L.P.; Wardbegnoche, W.L.; Lindquist, D.M.; Glasier, C.M.; Tuzcu, V.; Prodhan, P.; Dyamenahalli, U. Ketamine as a neuroprotective and anti-inflammatory agent in children undergoing surgery on cardiopulmonary bypass: A pilot randomized, double-blind, placebo-controlled trial. Pediatr. Crit. Care Med 2012, 13, 328–337. [Google Scholar]
- Liu, F.; Paule, M.G.; Ali, S.; Wang, C. Ketamine-induced neurotoxicity and changes in gene expression in the developing rat brain. Curr. Neuropharmacol 2011, 9, 256–261. [Google Scholar]
- Forcelli, P.A.; Kim, J.; Kondratyev, A.; Gale, K. Pattern of antiepileptic drug–induced cell death in limbic regions of the neonatal rat brain. Epilepsia 2011, 52, e207–e211. [Google Scholar]
- Stefovska, V.G.; Uckermann, O.; Czuczwar, M.; Smitka, M.; Czuczwar, P.; Kis, J.; Kaindl, A.M.; Turski, L.; Turski, W.A.; Ikonomidou, C. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann. Neurol 2008, 64, 434–445. [Google Scholar]
- Zhou, Z.W.; Shu, Y.; Li, M.; Guo, X.; Pac-Soo, C.; Maze, M.; Ma, D. The glutaminergic, GABAergic, dopaminergic but not cholinergic neurons are susceptible to anaesthesia-induced cell death in the rat developing brain. Neuroscience 2011, 174, 64–70. [Google Scholar]
- Larsen, M.; Langmoen, I. The effect of volatile anaesthetics on synaptic release and uptake of glutamate. Toxicol. Lett 1998, 100, 59–64. [Google Scholar]
- Kulak, A.; Duarte, J.; Do, K.Q.; Gruetter, R. Neurochemical profile of the developing mouse cortex determined by in vivo 1H NMR spectroscopy at 14.1 T and the effect of recurrent anaesthesia. J. Neurochem 2010, 115, 1466–1477. [Google Scholar]
- Zhang, G.; Dong, Y.; Zhang, B.; Ichinose, F.; Wu, X.; Culley, D.J.; Crosby, G.; Tanzi, R.E.; Xie, Z. Isoflurane-induced caspase-3 activation is dependent on cytosolic calcium and can be attenuated by memantine. J. Neurosci 2008, 28, 4551–4560. [Google Scholar]
- Sanders, R.D.; Xu, J.; Shu, Y.; Januszewski, A.; Halder, S.; Fidalgo, A.; Sun, P.; Hossain, M.; Ma, D.; Maze, M. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology 2009, 110, 1077–1085. [Google Scholar]
- Yon, J.H.; Daniel-Johnson, J.; Carter, L.B.; Jevtovic-Todorovic, V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience 2005, 135, 815–827. [Google Scholar]
- Zhang, Y.; Dong, Y.; Wu, X.; Lu, Y.; Xu, Z.; Knapp, A.; Yue, Y.; Xu, T.; Xie, Z. The mitochondrial pathway of anesthetic isoflurane-induced apoptosis. J. Bio. Chem 2010, 285, 4025–4037. [Google Scholar]
- Sanchez, V.; Feinstein, S.D.; Lunardi, N.; Joksovic, P.M.; Boscolo, A.; Todorovic, S.M.; Jevtovic-Todorovic, V. General anesthesia causes long-term impairment of mitochondrial morphogenesis and synaptic transmission in developing rat brain. Anesthesiology 2011, 115, 992–1002. [Google Scholar]
- Boscolo, A.; Starr, J.; Sanchez, V.; Lunardi, N.; DiGruccio, M.; Ori, C.; Erisir, A.; Trimmer, P.; Bennett, J.; Jevtovic-Todorovic, V. The abolishment of anesthesia-induced cognitive impairment by timely protection of mitochondria in the developing rat brain: The importance of free oxygen radicals and mitochondrial integrity. Neurobiol. Dis. 2011, 45, 1031–1041. [Google Scholar]
- Ringler, S.L.; Aye, J.; Byrne, E.; Anderson, M.; Turner, C.P. Effects of disrupting calcium homeostasis on neuronal maturation: Early inhibition and later recovery. Cell Mol. Neurobiol 2008, 28, 389–409. [Google Scholar]
- Turner, C.P.; Pulciani, D.; Rivkees, S.A. Reduction in intracellular calcium levels induces injury in developing neurons. Exp. Neurol 2002, 178, 21–32. [Google Scholar]
- Turner, C.; Gutierrez, S.; Liu, C.; Miller, L.; Chou, J.; Finucane, B.; Carnes, A.; Kim, J.; Shing, E.; Haddad, T. Strategies to defeat ketamine-induced neonatal brain injury. Neuroscience 2012, 210, 384–392. [Google Scholar]
- Liu, F.; Paule, M.G.; Ali, S.; Wang, C. Ketamine-induced neurotoxicity and changes in gene expression in the developing rat brain. Curr. Neuropharmacol 2011, 9, 256–261. [Google Scholar]
- Allene, C.; Cossart, R. Early NMDA receptor-driven waves of activity in the developing neocortex: Physiological or pathological network oscillations? J. Physiol 2010, 588, 83–91. [Google Scholar]
- Sinner, B.; Friedrich, O.; Zink, W.; Zausig, Y.; Graf, B.M. The toxic effects of S (+)-ketamine on differentiating neurons in vitro as a consequence of suppressed neuronal Ca2+ oscillations. Anesth. Analg 2011, 113, 1161–1169. [Google Scholar]
- Greer, P.L.; Greenberg, M.E. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron 2008, 59, 846–860. [Google Scholar]
- Spitzer, N.C. Activity-dependent neuronal differentiation prior to synapse formation: The functions of calcium transients. J. Physiol. Paris 2002, 96, 73–80. [Google Scholar]
- De Koninck, P.; Schulman, H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 1998, 279, 227–230. [Google Scholar]
- Sinner, B.; Friedrich, O.; Zausig, Y.; Bein, T.; Graf, B.M. Toxic effects of midazolam on differentiating neurons in vitro as a consequence of suppressed neuronal Ca(2+)-oscillations. Toxicology 2011, 290, 96–101. [Google Scholar]
- Nikizad, H.; Yon, J.H.; Carter, L.B.; Jevtovic-Todorovic, V. Early exposure to general anesthesia causes significant neuronal deletion in the developing rat brain. Ann. N. Y. Acad. Sci 2007, 1122, 69–82. [Google Scholar]
- Wu, X.; Lu, Y.; Dong, Y.; Zhang, G.; Zhang, Y.; Xu, Z.; Culley, D.J.; Crosby, G.; Marcantonio, E.R.; Tanzi, R.E. The inhalation anesthetic isoflurane increases levels of proinflammatory TNF-[alpha], IL-6, and IL-1[beta]. Neurobiol. Aging 2010, 33, 1364–1378. [Google Scholar]
- Lu, Y.; Wu, X.; Dong, Y.; Xu, Z.; Zhang, Y.; Xie, Z. Anesthetic sevoflurane causes neurotoxicity differently in neonatal naïve and alzheimer’s disease transgenic mice. Anesthesiology 2010, 112, 1404–1416. [Google Scholar]
- Terrando, N.; Monaco, C.; Ma, D.; Foxwell, B.M.J.; Feldmann, M.; Maze, M. From the Cover: Tumor necrosis factor-{alpha} triggers a cytokine cascade yielding postoperative cognitive decline. Proc. Natl. Acad. Sci. USA 2010, 107, 20518–20522. [Google Scholar]
- Wan, Y.; Xu, J.; Ma, D.; Zeng, Y.; Cibelli, M.; Maze, M. Postoperative impairment of cognitive function in rats: A possible role for cytokine-mediated inflammation in the hippocampus. Anesthesiology 2007, 106, 436–443. [Google Scholar]
- Fidalgo, A.R.; Cibelli, M.; White, J.P.M.; Nagy, I.; Maze, M.; Ma, D. Systemic inflammation enhances surgery-induced cognitive dysfunction in mice. Neurosci. Lett 2011, 498, 63–66. [Google Scholar]
- Cibelli, M.; Fidalgo, A.R.; Terrando, N.; Ma, D.; Monaco, C.; Feldmann, M.; Takata, M.; Lever, I.J.; Nanchahal, J.; Fanselow, M.S. Role of interleukin-1β in postoperative cognitive dysfunction. Ann. Neurol 2010, 68, 360–368. [Google Scholar]
- Shu, Y.; Zhou, Z.; Wan, Y.; Sanders, R.D.; Li, M.; Pac-Soo, C.K.; Maze, M.; Ma, D. Nociceptive stimuli enhance anesthetic-induced neuroapoptosis in the rat developing brain. Neurobiol. Dis 2011, 45, 743–750. [Google Scholar]
- Lu, L.X.; Yon, J.H.; Carter, L.B.; Jevtovic-Todorovic, V. General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis 2006, 11, 1603–1615. [Google Scholar]
- Lu, B. Pro-region of neurotrophins: Role in synaptic modulation. Neuron 2003, 39, 735–738. [Google Scholar]
- Baranes, D.; Lederfein, D.; Huang, Y.Y.; Chen, M.; Bailey, C.H.; Kandel, E.R. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron 1998, 21, 813–825. [Google Scholar]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [Google Scholar]
- Gascon, E.; Vutskits, L.; Zhang, H.; Barral-Moran, M.; Kiss, P.; Mas, C.; Kiss, J.Z. Sequential activation of p75 and TrkB is involved in dendritic development of subventricular zone-derived neuronal progenitors in vitro. Eur. J. Neurosci 2005, 21, 69–80. [Google Scholar]
- Lemkuil, B.P.; Head, B.P.; Pearn, M.L.; Patel, H.H.; Drummond, J.C.; Patel, P.M. Isoflurane neurotoxicity is mediated by p75NTR-RhoA activation and actin depolymerization. Anesthesiology 2011, 114, 49–57. [Google Scholar]
- Gualandris, A.; Jones, T.E.; Strickland, S.; Tsirka, S.E. Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J. Neurosci 1996, 16, 2220–2225. [Google Scholar]
- Head, B.P.; Patel, H.H.; Niesman, I.R.; Drummond, J.C.; Roth, D.M.; Patel, P.M. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology 2009, 110, 813–825. [Google Scholar]
- Patel, P.; Head, B.; Patel, H.; Drummond, J.; Roth, D. Tpa reduces isoflurane induced neuronal apoptosis and dendritic spines loss in rat neonatal neurons. Anesthesiology 2008, 109, A1413. [Google Scholar]
- Pearn, M.L.; Hu, Y.; Niesman, I.R.; Patel, H.H.; Drummond, J.C.; Roth, D.M.; Akassoglou, K.; Patel, P.M.; Head, B.P. Propofol neurotoxicity is mediated by p75 neurotrophin receptor activation. Anesthesiology 2012, 116, 352–361. [Google Scholar]
- Straiko, M.M.W.; Young, C.; Cattano, D.; Creeley, C.E.; Wang, H.; Smith, D.J.; Johnson, S.A.; Li, E.S.; Olney, J.W. Lithium protects against anesthesia-induced developmental neuroapoptosis. Anesthesiology 2009, 110, 862–868. [Google Scholar]
- Cui, Y.; Ling-Shan, G.; Yi, L.; Xing-Qi, W.; Xue-Mei, Z.; Xiao-Xing, Y. Repeated administration of propofol upregulated the expression of c-Fos and cleaved-caspase-3 proteins in the developing mouse brain. Indian J. Pharmacol 2011, 43, 648–651. [Google Scholar]
- Jiang, H.; Huang, Y.; Xu, H.; Sun, Y.; Han, N.; Li, Q.F. Hypoxia inducible factor-1α is involved in the neurodegeneration induced by isoflurane in the brain of neonatal rats. J. Neurochem 2012, 120, 453–460. [Google Scholar]
- Shu, Y.; Patel, S.M.; Pac-Soo, C.; Fidalgo, A.R.; Wan, Y.; Maze, M.; Ma, D. Xenon pretreatment attenuates anesthetic-induced apoptosis in the developing brain in comparison with nitrous oxide and hypoxia. Anesthesiology 2010, 113, 360–368. [Google Scholar]
- Hu, M.; Sun, Y.J.; Zhou, Q.G.; Auberson, Y.P.; Chen, L.; Hu, Y.; Luo, C.X.; Wu, J.Y.; Zhu, D.Y.; Li, L.X. Reduced spatial learning in mice treated with NVP-AAM077 through down-regulating neurogenesis. Eur. J. Pharmacol 2009, 622, 37–44. [Google Scholar]
- Hirasawa, T.; Wada, H.; Kohsaka, S.; Uchino, S. Inhibition of NMDA receptors induces delayed neuronal maturation and sustained proliferation of progenitor cells during neocortical development. J. Neurosci. Res 2003, 74, 676–687. [Google Scholar]
- Toriumi, K.; Mouri, A.; Narusawa, S.; Aoyama, Y.; Ikawa, N.; Lu, L.; Nagai, T.; Mamiya, T.; Kim, H.C.; Nabeshima, T. Prenatal NMDA receptor antagonism impaired proliferation of neuronal progenitor, leading to fewer glutamatergic neurons in the prefrontal cortex. Neuropsychopharmacology 2012, 37, 1387–1396. [Google Scholar]
- Tung, A.; Herrera, S.; Fornal, C.A.; Jacobs, B.L. The effect of prolonged anesthesia with isoflurane, propofol, dexmedetomidine, or ketamine on neural cell proliferation in the adult rat. Anesth. Analg 2008, 106, 1772–1777. [Google Scholar]
- Erasso, D.M.; Chaparro, R.E.; del Rio, C.E.Q.; Karlnoski, R.; Camporesi, E.M.; Saporta, S. Quantitative assessment of new cell proliferation in the dentate gyrus and learning after isoflurane or propofol anesthesia in young and aged rats. Brain Res 2011, 1441, 38–46. [Google Scholar]
- Stratmann, G.; Sall, J.W.; Bell, J.S.; Alvi, R.S.; May, L.D.V.; Ku, B.; Dowlatshahi, M.; Dai, R.; Bickler, P.E.; Russell, I.; et al. Isoflurane does not affect brain cell death, hippocampal neurogenesis, or long-term neurocognitive outcome in aged rats. Anesthesiology 2010, 112, 305–315. [Google Scholar]
- Lasarzik, I.; Winkelheide, U.; Stallmann, S.; Orth, C.; Schneider, A.; Tresch, A.; Werner, C.; Engelhard, K. Assessment of postischemic neurogenesis in rats with cerebral ischemia and propofol anesthesia. Anesthesiology 2009, 110, 529–537. [Google Scholar]
- Winkelheide, U.; Lasarzik, I.; Kaeppel, B.; Winkler, J.; Werner, C.; Kochs, E.; Engelhard, K. Dose-dependent effect of S(+) ketamine on post-ischemic endogenous neurogenesis in rats. Acta Anaesth. Scand 2009, 53, 528–533. [Google Scholar]
- Sall, J.W.; Stratmann, G.; Leong, J.; McKleroy, W.; Mason, D.; Shenoy, S.; Pleasure, S.J.; Bickler, P.E. Isoflurane inhibits growth but does not cause cell death in hippocampal neural precursor cells grown in culture. Anesthesiology 2009, 110, 826–833. [Google Scholar]
- Culley, D.J.; Boyd, J.D.; Palanisamy, A.; Xie, Z.; Kojima, K.; Vacanti, C.A.; Tanzi, R.E.; Crosby, G. Isoflurane decreases self-renewal capacity of rat cultured neural stem cells. Anesthesiology 2011, 115, 754–763. [Google Scholar]
- Crampton, S.J.; Collins, L.M.; Toulouse, A.; Nolan, Y.M.; O’Keeffe, G.W. Exposure of foetal neural progenitor cells to IL-1β impairs their proliferation and alters their differentiation—A role for maternal inflammation? J. Neurochem 2011, 120, 964–973. [Google Scholar]
- Ziv, N.E.; Smith, S.J. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 1996, 17, 91–102. [Google Scholar]
- Tan, H.; Ren, R.; Xiong, Z.; Wang, Y. Effects of ketamine and midazolam on morphology of dendritic spines in hippocampal CA1 region of neonatal mice. Chin. Med. J. (Engl. ) 2009, 122, 455–459. [Google Scholar]
- de Roo, M.; Klauser, P.; Briner, A.; Nikonenko, I.; Mendez, P.; Dayer, A.; Kiss, J.Z.; Muller, D.; Vutskits, L. Anesthetics rapidly promote synaptogenesis during a critical period of brain development. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Briner, A.; de Roo, M.; Dayer, A.; Muller, D.; Habre, W.; Vutskits, L. Volatile anesthetics rapidly increase dendritic spine density in the rat medial prefrontal cortex during synaptogenesis. Anesthesiology 2010, 112, 546–556. [Google Scholar]
- Yang, G.; Chang, P.C.; Bekker, A.; Blanck, T.J.J.; Gan, W.B. Transient effects of anesthetics on dendritic spines and filopodia in the living mouse cortex. Anesthesiology 2011, 115, 718–726. [Google Scholar]
- Kirov, S.A.; Goddard, C.A.; Harris, K.M. Age-dependence in the homeostatic upregulation of hippocampal dendritic spine number during blocked synaptic transmission. Neuropharmacology 2004, 47, 640–648. [Google Scholar]
- Briner, A.; Nikonenko, I.; de Roo, M.; Dayer, A.; Muller, D.; Vutskits, L. Developmental stage-dependent persistent impact of propofol anesthesia on dendritic spines in the rat medial prefrontal cortex. Anesthesiology 2011, 115, 282–293. [Google Scholar]
- Vutskits, L.; Gascon, E.; Tassonyi, E.; Kiss, J.Z. Clinically relevant concentrations of propofol but not midazolam alter in vitro dendritic development of isolated γ-aminobutyric acid-positive interneurons. Anesthesiology 2005, 102, 970–976. [Google Scholar]
- Vutskits, L.; Gascon, E.; Potter, G.; Tassonyi, E.; Kiss, J.Z. Low concentrations of ketamine initiate dendritic atrophy of differentiated GABAergic neurons in culture. Toxicology 2007, 234, 216–226. [Google Scholar]
- Vutskits, L.; Gascon, E.; Tassonyi, E.; Kiss, J.Z. Effect of ketamine on dendritic arbor development and survival of immature GABAergic neurons in vitro. Toxicol. Sci 2006, 91, 540–549. [Google Scholar]
- Woodall, A.J.; Naruo, H.; Prince, D.J.; Feng, Z.P.; Winlow, W.; Takasaki, M.; Syed, N.I. Anesthetic treatment blocks synaptogenesis but not neuronal regeneration of cultured Lymnaea neurons. J. Neurophysiol 2003, 90, 2232–2239. [Google Scholar]
- Turina, D.; Loitto, V.; Björnström, K.; Sundqvist, T.; Eintrei, C. Propofol causes neurite retraction in neurones. Br. J. Anaesth 2008, 101, 374–379. [Google Scholar]
- Onizuka, S.; Takasaki, M.; Syed, N.I. Long-term exposure to local but not inhalation anesthetics affects neurite regeneration and synapse formation between identified lymnaea neurons. Anesthesiology 2005, 102, 353–363. [Google Scholar]
- Ullian, E.M.; Christopherson, K.S.; Barres, B.A. Role for glia in synaptogenesis. Glia 2004, 47, 209–216. [Google Scholar]
- Dallasen, R.M.; Bowman, J.D.; Xu, Y.; Hashimoto, M.; Katakura, M.; Nabika, T.; Tanabe, Y.; Hossain, S.; Tsuchikura, S.; Shido, O. Isoflurane does not cause neuroapoptosis but reduces astroglial processes in young adult mice. Med. Gas. Res 2011, 1. [Google Scholar] [CrossRef]
- Lunardi, N.; Hucklenbruch, C.; Latham, J.R.; Scarpa, J.; Jevtovic-Todorovic, V. Isoflurane impairs immature astroglia development in vitro: The role of actin cytoskeleton. J. Neuropathol. Exp. Neurol 2011, 70, 281–291. [Google Scholar]
- Goldberg, E.M.; Jeong, H.Y.; Kruglikov, I.; Tremblay, R.; Lazarenko, R.M.; Rudy, B. Rapid developmental maturation of neocortical FS cell intrinsic excitability. Cereb. Cortex 2011, 21, 666–682. [Google Scholar]
- Borsook, D.; George, E.; Kussman, B.; Becerra, L. Anesthesia and perioperative stress: Consequences on neural networks and postoperative behaviors. Prog. Neurobiol 2010, 92, 601–612. [Google Scholar]
- Shih, J.; May, L.D.V.; Gonzalez, H.E.; Lee, E.W.; Alvi, R.S.; Sall, J.W.; Rau, V.; Bickler, P.E.; Lalchandani, G.R.; Yusupova, M. Delayed environmental enrichment reverses sevoflurane-induced memory impairment in rats. Anesthesiology 2012, 116, 586–602. [Google Scholar]
- Konishi, Y.; Chui, D.H.; Hirose, H.; Kunishita, T.; Tabira, T. Trophic effect of erythropoietin and other hematopoietic factors on central cholinergic neurons in vitro and in vivo. Brain Res 1993, 609, 29–35. [Google Scholar]
- Tsuchimoto, T.; Ueki, M.; Miki, T.; Morishita, J.; Maekawa, N. Erythropoietin attenuates isoflurane-induced neurodegeneration and learning deficits in the developing mouse brain. Pediatr. Anesth 2011, 21, 1209–1213. [Google Scholar]
- Tanaka, T.; Kai, S.; Koyama, T.; Daijo, H.; Adachi, T.; Fukuda, K.; Hirota, K. General anesthetics inhibit erythropoietin Induction under Hypoxic Conditions in the mouse brain. PLoS One 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Liang, G.; Yang, H. Isoflurane preconditioning inhibited isoflurane-induced neurotoxicity. Neurosci. Lett 2007, 425, 59–62. [Google Scholar]
- Ma, D.; Williamson, P.; Januszewski, A.; Nogaro, M.C.; Hossain, M.; Ong, L.P.; Shu, Y.; Franks, N.P.; Maze, M. Xenon mitigates isoflurane-induced neuronal apoptosis in the developing rodent brain. Anesthesiology 2007, 106, 746–753. [Google Scholar]
- Cattano, D.; Williamson, P.; Fukui, K.; Avidan, M.; Evers, A.S.; Olney, J.W.; Young, C. Potential of xenon to induce or to protect against neuroapoptosis in the developing mouse brain. Can. J. Anesth 2008, 55, 429–436. [Google Scholar]
- Ieraci, A.; Herrera, D.G. Nicotinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS Med 2006, 3. [Google Scholar] [CrossRef]
- Ullah, N.; Ullah, I.; Lee, H.Y.; Naseer, M.I.; Seok, P.M.; Ahmed, J.; Kim, M.O. Protective function of nicotinamide against ketamine-induced apoptotic neurodegeneration in the infant rat brain. J. Mol. Neurosci 2012, 47, 67–75. [Google Scholar]
- Tome, C.; Bauer, C.; Nottingham, C.; Smith, C.; Blackstone, K.; Brown, L.; Hlavaty, C.; Nelson, C.; Daker, R.; Sola, R. MK801-induced caspase-3 in the postnatal brain: Inverse relationship with calcium binding proteins. Neuroscience 2006, 141, 1351–1363. [Google Scholar]
- Naseer, M.; Ullah, N.; Ullah, I.; Koh, P.; Lee, H.; Park, M.; Kim, M. Vitamin C protects against ethanol and PTZ-induced apoptotic neurodegeneration in prenatal rat hippocampal neurons. Synapse 2011, 65, 562–571. [Google Scholar]
- Men’shanov, P.N.; Bannova, A.V.; Il’inykh, F.A.; Dygalo, N.N. Negative regulation of caspase-3 expression in the neonatal cerebral cortex by alpha2A-adrenoceptors. Bull. Exp. Biol. Med 2007, 143, 277–279. [Google Scholar]
- Laudenbach, V.; Mantz, J.; Lagercrantz, H.; Desmonts, J.M.; Evrard, P.; Gressens, P. Effects of α2-adrenoceptor agonists on perinatal excitotoxic brain injury: Comparison of clonidine and dexmedetomidine. Anesthesiology 2002, 96, 134–141. [Google Scholar]
- Dygalo, N.N.; Bannova, A.V.; Kalinina, T.S.; Shishkina, G.T. Clonidine increases caspase-3 mRNA level and DNA fragmentation in the developing rat brainstem. Brain Res. Dev. Brain Res 2004, 152, 225–231. [Google Scholar]
- Liu, X.S.; Xue, Q.; Zeng, Q.W.; Li, Q.; Liu, J.; Feng, X.M.; Yu, B.W. Sevoflurane impairs memory consolidation in rats, possibly through inhibiting phosphorylation of glycogen synthase kinase-3[beta] in the hippocampus. Neurobiol. Learn Mem 2010, 94, 461–467. [Google Scholar]
- Shin, W.J.; Gwak, M.; Baek, C.H.; Kim, K.S.; Park, P.H. Neuroprotective effects of lithium treatment following hypoxic–ischemic brain injury in neonatal rats. Childs Nerv. Syst 2012, 28, 1–8. [Google Scholar]
- Tan, W.F.; Cao, X.Z.; Wang, J.K.; Lv, H.W.; Wu, B.Y.; Ma, H. Protective effects of lithium treatment for spatial memory deficits induced by tau hyperphosphorylation in splenectomized rats. Clin. Exp. Pharmacol. Physiol 2010, 37, 1010–1015. [Google Scholar]
- Zhao, L.D.; Wang, F.Z.; Gui, B.; Hua, F.Z.; Qian, Y.N. Prophylactic lithium alleviates postoperative cognition impairment by phosphorylating hippocampal glycogen synthase kinase-3β (Ser9) in aged rats. Exp. Gerontol 2011, 46, 1031–1036. [Google Scholar]
- Leyhe, T.; Eschweiler, G.W.; Stransky, E.; Gasser, T.; Annas, P.; Basun, H.; Laske, C. Increase of BDNF serum concentration in lithium treated patients with early Alzheimer’s disease. J. Alzheimers. Dis 2009, 16, 649–656. [Google Scholar]
- Kumral, A.; Yesilirmak, D.C.; Sonmez, U.; Baskin, H.; Tugyan, K.; Yilmaz, O.; Genc, S.; Gokmen, N.; Genc, K.; Duman, N. Neuroprotective effect of the peptides ADNF-9 and NAP on hypoxic-ischemic brain injury in neonatal rats. Brain. Res 2006, 1115, 169–178. [Google Scholar]
- Yon, J.H.; Carter, L.B.; Reiter, R.J.; Jevtovic-Todorovic, V. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol. Dis 2006, 21, 522–530. [Google Scholar]
- Cetinkaya, M.; Alkan, T.; Ozyener, F.; Kafa, I.; Kurt, M.; Koksal, N. Possible neuroprotective effects of magnesium sulfate and melatonin as both pre-and post-treatment in a neonatal hypoxic-ischemic rat model. Neonatology 2011, 99, 302–310. [Google Scholar]
- Zou, X.; Sadovova, N.; Patterson, T.; Divine, R.; Hotchkiss, C.; Ali, S.; Hanig, J.; Paule, M.; Slikker, W., Jr; Wang, C. The effects of l-carnitine on the combination of inhalation anesthetic-induced developmental, neuronal apoptosis in the rat frontal cortex. Neuroscience 2008, 151, 1053–1065. [Google Scholar]
- Scafidi, S.; Racz, J.; Hazelton, J.; McKenna, M.C.; Fiskum, G. Neuroprotection by acetyl-l-carnitine after traumatic injury to the immature rat brain. Dev. Neurosci 2010, 32, 480–487. [Google Scholar]
- Boscolo, A.; Starr, J.A.; Sanchez, V.; Lunardi, N.; DiGruccio, M.R.; Ori, C.; Erisir, A.; Trimmer, P.; Bennett, J.; Jevtovic-Todorovic, V. The abolishment of anesthesia-induced cognitive impairment by timely protection of mitochondria in the developing rat brain: The importance of free oxygen radicals and mitochondrial integrity. Neurobiol. Dis 2012, 45, 1031–1041. [Google Scholar]
- Vizcaychipi, M.P.; Xu, L.; Barreto, G.E.; Ma, D.; Maze, M.; Giffard, R.G. Heat shock protein 72 overexpression prevents early postoperative memory decline after orthopedic surgery under general anesthesia in mice. Anesthesiology 2011, 114, 891–900. [Google Scholar]
- Zhang, J.; Zhou, W.; Qiao, H. Bioenergetic homeostasis decides neuroprotection or neurotoxicity induced by volatile anesthetics: A uniform mechanism of dual effects. Med. Hypotheses 2011, 77, 223–229. [Google Scholar]
- Berns, M.; Zacharias, R.; Seeberg, L.; Schmidt, M.; Kerner, T. Effects of sevoflurane on primary neuronal cultures of embryonic rats. Eur. J. Anaesth 2009, 26, 597–602. [Google Scholar]
- Palanisamy, A.; Baxter, M.G.; Keel, P.K.; Xie, Z.; Crosby, G.; Culley, D.J. Rats exposed to isoflurane in utero during early gestation are behaviorally abnormal as adults. Anesthesiology 2011, 114, 521–528. [Google Scholar]
- Sprung, J.; Flick, R.P.; Katusic, S.K.; Colligan, R.C.; Barbaresi, W.J.; Bojanić, K.; Welch, T.L.; Olson, M.D.; Hanson, A.C.; Schroeder, D.R. Attention-deficit/hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clin. Proc 2012, 87, 120–129. [Google Scholar]
- Meyer, P.; Langlois, C.; Soėte, S.; Leydet, J.; Echenne, B.; Rivier, F.; Bonafé, A.; Roubertie, A. Unexpected neurological sequelae following propofol anesthesia in infants: Three case reports. Brain Dev 2010, 32, 872–878. [Google Scholar]
- Felderhoff-Mueser, U.; Bittigau, P.; Sifringer, M.; Jarosz, B.; Korobowicz, E.; Mahler, L.; Piening, T.; Moysich, A.; Grune, T.; Thor, F. Oxygen causes cell death in the developing brain. Neurobiol. Dis 2004, 17, 273–282. [Google Scholar]
- Xie, Z.; Moir, R.D.; Romano, D.M.; Tesco, G.; Kovacs, D.M.; Tanzi, R.E. Hypocapnia induces caspase-3 activation and increases Aβ production. Neurodegener. Dis 2004, 1, 29–37. [Google Scholar]
- Morgan, P.G.; Sedensky, M. A new phase in anesthetic-induced neurotoxicity research. Anesthesiology 2011, 114, 10–11. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lei, X.; Guo, Q.; Zhang, J. Mechanistic Insights into Neurotoxicity Induced by Anesthetics in the Developing Brain. Int. J. Mol. Sci. 2012, 13, 6772-6799. https://doi.org/10.3390/ijms13066772
Lei X, Guo Q, Zhang J. Mechanistic Insights into Neurotoxicity Induced by Anesthetics in the Developing Brain. International Journal of Molecular Sciences. 2012; 13(6):6772-6799. https://doi.org/10.3390/ijms13066772
Chicago/Turabian StyleLei, Xi, Qihao Guo, and Jun Zhang. 2012. "Mechanistic Insights into Neurotoxicity Induced by Anesthetics in the Developing Brain" International Journal of Molecular Sciences 13, no. 6: 6772-6799. https://doi.org/10.3390/ijms13066772