Anti-TNF-α Activity of Portulaca oleracea in Vascular Endothelial Cells

Abstract
:1. Introduction
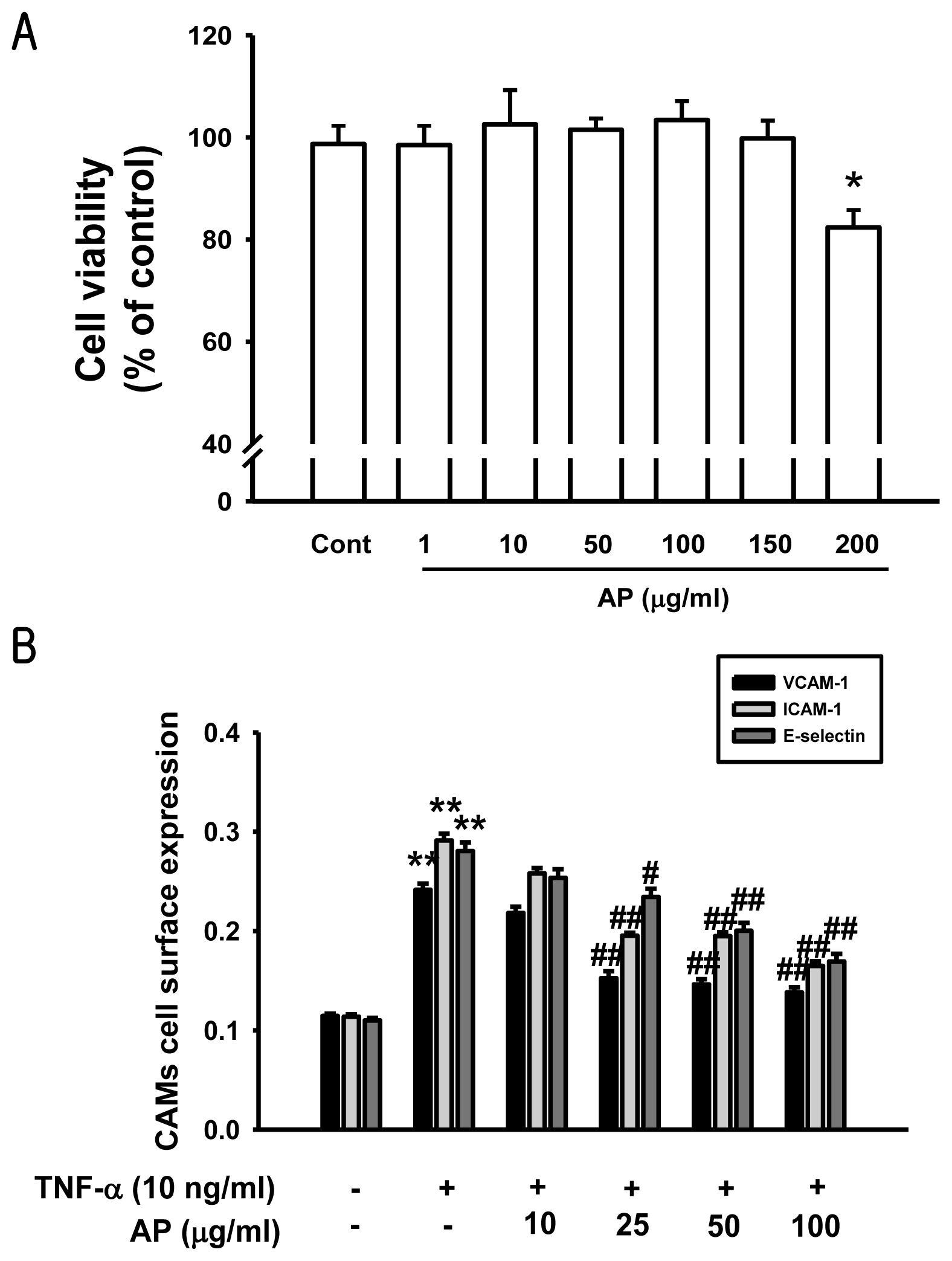
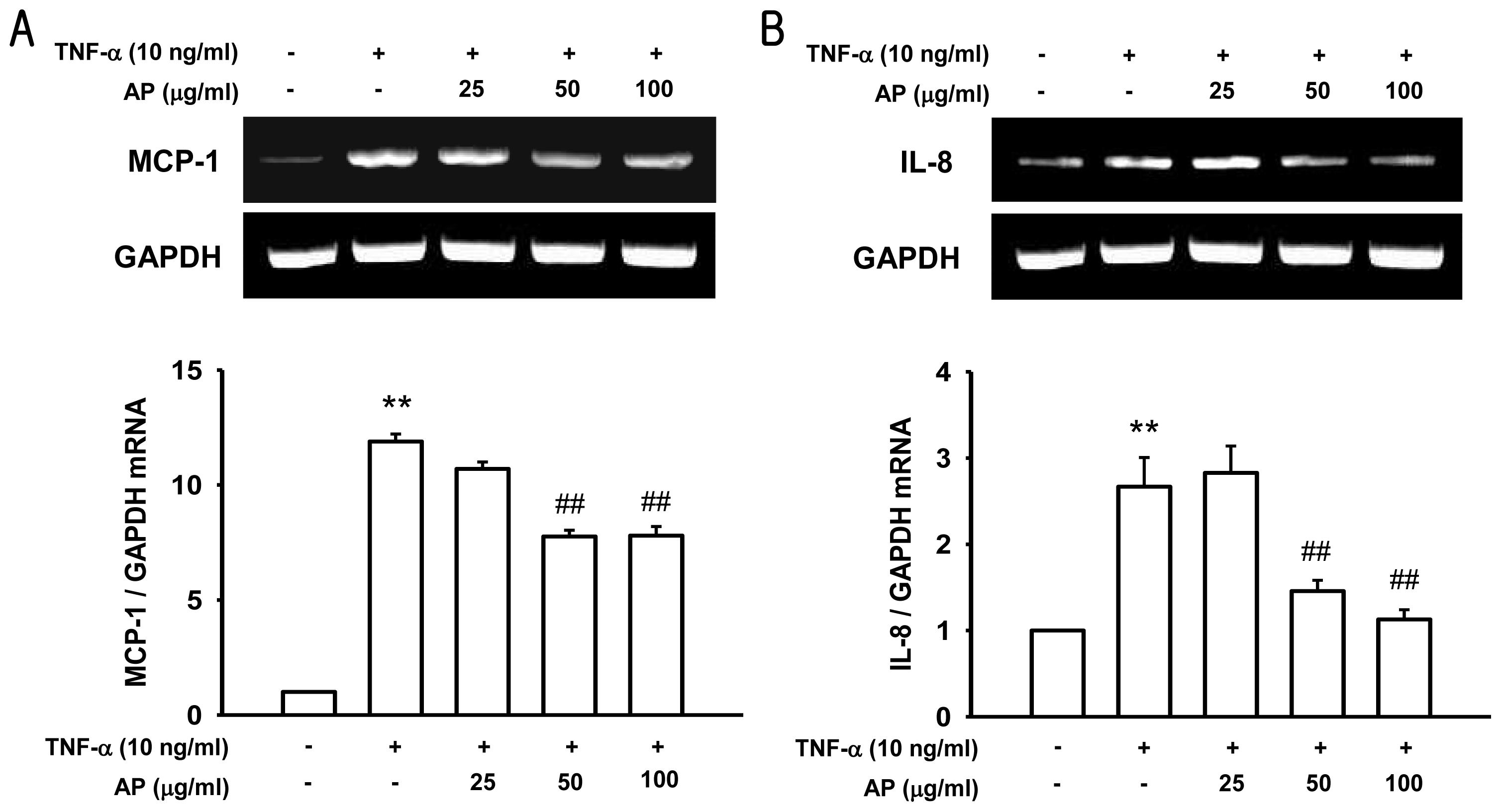
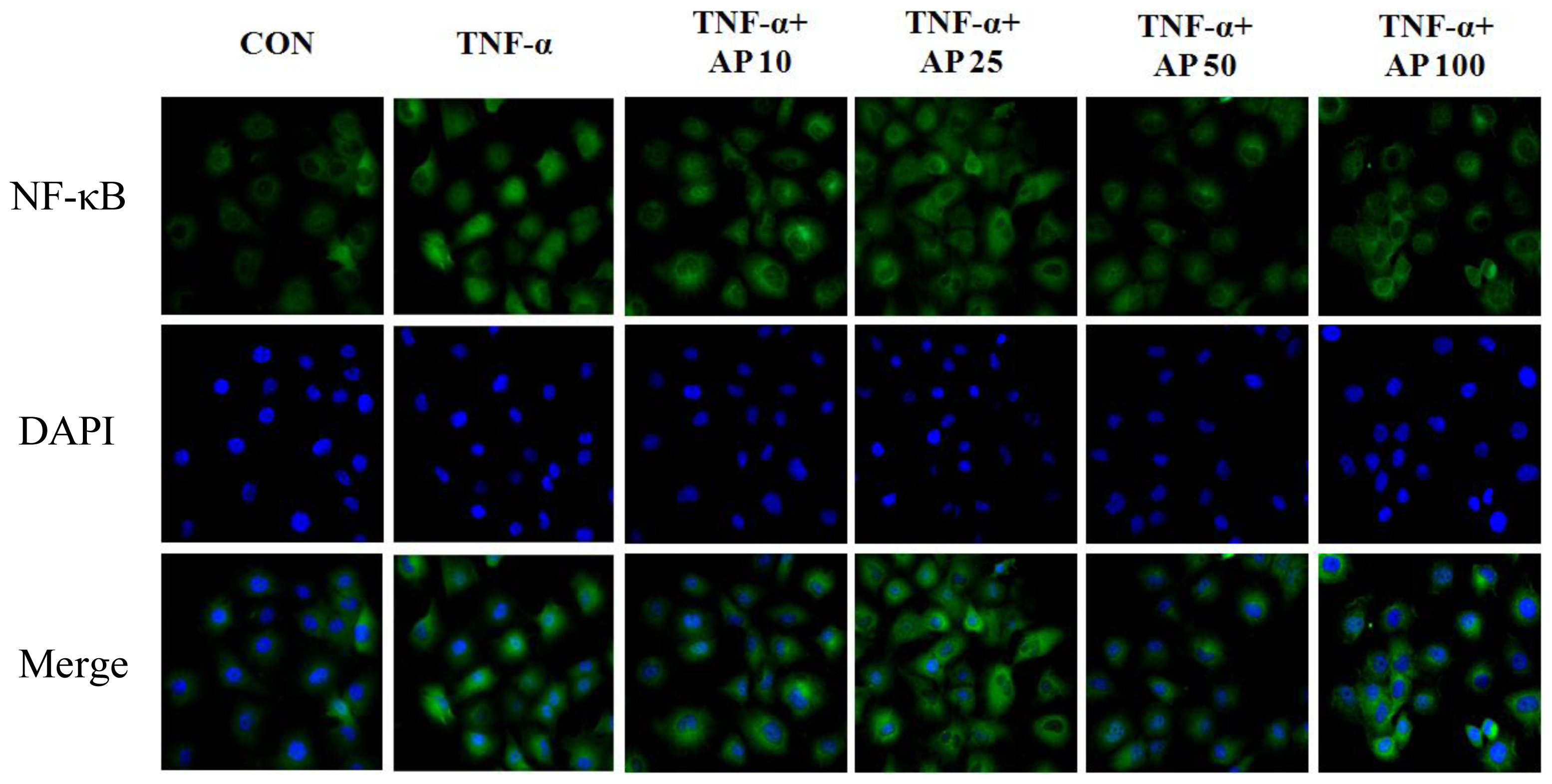
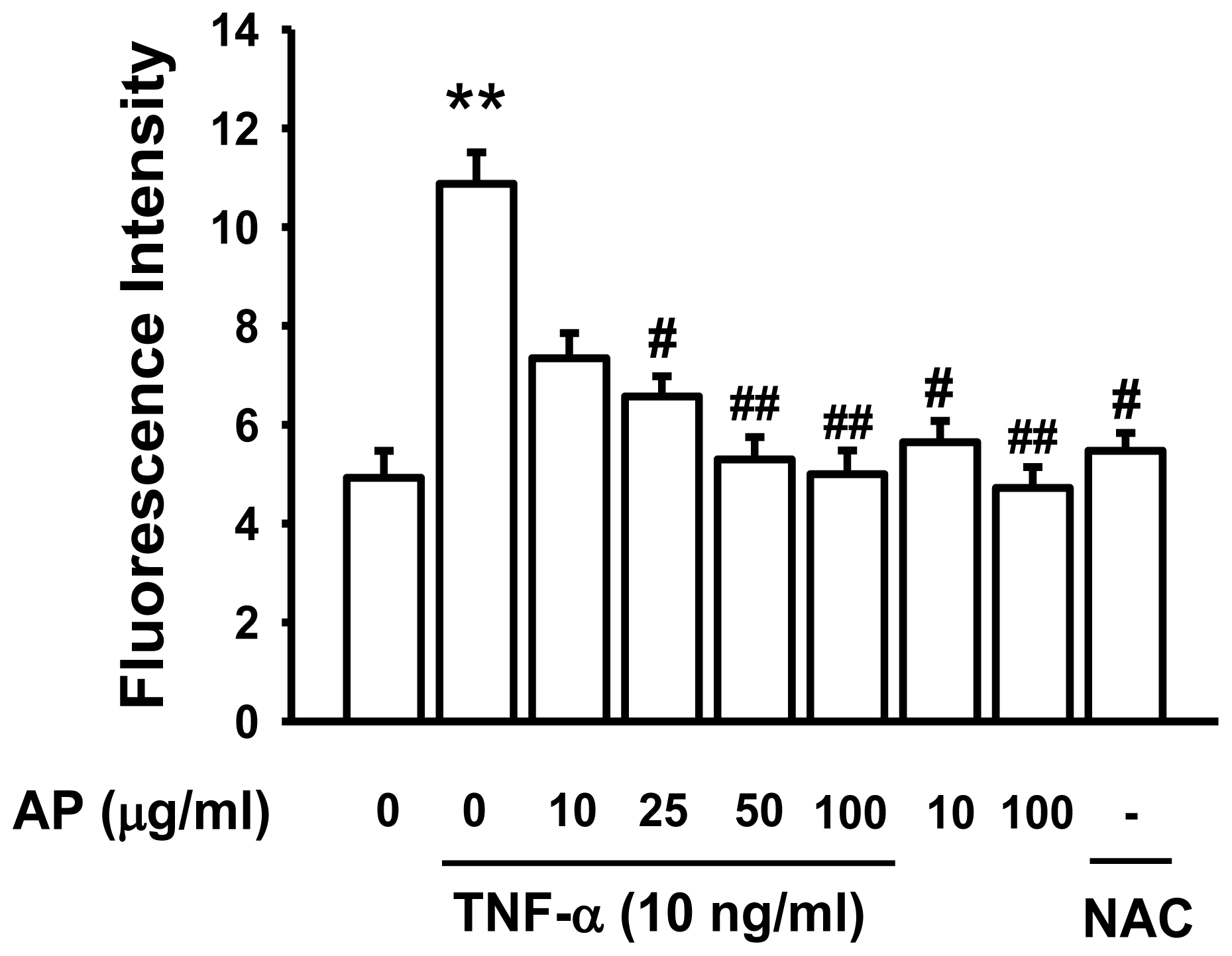
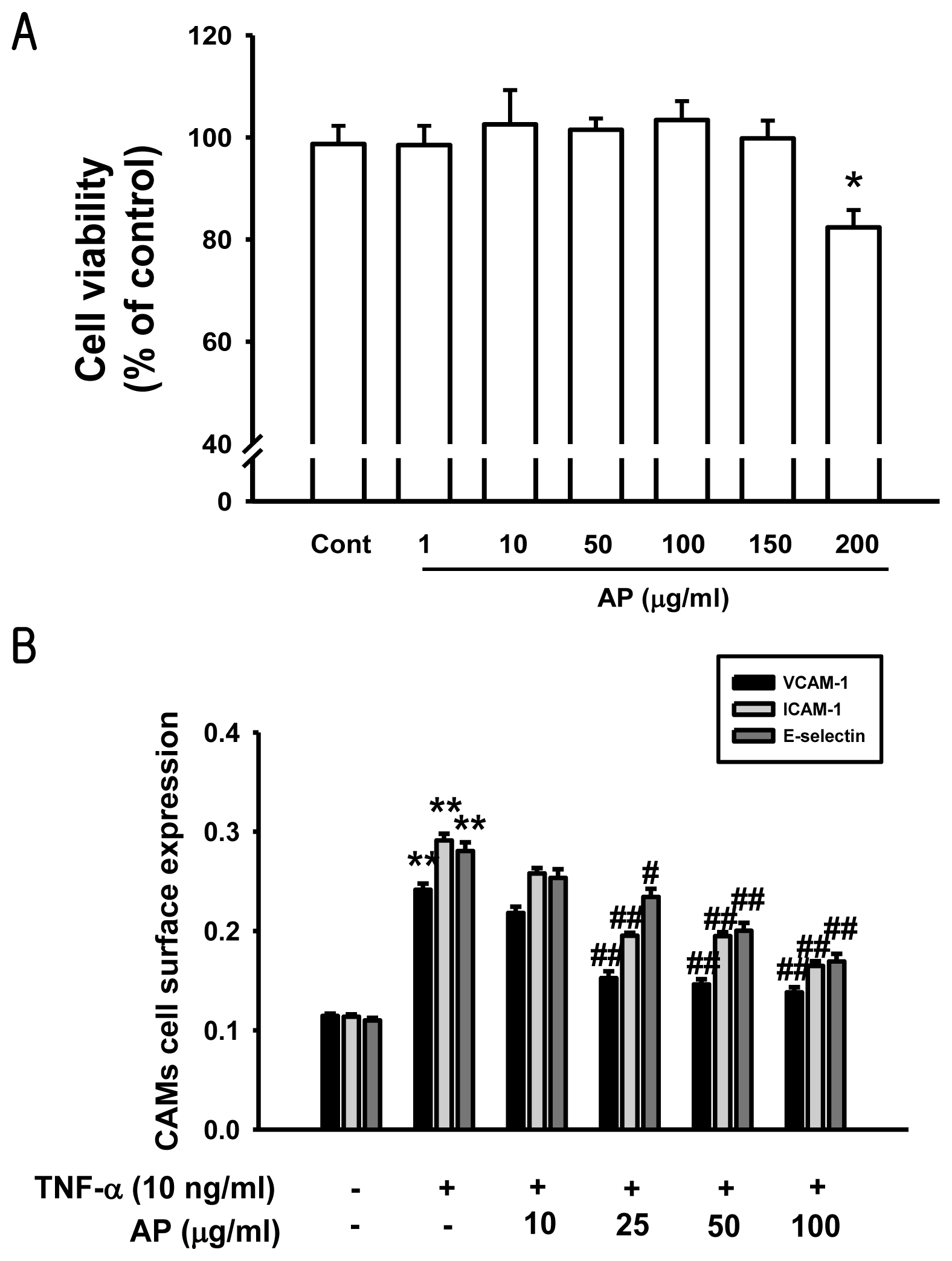
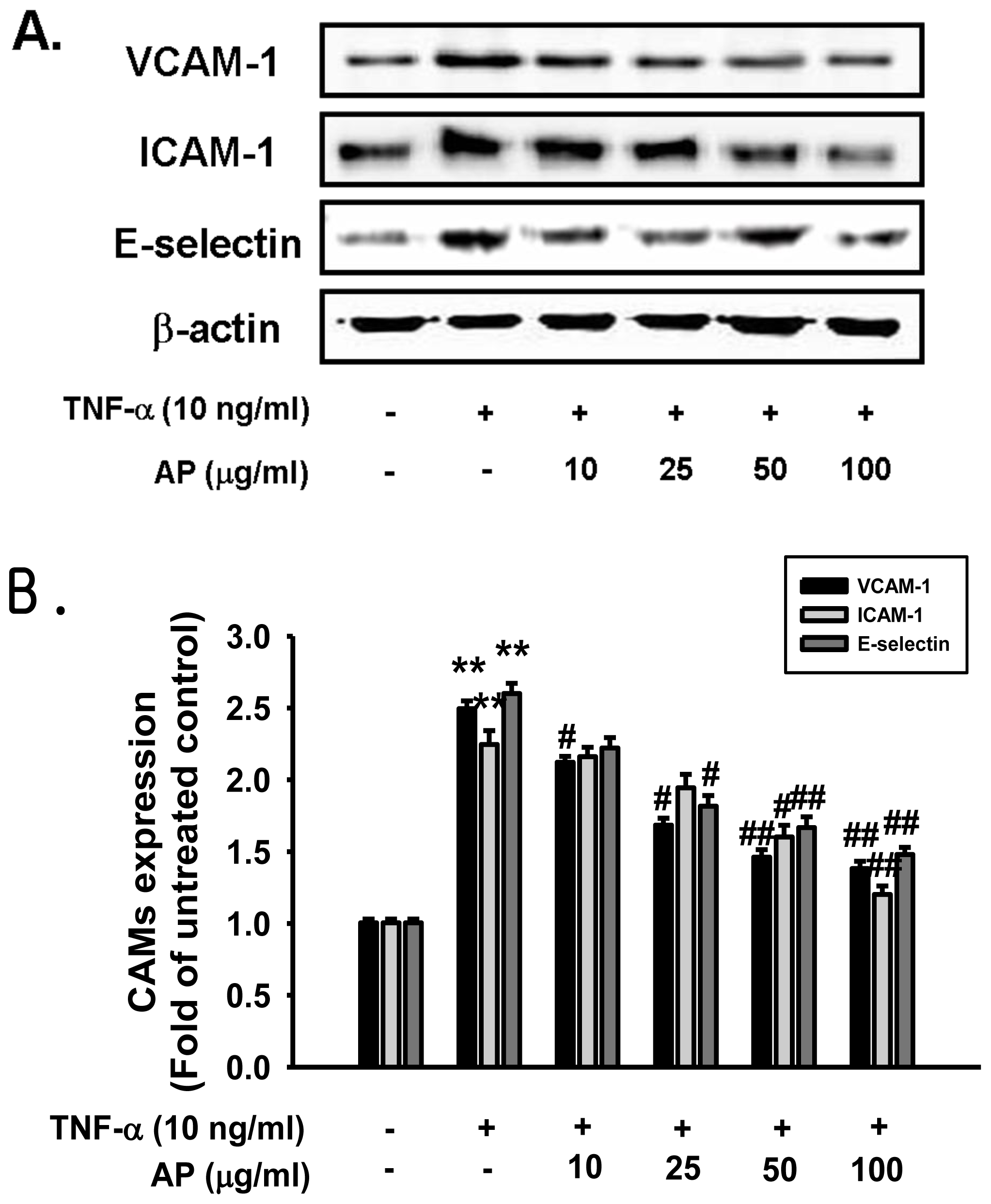
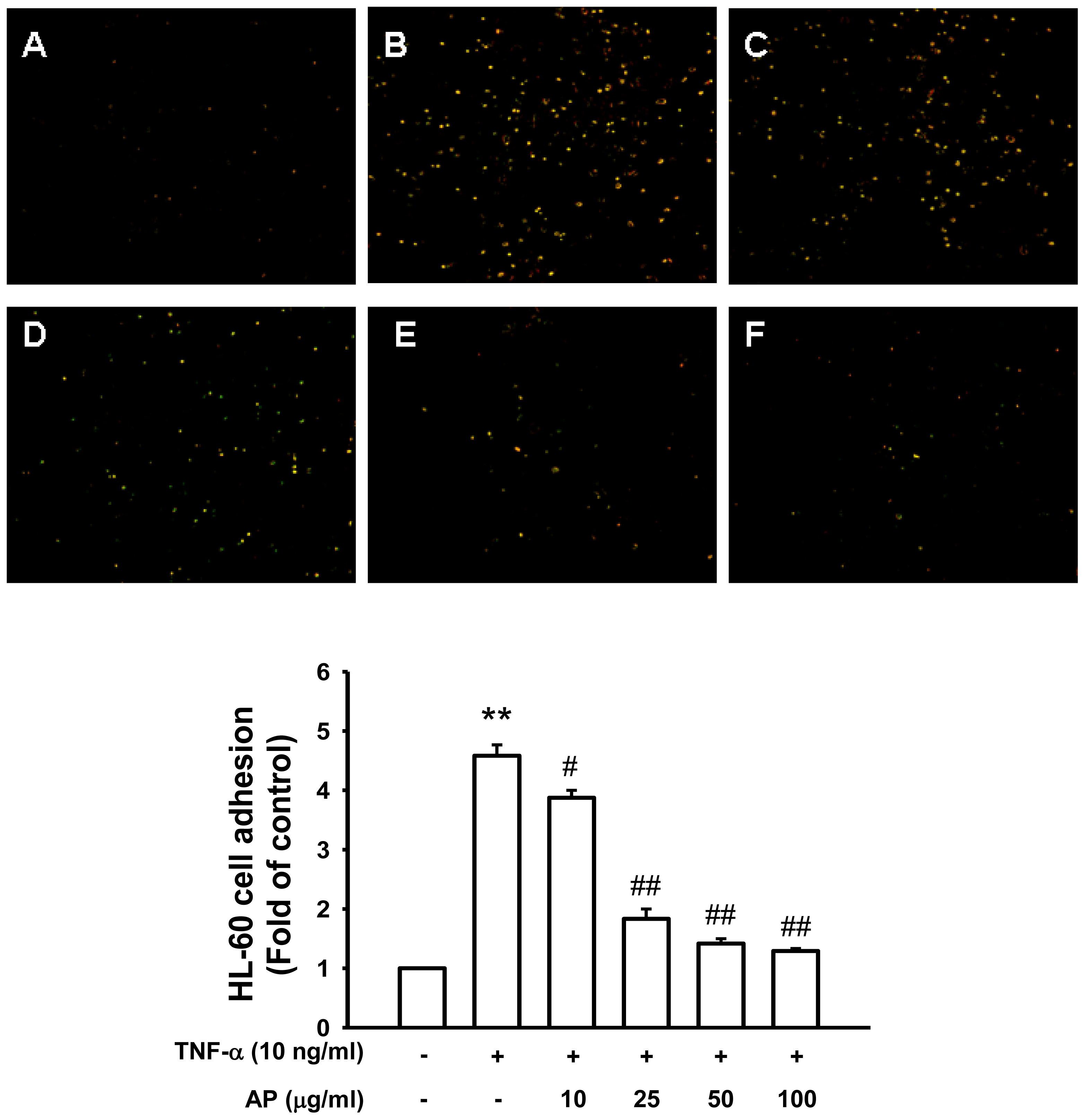
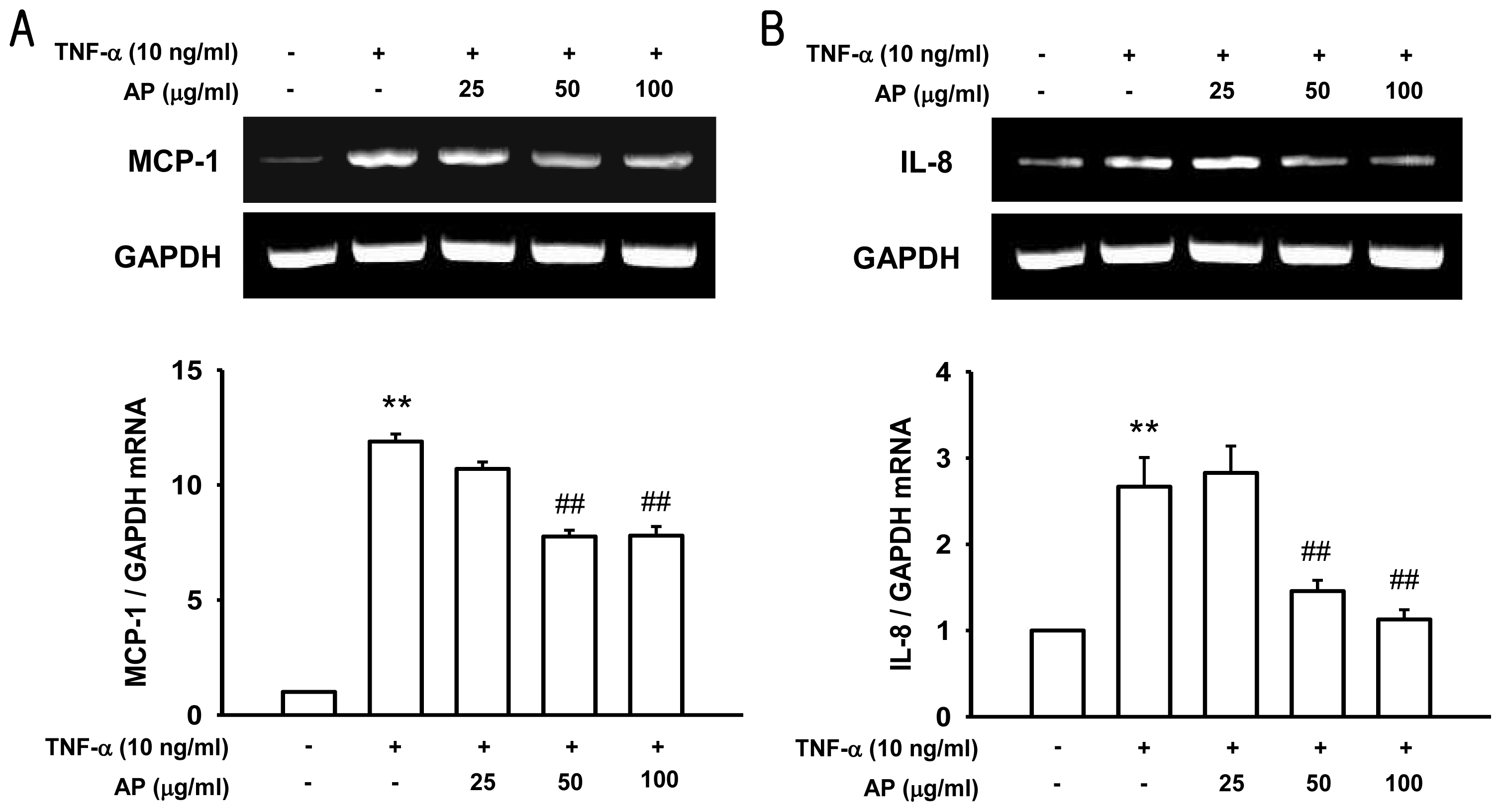
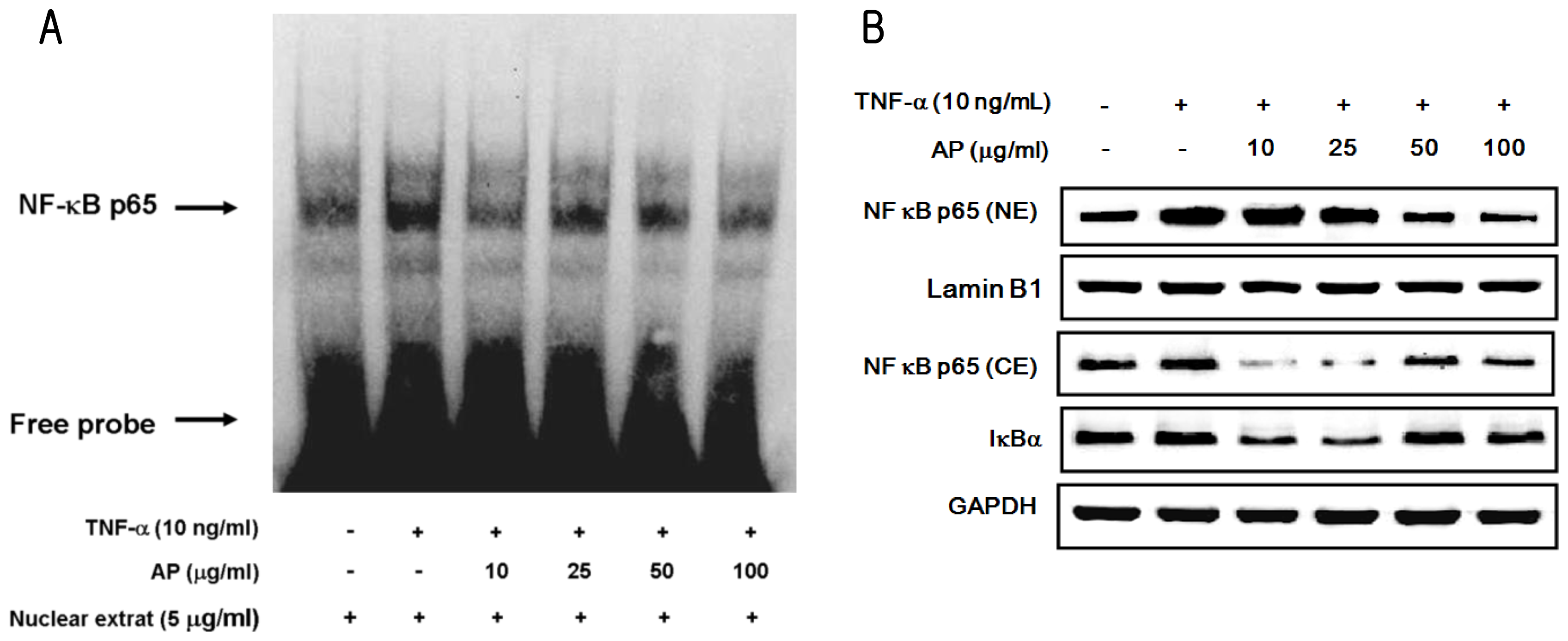
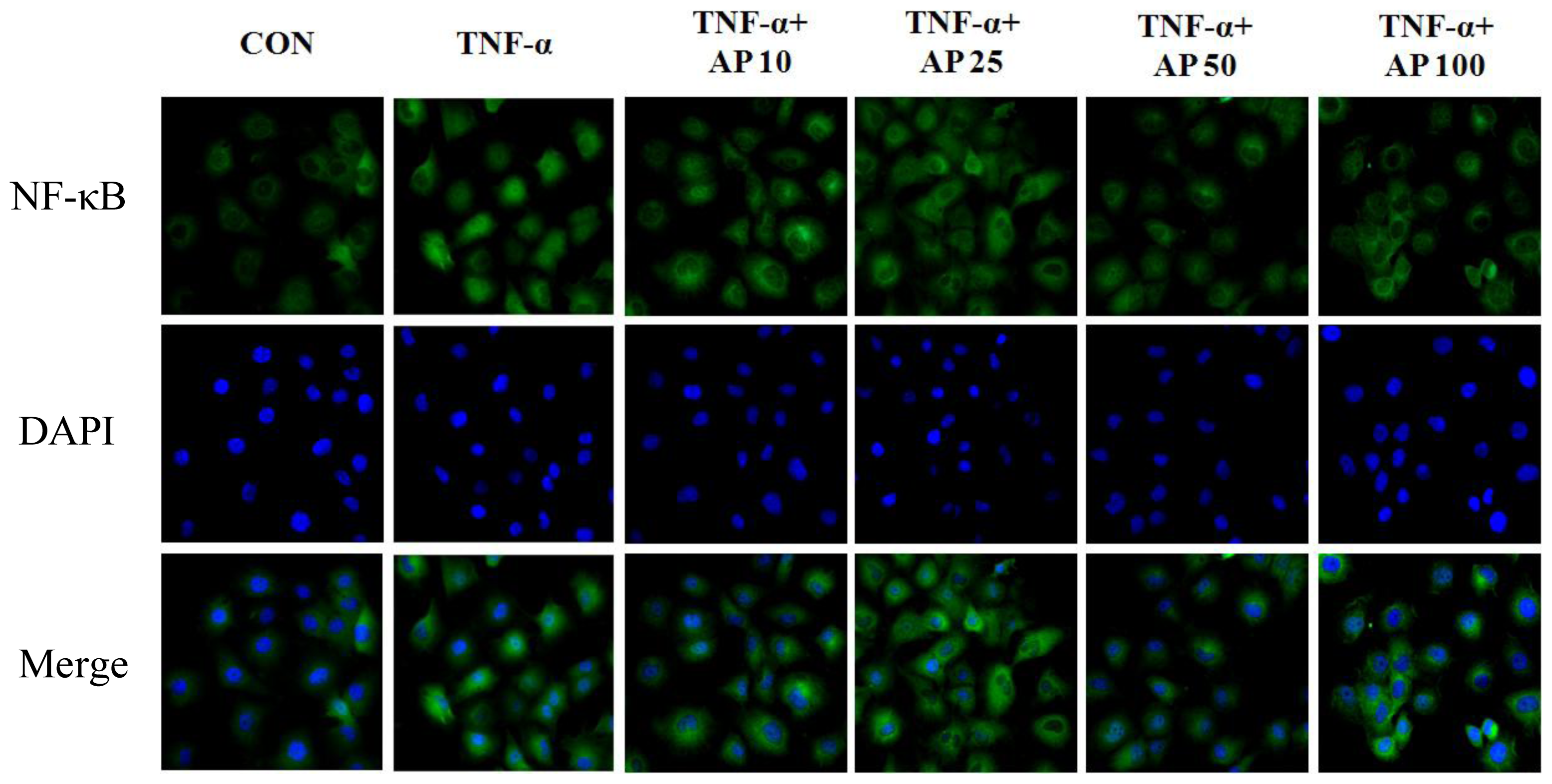
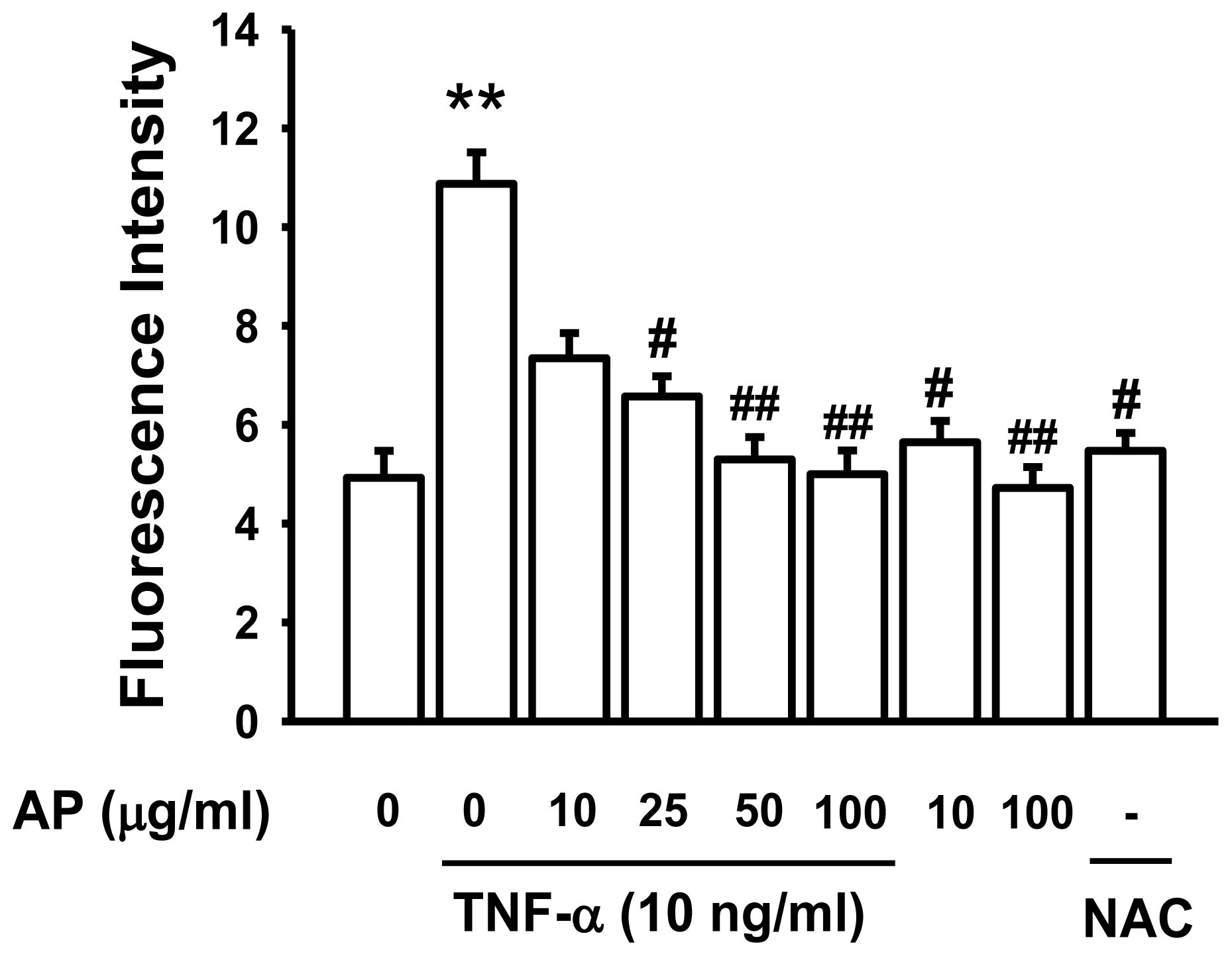
2. Results and Discussion
3. Experimental Section
3.1. Extraction of Portulaca oleracea
3.2. Cell Culture
3.3. Determination of Cell Based ELISA
3.4. Monocyte-Endothelial Cell Adhesion Assay
3.5. Preparation of Cytoplasmic and Nuclear Extracts
3.6. Protein Extraction and Western Blot Analysis
3.7. Electrophoretic Mobility Shift Assay
3.8. RNA Isolation and RT-PCR Analysis
3.9. Immunofluorescence Microscopy
3.10. Intracellular ROS Production Assay
3.11. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Libby, P.; Sukhova, G.; Lee, R.T.; Galis, Z.S. Cytokines regulate vascular functions related to stability of the atherosclerotic plaque. J. Cardiovasc. Pharmacol 1995, 2, 9–12. [Google Scholar]
- Ross, R. Atherosclerosis: An inflammatory disease. N. Engl. J. Med 1999, 340, 115–126. [Google Scholar]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; da Ros, R.; Maier, A.; Zuodar, G.; Ceriello, A. Intermittent HG enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical vein endothelial cells in culture: The distinct role of protein kinase C and mitochondrial superoxide production. Atherosclerosis 2005, 183, 259–267. [Google Scholar]
- Minami, T.; Aird, W.C. Endothelial cell gene regulation. Trends Cardiovasc. Med 2005, 15, 174–184. [Google Scholar]
- Madge, L.A.; Pober, J.S. TNF signaling in vascular endothelial cells. Exp. Mol. Pathol 2001, 70, 317–325. [Google Scholar]
- Ahmad, M.; Zhang, Y.; Parpharalambus, C.; Alexander, R.W. Role of isoprenylcysteine carboxyl methyltransferase in tumor necrosis factor-α stimulation of expression of vascular cell adhesion molecule-1 in ECs. Arterioscler. Thromb. Vasc. Biol 2001, 22, 759–764. [Google Scholar]
- Ramana, K.V.; Bhatnagar, A.; Srivastava, S.K. Inhibition of aldose reductase attenuates TNF-α-induced expression of adhesion molecules in endothelial cells. FASEB J 2004, 18, 1209–1218. [Google Scholar]
- Lockyer, J.M.; Colladay, J.S.; Alprin-Lea, W.L.; Hammond, T.; Buda, A.J. Inhibition of nuclear factor κB-mediated adhesion molecule expression in human endothelial cells. Circ. Res 1998, 82, 314–320. [Google Scholar]
- Lee, Y.J.; Kang, D.G.; Kim, J.S.; Lee, H.S. Lycopus lucidus inhibits high glucose-induced vascular inflammation in human umbilical vein endothelial cells. Vascul. Pharmacol 2008, 48, 38–46. [Google Scholar]
- Kim, H.J.; Park, K.G.; Yoo, E.K.; Kim, Y.H.; Kim, Y.N.; Kim, H.S.; Kim, H.T.; Park, J.Y.; Lee, K.U.; Jang, W.G.; et al. Effects of PGC-1α on TNF-α-induced MCP-1 and VCAM-1 expression and NF-κB activation in human aortic smooth muscle and endothelial cells. Antioxid. Redox. Signal 2007, 9, 301–307. [Google Scholar]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol 2003, 91, 7A–11A. [Google Scholar]
- Chen, C.C.; Chow, M.P.; Huang, W.C.; Lin, Y.C.; Chang, Y.J. Flavonoids inhibit tumor necrosis factor-α-induced up-regulation of intercellular adhesion molecule-1 (ICAM-1) in respiratory epithelial cells through activator protein-1 and nuclear factor-κB: Structure-activity relationships. Mol. Pharmacol 2004, 66, 683–693. [Google Scholar]
- Yoon, J.J.; Lee, Y.J.; Kim, J.S.; Kang, D.G.; Lee, H.S. Protective role of betulinic acid on TNF-α-induced cell adhesion molecules in vascular endothelial cells. Biochem. Biophys. Res. Commun 2010, 391, 96–101. [Google Scholar]
- Mohanapriya, S.; Senthilkumar, P.; Sivakumar, S.; Dineshkumar, M.; Subbhuraam, C.V. Effects of copper sulfate and copper nitrate in aquatic medium on the restoration potential and accumulation of copper in stem cuttings of the terrestrial medicinal plant, Portulaca oleracea Linn. Environ. Monit. Assess 2006, 121, 233–244. [Google Scholar]
- Rasheed, A.N.; Afifi, F.U.; Shaedah, M.; Taha, M.O. Investigation of the active constituents of Portulaca oleracea L. (Portulacaceae) growing in Jordan. Pak. J. Pharm. Sci 2004, 17, 37–45. [Google Scholar]
- Zhang, X.J.; Ji, Y.B.; Qu, Z.h.Y.; Xia, J.C.h.; Wang, L. Experimental studies on antibiotic functions of Portulaca oleracea L. in vitro. Chin. J. Microecol 2002, 14, 277–280. [Google Scholar]
- Chan, K.; Islam, M.W.; Kamil, M.; Radhakrishnan, R.; Zakaria, M.N.M.; Habibullah, M.; Attas, A. The analgesic and anti-inflammatory effects of Portulaca oleracea L. subsp. sativa (Haw.) Celak. J. Ethnopharmacol 2000, 73, 445–451. [Google Scholar]
- Parry, O.; Marks, J.A.; Okwuasaba, F. The skeletal muscle relaxant action of Portulaca oleracea: Role of potassium ions. J. Ethnopharmacol 1993, 40, 187–194. [Google Scholar]
- Rasheed, A.N.; Afifi, F.U.; Disi, A.M. Simple evaluation of the wound healing activity of a crude extract of Portulaca oleracea L. (growing in Jordan) in Mus musculus JVI-1. J. Ethnopharmacol 2003, 88, 131–136. [Google Scholar]
- Awad, N.E. Lipid content and antimicrobial activity of phenolic constituents of cultivated Portulaca oleracea L. Bull. Fac. Pharm 1994, 32, 137–142. [Google Scholar]
- Sakai, N.; Inada, K.; Okamoto, M.; Shizuri, Y.; Fukuyama, Y.; Portuloside, A. a monoterpene glucoside from Portulaca oleracea. Phytochemistry 1996, 42, 1625–1628. [Google Scholar]
- Liu, L.; Howe, P.; Zhou, Y.F.; Xu, Z.Q.; Hocart, C.; Zhan, R. Fatty acids and beta-carotene in australian purslane (Portulaca oleracea) varieties. J. Chromatogr. A 2000, 893, 207–213. [Google Scholar]
- Che, W.; Lerner-Marmarosh, N.; Huang, Q.; Osawa, M.; Ohta, S.; Yoshizumi, M. Insulin-like growth factor-1 enhances inflammatory responses in ECs: Role of Gab1 and MEKK3 in TNF-α-induced c-Jun and NF-κB activation and adhesion molecule expression. Circ. Rec 2002, 90, 1222–1230. [Google Scholar]
- Abou-Raya, A.; Abou-Raya, S. Inflammation: A pivotal link between autoimmune diseases and atherosclerosis. Autoimmun. Rev 2006, 5, 331–337. [Google Scholar]
- Sneddon, A.A.; McLeod, E.; Wahle, K.W.; Arthur, J.R. Cytokine-induced monocyte adhesion to endothelial cells involves platelet-activating factor: Suppression by conjugated linoleic acid. Biochim. Biophys. Acta 2006, 1761, 793–801. [Google Scholar]
- Lee, D.K.; Nathan; Grantham, R.; Trachte, A.L.; Mannion, J.D.; Wilson, C.L. Activation of the canonical Wnt/beta-catenin pathway enhances monocyte adhesion to endothelial cells. Biochem. Biophys. Res. Commun 2006, 347, 109–116. [Google Scholar]
- Gerszten, R.E.; Garcia-Zepeda, E.A.; Lim, Y.C. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999, 398, 718–723. [Google Scholar]
- Yang, Y.Y.; Hu, C.J.; Chang, S.M.; Tai, T.Y.; Leu, S.J. Aspirin inhibits monocyte chemoattractant protein-1 and interleukin-8 expression in TNF-α stimulated human umbilical vein endothelial cells. Atherosclerosis 2004, 174, 207–213. [Google Scholar]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-κB and cytokine inducible enhancers. FASEB J 1995, 9, 899–909. [Google Scholar]
- Murase, T.; Kume, N.; Hase, T.; Shibuya, Y.; Nishizawa, Y.; Tokimitsu, I.; Kita, T. Gallates inhibit cytokine-induced nuclear translocation of NF-κB and expression of leukocyte adhesion molecules in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol 1999, 19, 1412–1420. [Google Scholar]
- Kanters, E.; Pasparakis, M.; Gijbels, M.J.; Vergouwe, M.N.; Partouns-Hendriks, I.; Fijneman, R.J.; Clausen, B.E.; Förster, I.; Kockx, M.M.; Rajewsky, K.; et al. Inhibition of NF-κB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J. Clin. Invest 2003, 112, 1176–1185. [Google Scholar]
- Trivedi, C.M.; Patel, R.C.; Patel, C.V. Homeobox gene HOXA9 inhibits nuclear factor-κB dependent activation of endothelium. Atherosclerosis 2007, 195, e50–e60. [Google Scholar]
- Hajra, L.; Evans, A.I.; Chen, M.; Hyduk, S.J.; Collins, T.; Cybulsky, M.I. The NF-κB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. USA 2000, 97, 9052–9057. [Google Scholar]
- Kumar, S.; Sharma, A.; Madan, B.; Singhal, V.; Ghosh, B. Isoliquiritigenin inhibits IκB kinase activity and ROS generation to block TNF-α induced expression of cell adhesion molecules on human endothelial cells. Biochem. Pharmacol 2007, 73, 1602–1612. [Google Scholar]
- Bonizzi, G.; Piette, J.; Merville, M.P.; Bours, V. Cell type-specific role for reactive oxygen species in nuclear factor-κB activation by interleukin-1. Biochem. Pharmacol 2000, 59, 7–11. [Google Scholar]
- Schoonbroodt, S.; Piette, J. Oxidative stress interference with the nuclear factor-κB activation pathways. Biochem. Pharmacol 2000, 60, 1075–1083. [Google Scholar]
- Calixto, J.B.; Campos, M.M.; Otuki, M.F.; Santos, A.R. Antiinflammatory compounds of plant origin. Part II. Modulation of pro-inflammatory cytokines, chemokines and adhesion molecules. Planta Med 2004, 70, 93–103. [Google Scholar]
- Hayashi, K.; Takahata, H.; Kitagawa, N. N-acetylcysteine inhibited nuclear factor-κB expression and the intimal hyperplasia in rat carotid arterial injury. Neurol. Res 2001, 23, 731–738. [Google Scholar]
- Lee, A.S.; Lee, Y.J.; Lee, S.M.; Yoon, J.J.; Kim, J.S.; Kang, D.G.; Lee, H.S. Portulaca oleracea ameliorates diabetic vascular inflammation and endothelial dysfunction in db/db mice. Evid. Based Complement Alternat Med 2012, 2012. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Zakaria, M.N.; Islam, M.W.; Chen, H.B.; Kamil, M.; Chan, K. Neuropharmacological actions of Portulaca oleracea L v. sativa (Hawk). J. Ethnopharmacol 2001, 76, 171–176. [Google Scholar]
- Cai, Y.; Luo, Q.; Sun, M.; Corke, H. Antioxidant activity and phenolic compounds of 112 traditional Chinese medicinal plants associated with anticancer. Life Sci 2004, 74, 2157–2184. [Google Scholar]
- Gao, D.; Li, Q.; Fan, Y. Hypoglycemic effects and mechanisms of Portulaca oleracea L. in alloxan-induced diabetic rats. J. Med. Plants Res 2010, 4, 1996–2003. [Google Scholar]
- Wang, C.Q.; Yang, G.Q. Betacyanins from Portulaca oleracea L. ameliorate cognition deficits and attenuate oxidative damage induced by D-galactose in the brains of senescent mice. Phytomedicine 2010, 17, 527–532. [Google Scholar]
- Zhao, R.; Li, Q.; Xiao, B. Effect of Lycium barbarum polysaccharide on the improvement of insulin resistance in NIDDM rats. Yakugaku Zassh 2006, 125, 981–988. [Google Scholar]
- Singab, A.N.; El-Beshbishy, H.A.; Yonekawa, M.; Nomura, T.; Fukai, T. Hypoglycemic effect of Egyptian Morus alba root bark extract: Effect on diabetes and lipid peroxidation of streptozotocin-induced diabetic rats. J. Ethnopharmacol 2005, 100, 333–338. [Google Scholar]
- Manduteanu, I.; Voinea, M.; Antohe, F.; Dragomir, E.; Capraru, M.; Radulescu, L.; Simionescu, M. Effect of enoxaparin on high glucose-induced activation of endothelial cells. Eur. J. Pharmacol 2003, 477, 269–276. [Google Scholar]
- De Clerck, L.S.; Bridts, C.H.; Mertens, A.M.; Moens, M.M.; Stevens, W.J. Use of fluorescent dyes in the determination of adherence of human leucocytes to endothelial cells and the effect of fluorochromes on cellular function. J. Immunol. Methods 1994, 172, 115–124. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| VCAM-1 | sense: CAAATCCTTGATACTGCTCATC |
| anti-sense: TTGACTTCTTGCTCACAGC | |
| MCP-1 | sense: CAGCCAGATGCAATCAATGC |
| anti-sense: GTGGTCCATGGAATCCTGAA | |
| IL-8 | sense: GCATAAAGACATACTCCAAACC |
| anti-sense: ACTTCTCCACAACCCTCTG | |
| GAPDH | sense: GCACCGTCAAGGCTGAGAAC |
| anti-sense: TGGTGGTGAAGACGCCAGT |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, A.S.; Kim, J.S.; Lee, Y.J.; Kang, D.G.; Lee, H.S. Anti-TNF-α Activity of Portulaca oleracea in Vascular Endothelial Cells. Int. J. Mol. Sci. 2012, 13, 5628-5644. https://doi.org/10.3390/ijms13055628
Lee AS, Kim JS, Lee YJ, Kang DG, Lee HS. Anti-TNF-α Activity of Portulaca oleracea in Vascular Endothelial Cells. International Journal of Molecular Sciences. 2012; 13(5):5628-5644. https://doi.org/10.3390/ijms13055628
Chicago/Turabian StyleLee, An Sook, Jin Sook Kim, Yun Jung Lee, Dae Gill Kang, and Ho Sub Lee. 2012. "Anti-TNF-α Activity of Portulaca oleracea in Vascular Endothelial Cells" International Journal of Molecular Sciences 13, no. 5: 5628-5644. https://doi.org/10.3390/ijms13055628