Microsatellite Development for an Endangered Bream Megalobrama pellegrini (Teleostei, Cyprinidae) Using 454 Sequencing


Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. 454 Sequencing Results
2.1.2. Microsatellite Development
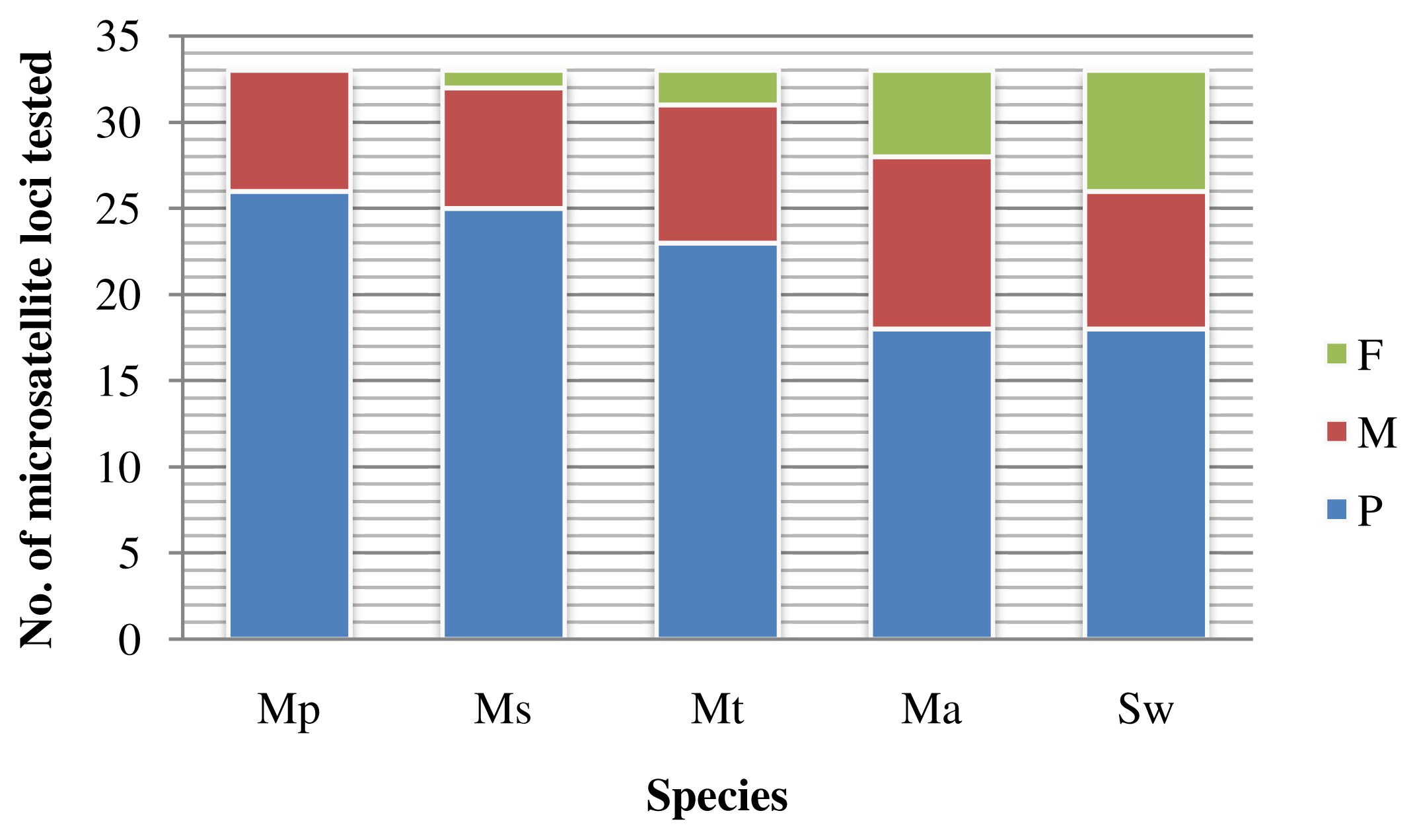
2.1.3. Cross-species Amplification
2.2. Discussion
2.2.1. 454 Sequencing Results
2.2.2. The Microsatellite Development from M. pellegrini
2.2.3. High Level of Cross-species Amplification
3. Experimental Section
3.1. Sample Collection and 454 Sequencing
3.2. Microsatellite Discovery and Primer Screening
3.3. Marker Testing and Cross-species Amplification
3.4. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Luo, Y.L. A revision of fishes of the cyprinid genus Megalobrama. Acta Hydrobiol. Sin 1990, 14, 159–165. [Google Scholar]
- Chen, Y.Y. Fauna Sinica, Osteichthyes Cypriniformes II (in Chinese); Science Press: Beijing, China, 1998. [Google Scholar]
- Park, Y.S.; Chang, J.B.; Lek, S.; Cao, W.X.; Brosse, S. Conservation strategies for endemic fish species threatened by the Three Gorges Dam. Conserv. Biol 2003, 17, 1748–1758. [Google Scholar]
- Liu, J. A quantitative analysis on threat and priority of conservation order of the endemic fishes in upper reaches of the Yangtze River. China Environ. Sci 2004, 24, 395–399. [Google Scholar]
- Li, W.J.; Wang, J.W.; Xie, C.X.; Tan, D.Q. Reproductive biology and spawning habitats of Megalobrama pellegrini, an endemic fish in upper-reaches of Yangtze River basin. Acta Ecol. Sin 2007, 27, 1917–1925. [Google Scholar]
- Gao, X.; Tan, D.Q.; Liu, H.Z.; Wang, J.W. Exploitation status and conservation of a population of Megalobrama pellegrini in Longxi river in the upper Yangtze River basin. Sichuan J. Zool 2009, 28, 329–333. [Google Scholar]
- Wang, J.W.; Tan, D.Q.; Li, W.J. Preliminary studies on artificial propagation and embryonic development of Megalobrama pellegrini. Acta Hydrobiol. Sin 2005, 29, 130–136. [Google Scholar]
- Li, W.J.; Wang, J.W.; Tan, D.Q.; Dan, S.G. Observation on postembryonic development of Megalobrama pellegrini. J. Fish. China 2005, 29, 729–737. [Google Scholar]
- Gao, X.; Liu, H.Z.; Wang, J.W. Applicaiton of logistic regression analysis on study of life history pattern of Megalobrama pellegrini. Sichuan. J. Zool 2008, 27, 506–509. [Google Scholar]
- Zhu, Z.Q.; Li, Y.; Zheng, K.D.; Zhu, X.Z.; Liu, B.; Zhang, L. Cloning and sequence analysis of encoding cDNA sequence of Neuropeptide Y in Megalobrama pellegrini. Freshw. Fish 2009, 39, 14–17. [Google Scholar]
- Liu, H.Z.; Wang, Y.P. Studies on genetic structure and null allele in a natural population of Megalobrama pellegrini. Acta Hydrobiol. Sin 1997, 21, 194–196. [Google Scholar]
- Xu, W.; Xiong, B.X. Advances in the research on genus Megalobrama in China. J. Hydroecol 2008, 1, 7–11. [Google Scholar]
- Cai, M.J.; Zhang, M.Y.; Zeng, Q.L.; Liu, H.Z. A study on the morphological of the genus Megalobrama. Acta Hydrobiol. Sin 2001, 25, 631–635. [Google Scholar]
- Mittal, N.; Dubey, A. Microsatellite markers-a new practice of DNA based markers in molecular genetics. Phcog. Rev 2009, 3, 235–246. [Google Scholar]
- Jones, A.G.; Small, C.M.; Paczolt, K.A.; Ratterman, N.L. A practical guide to methods of parentage analysis. Mol. Ecol. Resour 2010, 10, 6–30. [Google Scholar]
- Goldstein, D.B.; Schlotterer, C. Microsatellites, Evolution and Applications; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Ellegren, H. Microsatellites, simple sequences with complex evolution. Nat. Rev. Genet 2004, 5, 435–445. [Google Scholar]
- Glenn, T.C.; Schable, N.A. Isolating Microsatellite DNA Loci. In Methods in Enzymology, Molecular Evolution, Producing the Biochemical Data, Part B; Zimmer, E.A., Roalson, E.H., Eds.; Academic Press: San Diego, CA USA, 2005; pp. 202–222. [Google Scholar]
- Abdelkrim, J.; Robertson, B.C.; Stanton, J.-A.L.; Gemmell, N.J. Fast, cost-effective development of species-specific microsatellite markers by genomic sequencing. BioTechniques 2009, 46, 185–191. [Google Scholar]
- Allentoft, M.E.; Schuster, S.C.; Holdaway, R.N.; Hale, M.L.; McLay, E.; Oskam, C.; Gilbert, T.P.; Spencer, P.; Willerslev, E.; Bunce, M. Identification of microsatellites from an extinct moa species using high-throughput (454) sequence data. BioTechniques 2009, 46, 195–200. [Google Scholar]
- Santana, Q.C.; Coetzee, M.P.A.; Steenkamp, E.T.; Mlonyeni, O.X.; Hammond, G.N.A.; Wingfield, M.J.; Wingfield, B.D. Microsatellite discovery by deep sequencing of enriched genomic libraries. BioTechniques 2009, 46, 217–223. [Google Scholar]
- Castoe, T.A.; Poole, A.W.; Gu, W.J.; Jason de Koning, A.P.; Daza, J.M.; Smith, E.N.; Pollock, D.D. Rapid identification of thousands of copperhead snake (Agkistrodon contortrix) microsatellite loci from modest amounts of 454 shotgun genome sequence. Mol. Ecol. Resour 2009, 10, 341–347. [Google Scholar]
- Csencsics, D.; Brodbeck, S.; Holderegger, R. Cost-effective, species-specific microsatellite development for the endangered dwarf bulrush (Typha minima) uing next-generation sequencing technology. J. Hered 2010, 101, 789–793. [Google Scholar]
- Saarinen, E.V.; Austin, J.D. When technology meets conservation, increased microsatellite marker production using 454 genome sequencing on the endangered okaloosa darter (Etheostoma okaloosae). J. Hered 2010, 101, 784–788. [Google Scholar]
- Gardner, M.G.; Fitch, A.J.; Bertozzi, T.; Lowe, A.J. Rise of the machines—Recommendations for ecologists when using next generation sequencing for microsatellite development. Mol. Ecol. Resour 2011, 11, 1093–1101. [Google Scholar]
- McCulloch, E.S.; Stevens, R.D. Rapid development and screening of microsatellite loci for Artibeus lituratus and their utility for six related species within Phyllostomidae. Mol. Ecol. Resour 2011, 11, 903–913. [Google Scholar]
- Perry, J.C.; Rowe, L. Rapid microsatellite development for water striders by next-generation sequencing. J. Hered 2011, 102, 125–129. [Google Scholar]
- Wood, R.; Weyeneth, N.; Appleton, B. Development and characterisation of 20 microsatellite loci isolated from the large bent-wing bat, Miniopterus schreibersii (Chiroptera, Miniopteridae) and their cross-taxa utility in the family Miniopteridae. Mol. Ecol. Resour 2011, 11, 675–685. [Google Scholar]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Leger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; Petit, R.J. Current trends in microsatellite genotyping. Mol. Ecol. Resour 2011, 11, 591–611. [Google Scholar]
- Li, W.T.; Liao, X.L.; Yu, X.M.; Wang, D.; Tong, J.G. Isolation and characterization of polymorphic microsatellite loci in Wuchang bream (Megalobrama amblycephala). Mol. Ecol. Notes 2007, 7, 771–773. [Google Scholar]
- Lai, Y.; Sun, F. The relationship between microsatellite slippage mutation rate and the number of repeat units. Mol. Biol. Evol 2003, 20, 2123–2131. [Google Scholar]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet 1980, 32, 314–331. [Google Scholar]
- Aliah, R.S.; Takagi, M.; Dong, S.; Teoh, C.T.; Taniguchi, N. Isolation and inheritance of microsatelllite markers in the common carp Cyprinus carpio. Fish Sci 1999, 65, 235–239. [Google Scholar]
- Du, C.B.; Sun, X.W.; Lou, Y.D.; Shen, J.B. The genetic heterozygosity analysis to wild carp and two cultivated strains of common carp using microsatellite technique. J. Shanghai Fish Univ 2000, 9, 285–289. [Google Scholar]
- Wang, W.; You, F.; Gao, T.X.; Zhang, P.J. Genetic variations at ten microsatellite loci in natural and cultured stocks of left-eyed flounder Paralichthys olivaceu in Shandong coastal waters. Oceanol. Limnol. Sin 2004, 35, 530–537. [Google Scholar]
- Sumantadinata, K.; Taniguchi, N. Comparison of electrophoretic allele frequencies and genetic variability of common carp stocks from Indonesia and Japan. Aquaculture 1990, 88, 263–271. [Google Scholar]
- Liao, X.L.; Yu, X.M.; Chang, J.B.; Tong, J.G. Polymorphic microsatellites in largemouth bronze gudgeon (Coreius guichenoti) developed from repeat-enriched libraries and cross-species amplifications. Mol. Ecol. Notes 2007, 7, 1104–1107. [Google Scholar]
- Liao, X.L.; Wang, D.; Yu, X.M.; Li, W.T.; Cheng, L.; Wang, J.W.; Tong, J.G. Characterization of novel microsatellite loci in rare minnow (Gobiocypris rarus) and amplification in closely related species in Gobioninae. Conserv. Genet 2007, 8, 1003–1007. [Google Scholar]
- Yue, H.; Yuan, H.; Zhang, X.Y. Fifteen novel polymorphic microsatellites in rock carp, Procypris rabaudi (Tchang), an endemic fish species in the upper reaches of the Yangtze River drainage. Conserv. Genet 2009, 10, 539–542. [Google Scholar]
- Zhu, D.; Chang, J.B. Annual variations of biotic integrity in the upper Yangtze River using an adapted index of biotic integrity (IBI). Ecol. Indic 2008, 8, 564–572. [Google Scholar]
- Faircloth, B.C. Msatcommander, detection of microsatellite repeat arrays and automated, locus-specific primer design. Mol. Ecol. Resour 2008, 8, 92–94. [Google Scholar]
- Yeh, F.C.; Yang, R.C.; Boyle, T. POPGENE Microsoft Windows-based Freeware for Population Genetic Analysis Release 1.31; University of Alberta: Edmonton, Canada, 1999. [Google Scholar]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol. Bioinforma. Online 2005, 1, 47–50. [Google Scholar]
- Kalinowski, S.; Taper, M.; Marshall, T. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. Notes 2007, 16, 1099–1106. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER, software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar]

{kind=link}
| Locus | Repeat motif | Primer sequence (5′–3′) | Longxi River population (N = 30) | Cross-amplification (N = 5) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F | R | NA | HO | HE | PIC | GenBank Accession No. | Ms | Mt | Ma | Sw | ||
| MP1 | (CT)14 | GCCTCACTCTATCCCTACCT | CTCAGAAACAATCCATCCAG | 2 | 0.59 | 0.51 | 0.38 | JQ087470 | P | P | F | P |
| MP6 | (TATC) 10 | TATGATGGTGTTGCCTTGGT | GCTTTGTCCTGAGACTGTGG | 4 | 0.15 *** | 0.65 | 0.58 | JQ087471 | P | P | P | P |
| MP8 | (ATCT)12 | GGGGAAAATCAGAGGGAATG | GACGACGGATGGACAGACAG | 3 | 0.50 | 0.57 | 0.46 | JQ087472 | P | P | P | P |
| MP9 | (TTGA) 12 | GTTCCGTCGTTACCAATAGAG | ACCCAAGGTCGGTCACAT | 3 | 0.72 * | 0.52 | 0.44 | JQ087473 | M | M | M | M |
| MP11 | (AT)15 | TGGTGAGCAGACGAAACTT | TAACCAGCGAGAACGATGT | 2 | 0.70 ** | 0.46 | 0.35 | JQ087474 | M | M | M | P |
| MP14 | (AATA)9 | CTCGTGATGAAAGAAGAGTTAG | AATAGCCAACTGAACTGAGC | 3 | 0.63 | 0.57 | 0.49 | JQ087475 | P | P | M | M |
| MP15 | (TAA)10 | CGTGAGATTCCCGTCTCGTC | AAAGGCAGGTGTCCCAAAAC | 3 | 0.77 | 0.63 | 0.54 | JQ087476 | P | P | P | P |
| MP16 | (CTAT)15 | CACATTTCAGCATTTCAAGACT | TGGGTTGTTATTCTGTTTCTGA | 5 | 0.53 | 0.64 | 0.57 | JQ087477 | M | P | P | P |
| MP17 | (AAAC)11 | TGGGGATACGGTGGAGAAC | GGTGCTGCTTGATTATTGGAG | 4 | 0.66 | 0.68 | 0.60 | JQ087478 | P | F | P | F |
| MP22 | (AC)16 | CTGATATGAGCAAGGTAGCAA | ACTCCATTAACAATCGCACA | 2 | 0.37 | 0.41 | 0.32 | JQ087479 | P | P | P | P |
| MP24 | (TCAA)8 | CAGACAATAGAGGGGTACACAC | TTGAATACAAGTAAGCAAAGGTT | 2 | 0.47 | 0.51 | 0.37 | JQ087480 | P | P | F | F |
| MP26 # | (TCTA)16 | TGGCTGAACTCCAAAATAAG | TCACCTAAACGGGAAAATAC | 4 | 0.27 ** | 0.41 | 0.36 | JQ087481 | P | P | F | F |
| MP27 | (GATA)11 | GAGCCACTTCACTATCGTTTA | CGCTATTGGGTTCAACATT | 4 | 0.30 | 0.30 | 0.27 | JQ087482 | P | P | P | M |
| MP28 | (AGAT)16 | ATTCTTCCCACTGTCATTTC | CTACCCAAAACTGGCTGA | 4 | 0.50 | 0.51 | 0.42 | JQ087483 | P | P | P | P |
| MP29 | (TATT)11 | AAAATGTCATGTCTGCTGTAT | TAAACTCTTCAAGTGGCTCA | 3 | 0.20 | 0.22 | 0.20 | JQ087484 | P | P | P | P |
| MP30# | (GATA)11 | AAGAGCCACTTCACTATCGT | GCTATTGGGTTCTAACATTG | 4 | 0.29 | 0.36 | 0.32 | JQ087485 | P | P | P | P |
| MP32 | (TGTT)15 | GAGTCATTGAGTCCGTTTAGA | TCAGTTGAGGAGACATTTGC | 4 | 0.77 | 0.65 | 0.56 | JQ087486 | P | M | M | M |
| MP34 | (ATG)11 | TATTCAACTTCGTGCTCCTA | GTCCATGCTTTCTGTCTTAA | 2 | 0.77 ** | 0.48 | 0.36 | JQ087487 | P | P | M | M |
| MP40 | (AAT)13 | TATCCGTATTGCCCAAAC | TTGCTGGCATCTTACTTTC | 3 | 0.61 | 0.63 | 0.55 | JQ087488 | P | P | P | M |
| MP41 | (TTA)12 | GGCTACAGCAGGTTTTATTT | TTACCTTTTCACCAATTCCA | 2 | 0.14 *** | 0.48 | 0.36 | JQ087489 | P | P | P | P |
| MP42 | (TAA)12 | TAGGAACAATGAGGGAACT | CAAGGATTAAGCCTGGTC | 2 | 0.23 | 0.30 | 0.26 | JQ087490 | P | M | M | P |
| MP43 | (TTA)11 | GGGGACACCTTAGACTTAT | AAGTGTAAACCCTGAAGAAC | 2 | 0.47 | 0.49 | 0.37 | JQ087491 | P | P | P | P |
| MP45 | (AATA)8 | GCGTATTTTATCATCTTTTGTGT | TGGGAGTGAAATGGAGTGAC | 5 | 0.80 | 0.72 | 0.67 | JQ087492 | P | P | P | P |
| MP46 | (GATA)11 | GTGTGGAGTGTTTCCCTTTAGC | CCGCTATTGGGTTCAACATT | 2 | 0.07 *** | 0.50 | 0.37 | JQ087493 | P | P | P | F |
| MP48 | (GGAG)15 | ATGCTGTTCCAGGATCAAC | CCGCTTTTATAGCCTTTAGT | 4 | 0.53 | 0.59 | 0.51 | JQ087494 | P | M | M | F |
| MP55 | (TAT)12 | CATACTCTGTGCATCACTTTGGTC | TTTACGAGGGCTTATTAGGGC | 2 | 0.30 | 0.48 | 0.36 | JQ087495 | P | P | P | P |
| MP2 | (CA)18 | TGTCCTCATAAGTCACCCTC | ACACCGTCTAATCTGCCTAC | 1 | 0.00 | 0.00 | M | M | M | M | P | |
| MP3 | (AC)21 | ACAACACTTCACCACCCA | TAAGAACTACAAATACCCACTG | 1 | 0.00 | 0.00 | M | F | F | F | F | |
| MP5 | (AGAT)9 | AGTCCTCTGCCACCTCCTG | GCTACTTGACCCTTCATCATACA | 1 | 0.00 | 0.00 | M | P | P | P | P | |
| MP19 | (TAT) 10 | AAAGGGATAGCAGACAAGATAC | ACAACCCAAACAGGTGAAT | 1 | 0.00 | 0.00 | M | M | P | F | F | |
| MP23 | (AGA)11 | AGTTTTCGAGCACGTTTAGG | CCCACTGTTCACTGTTTTCC | 1 | 0.00 | 0.00 | M | M | M | M | M | |
| MP25 | (AAAC)14 | AACAGGGGAGGGGAAGATTA | CTATTGAAGAGGCAAAGCGAG | 1 | 0.00 | 0.00 | M | P | P | P | P | |
| MP50 | (TTTC)14 | CGTAATGTTTGGTGTCGTTTTG | AATGTGGGCGGGGTTTAGT | 1 | 0.00 | 0.00 | M | M | M | M | M | |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, J.; Yu, X.; Zhao, K.; Zhang, Y.; Tong, J.; Peng, Z. Microsatellite Development for an Endangered Bream Megalobrama pellegrini (Teleostei, Cyprinidae) Using 454 Sequencing. Int. J. Mol. Sci. 2012, 13, 3009-3021. https://doi.org/10.3390/ijms13033009
Wang J, Yu X, Zhao K, Zhang Y, Tong J, Peng Z. Microsatellite Development for an Endangered Bream Megalobrama pellegrini (Teleostei, Cyprinidae) Using 454 Sequencing. International Journal of Molecular Sciences. 2012; 13(3):3009-3021. https://doi.org/10.3390/ijms13033009
Chicago/Turabian StyleWang, Jinjin, Xiaomu Yu, Kai Zhao, Yaoguang Zhang, Jingou Tong, and Zuogang Peng. 2012. "Microsatellite Development for an Endangered Bream Megalobrama pellegrini (Teleostei, Cyprinidae) Using 454 Sequencing" International Journal of Molecular Sciences 13, no. 3: 3009-3021. https://doi.org/10.3390/ijms13033009