Computational Studies of Difference in Binding Modes of Peptide and Non-Peptide Inhibitors to MDM2/MDMX Based on Molecular Dynamics Simulations

Abstract
:1. Introduction
2. Results and Discussion
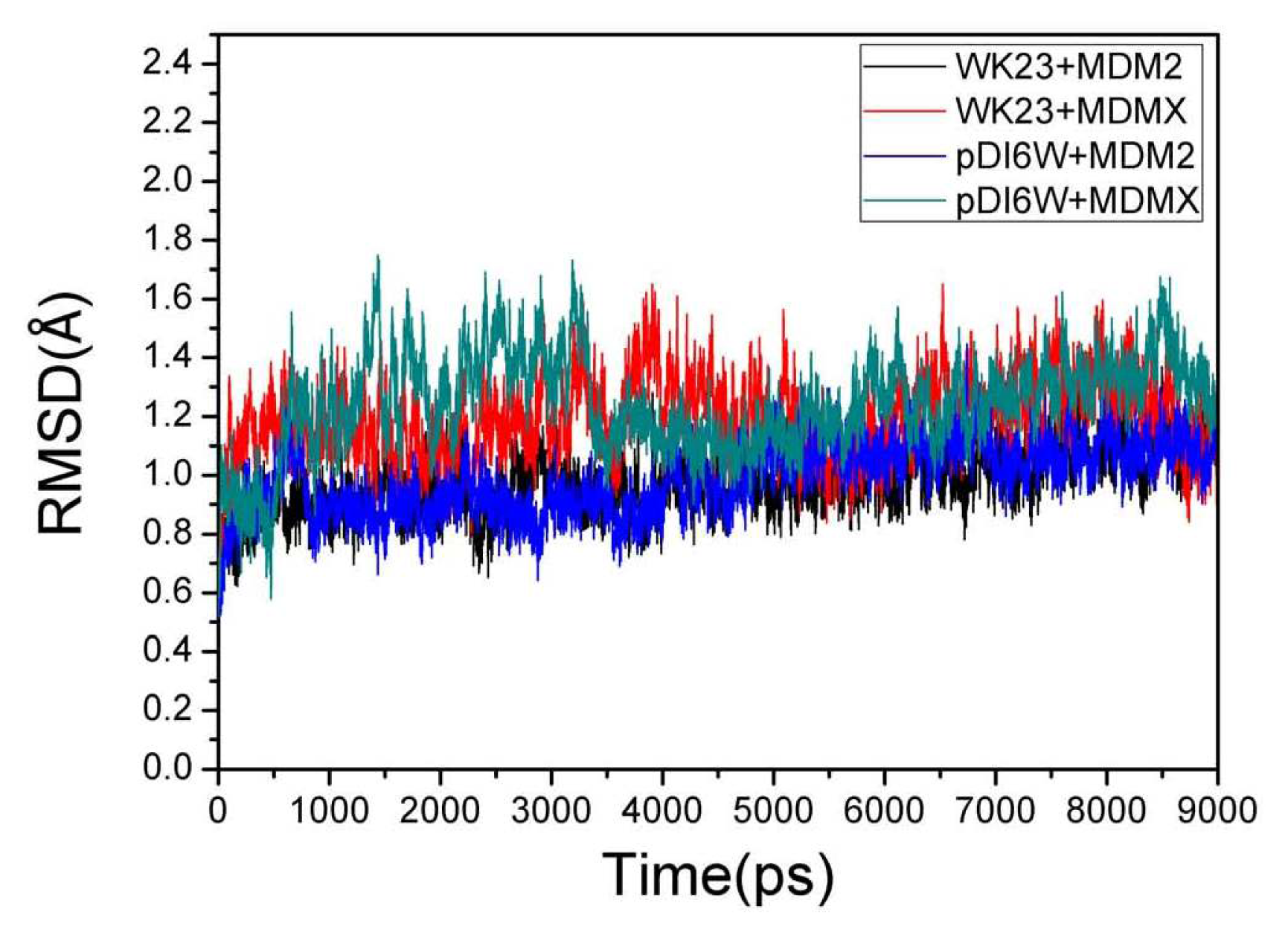
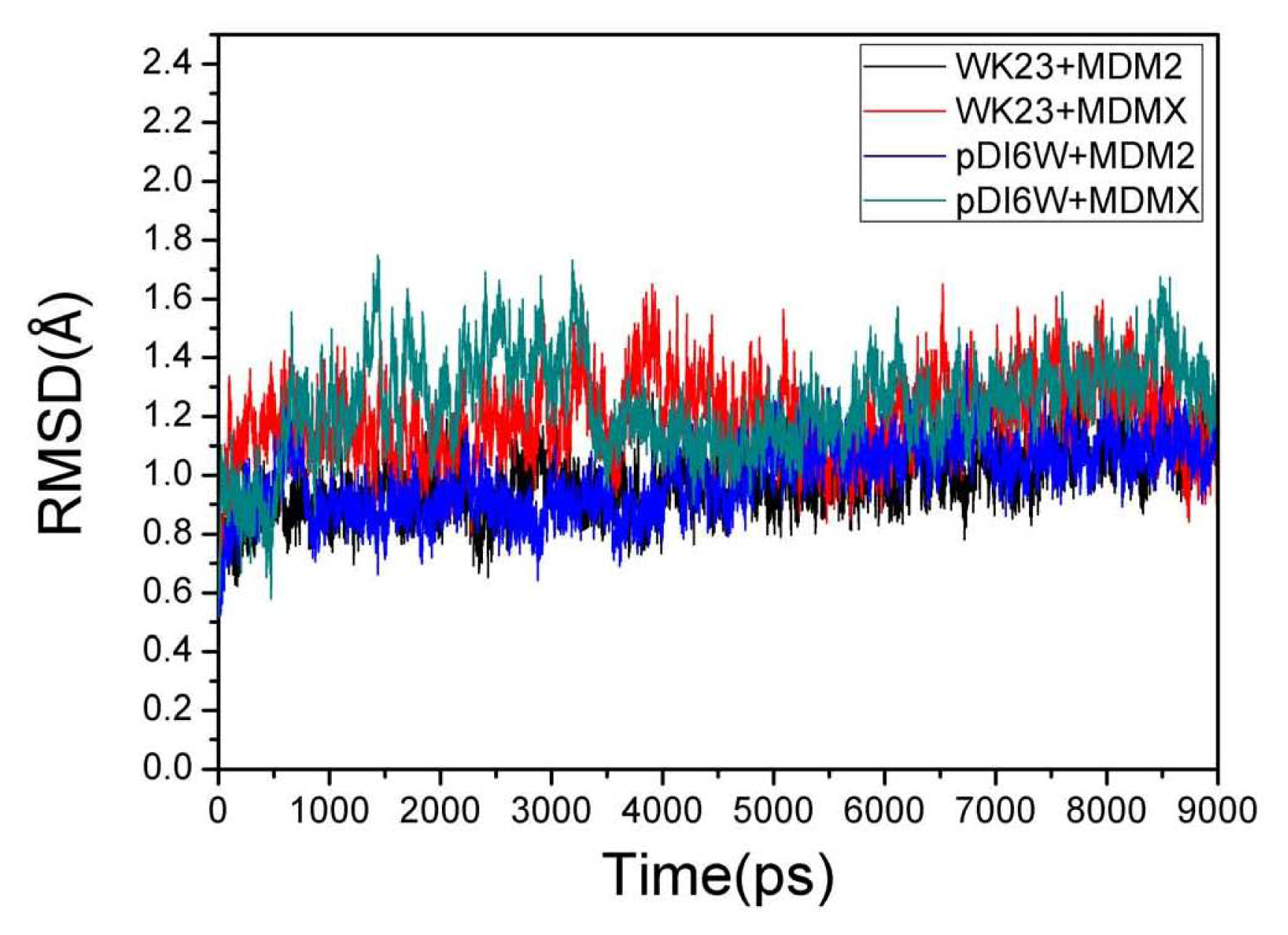
2.1. System Stability During MD Simulations
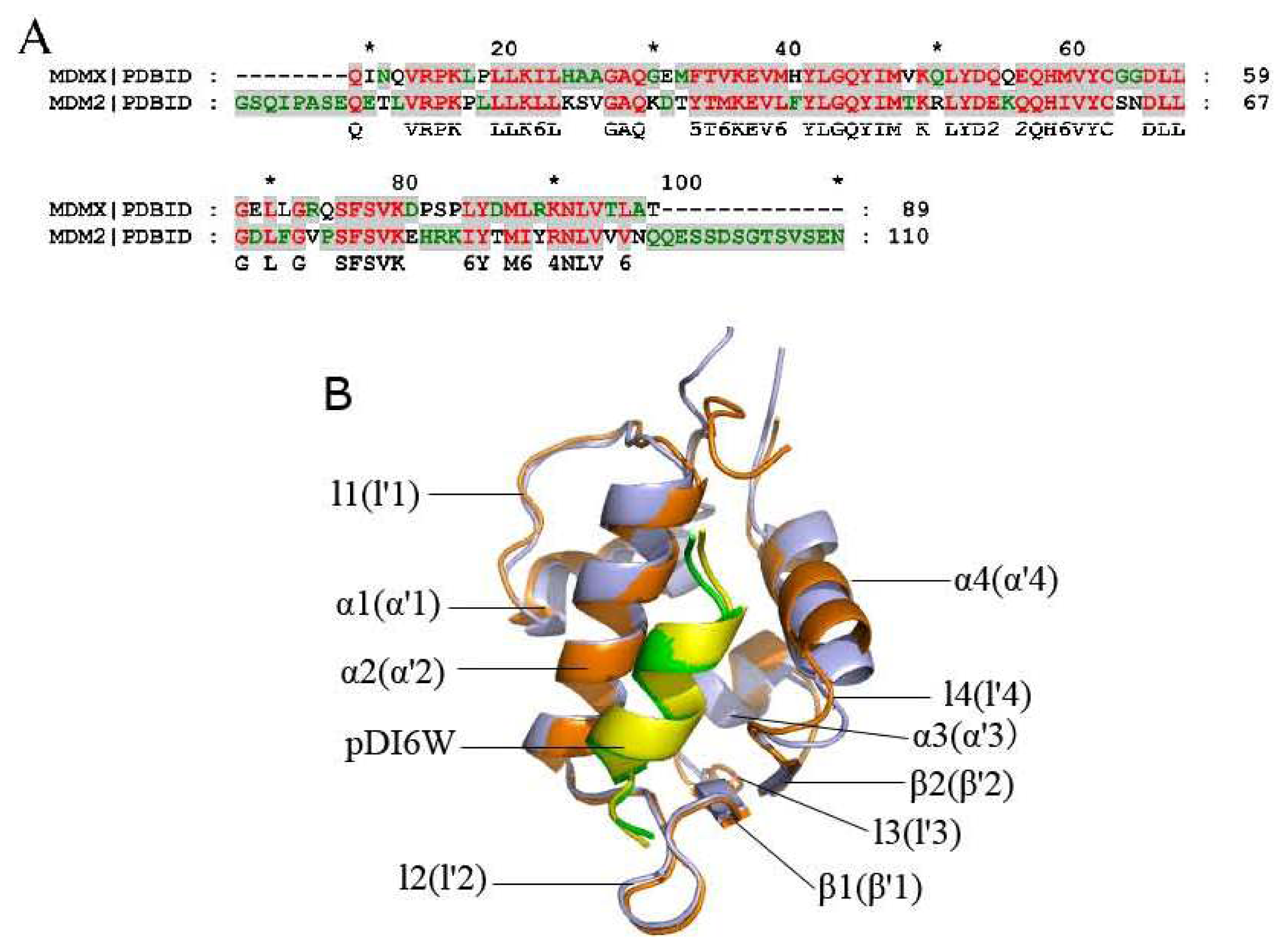
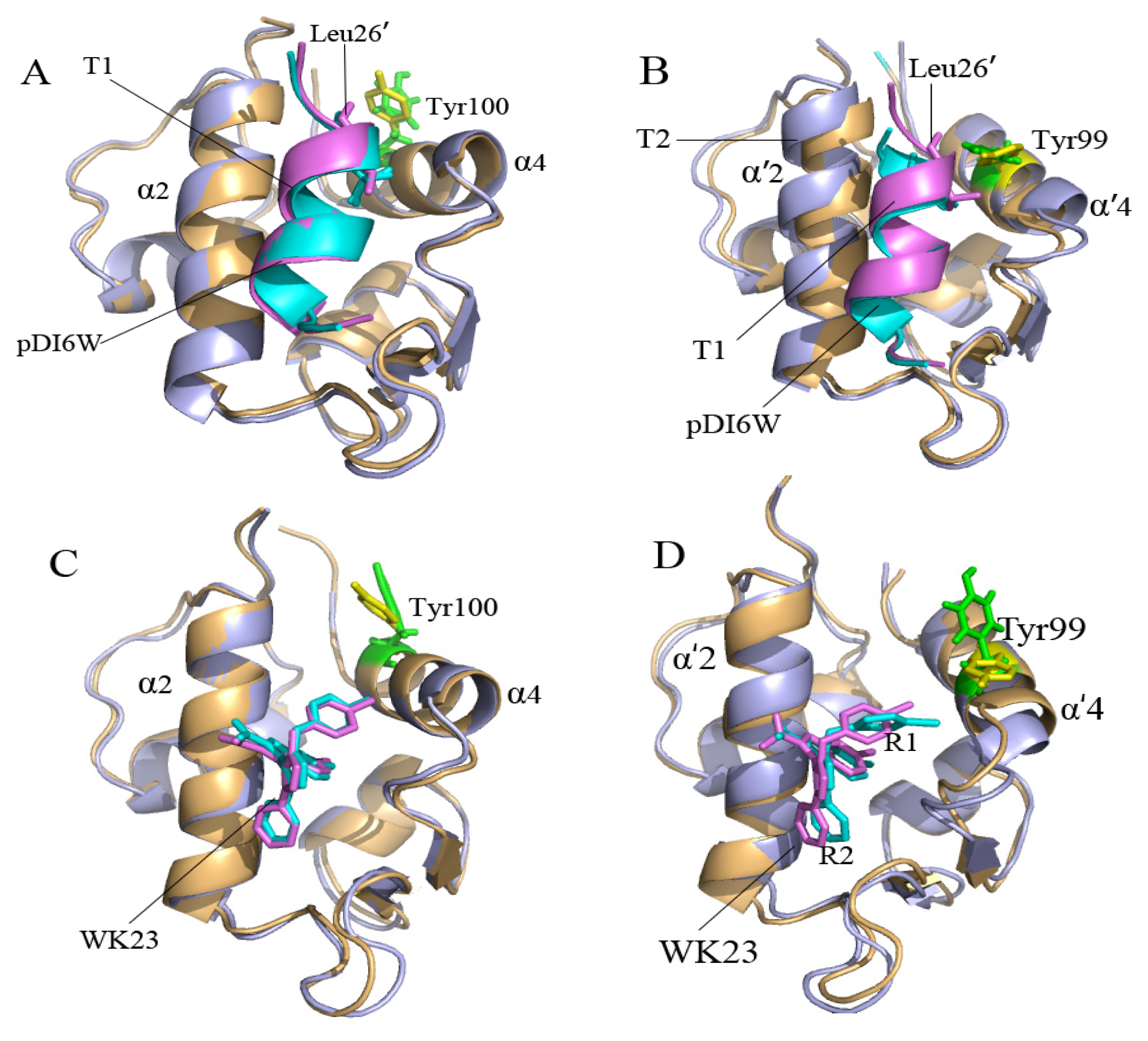
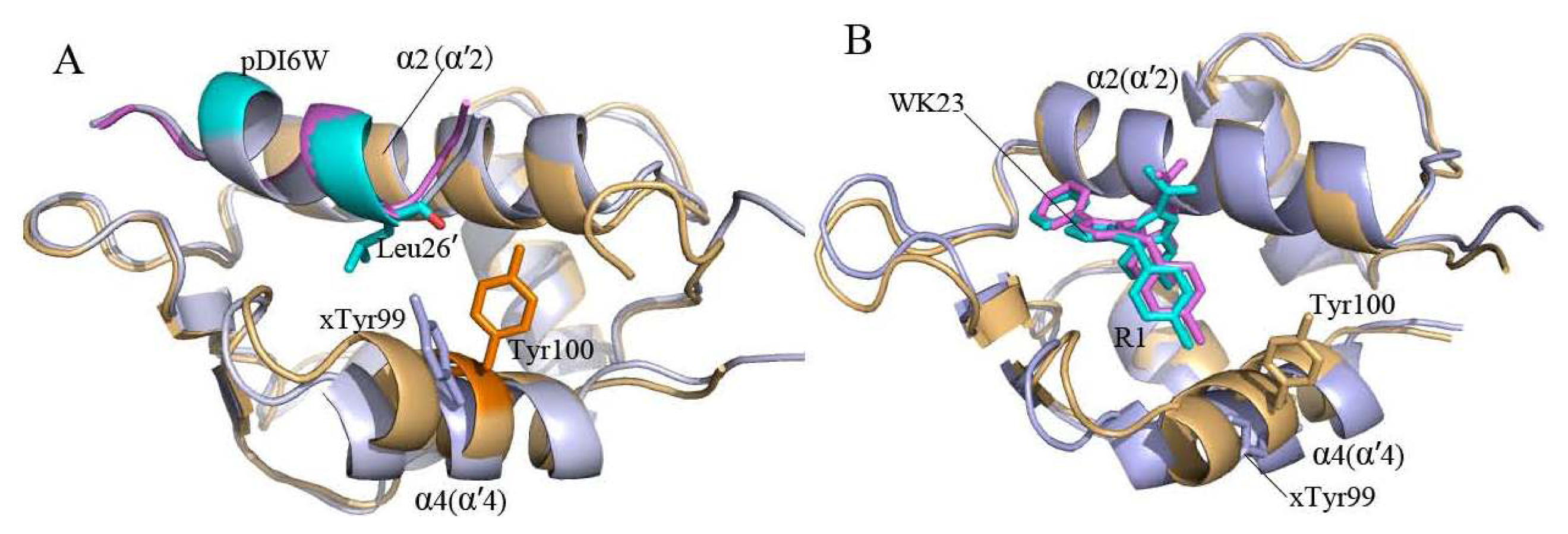
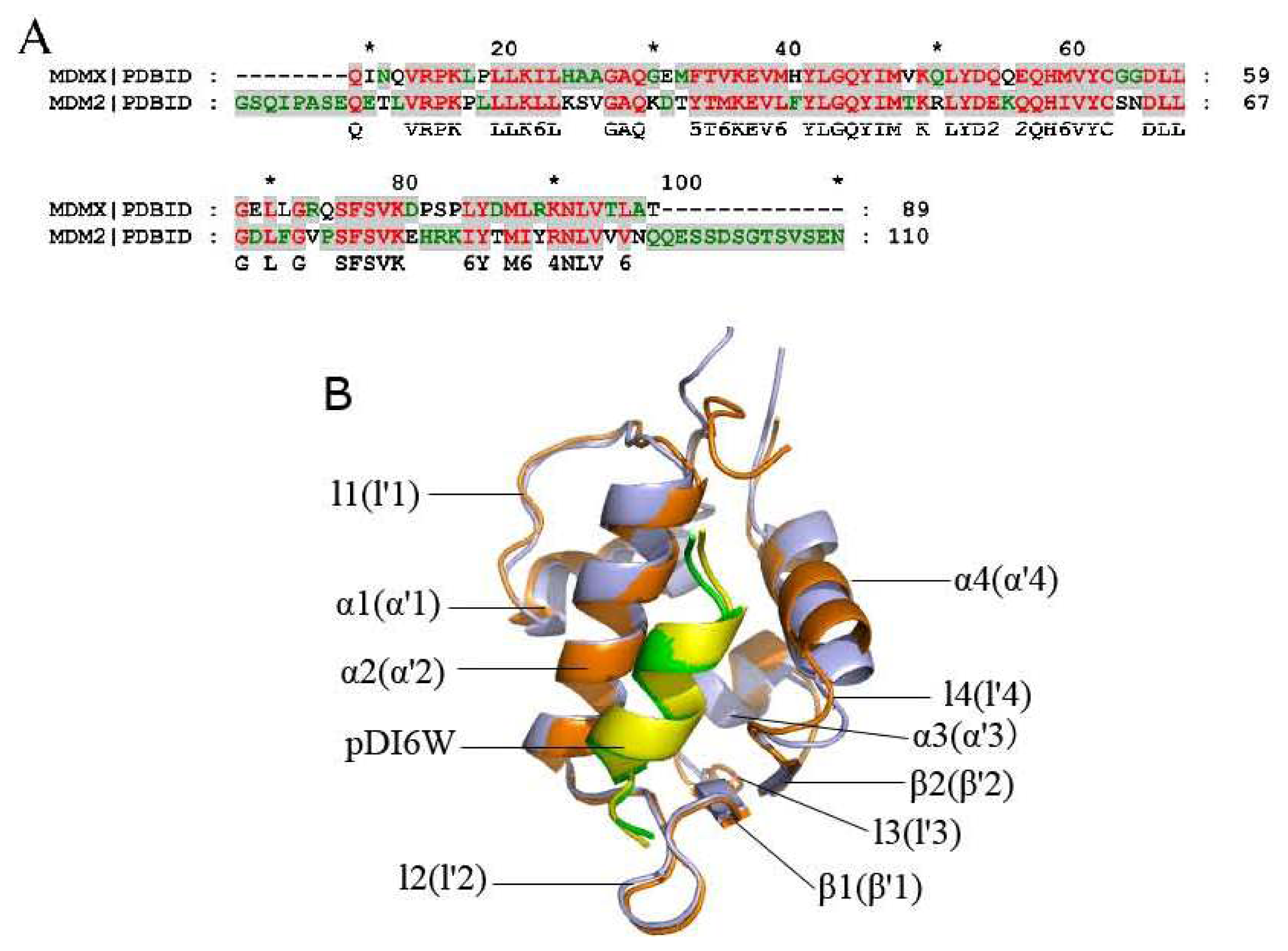
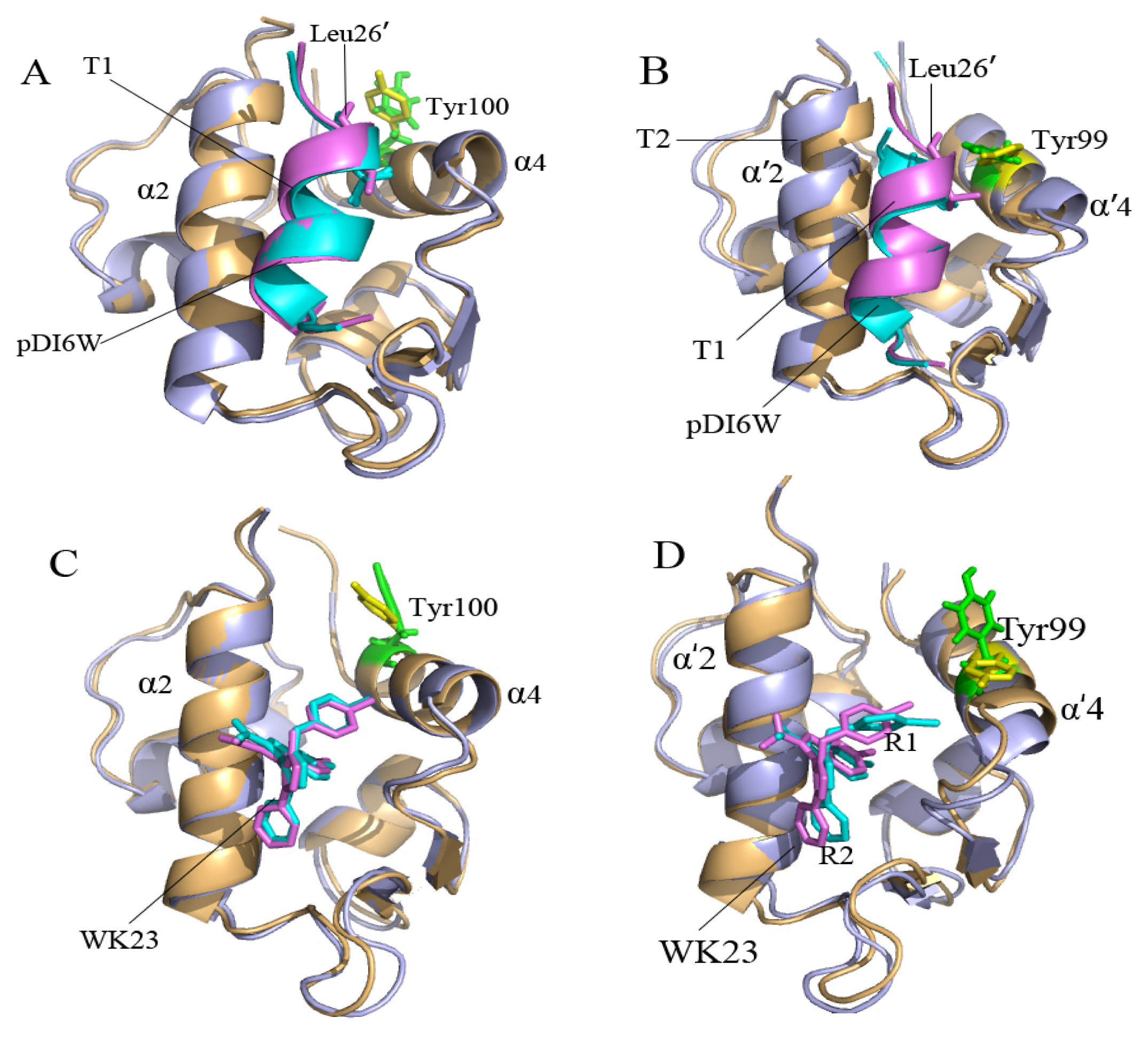
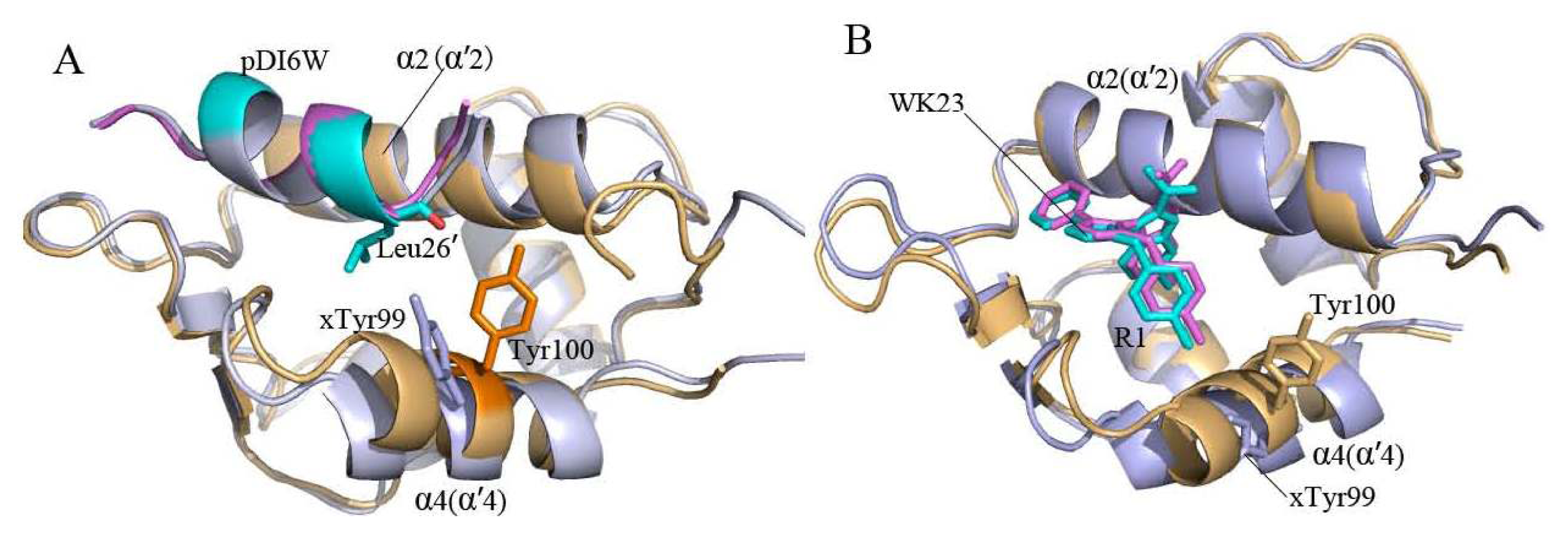
2.2. Superimposition Analyses
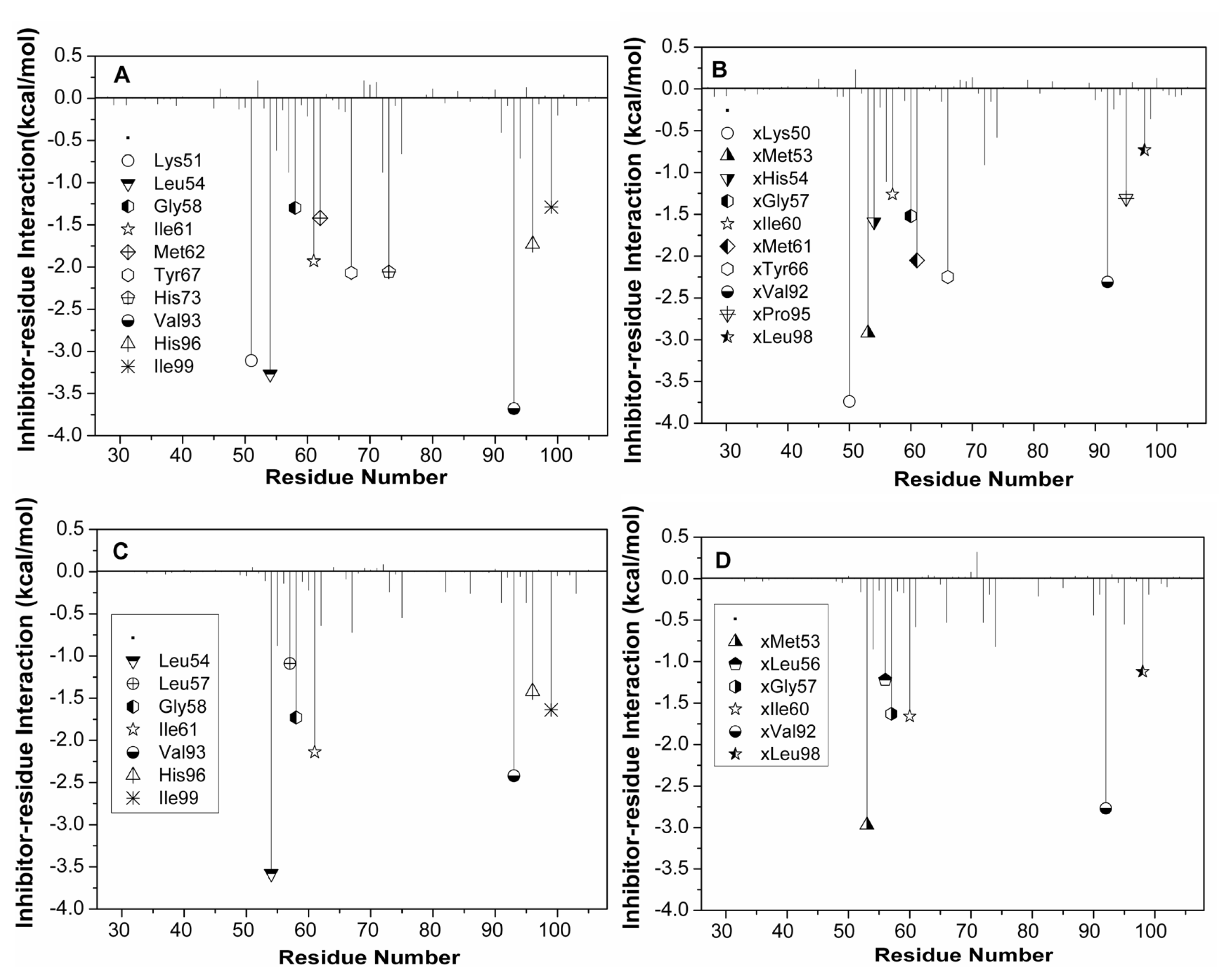
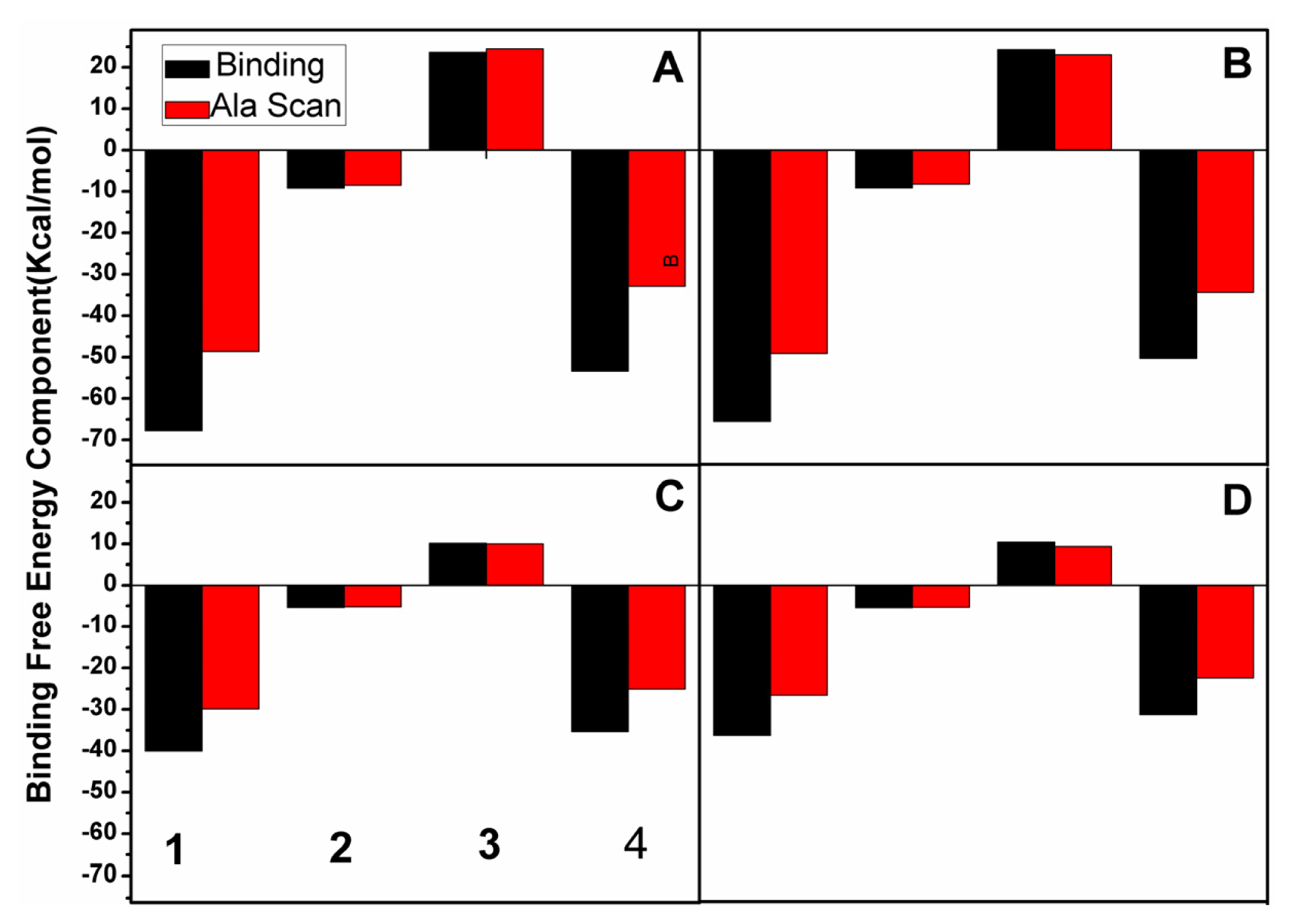
2.3. Calculations of Binding Free Energies
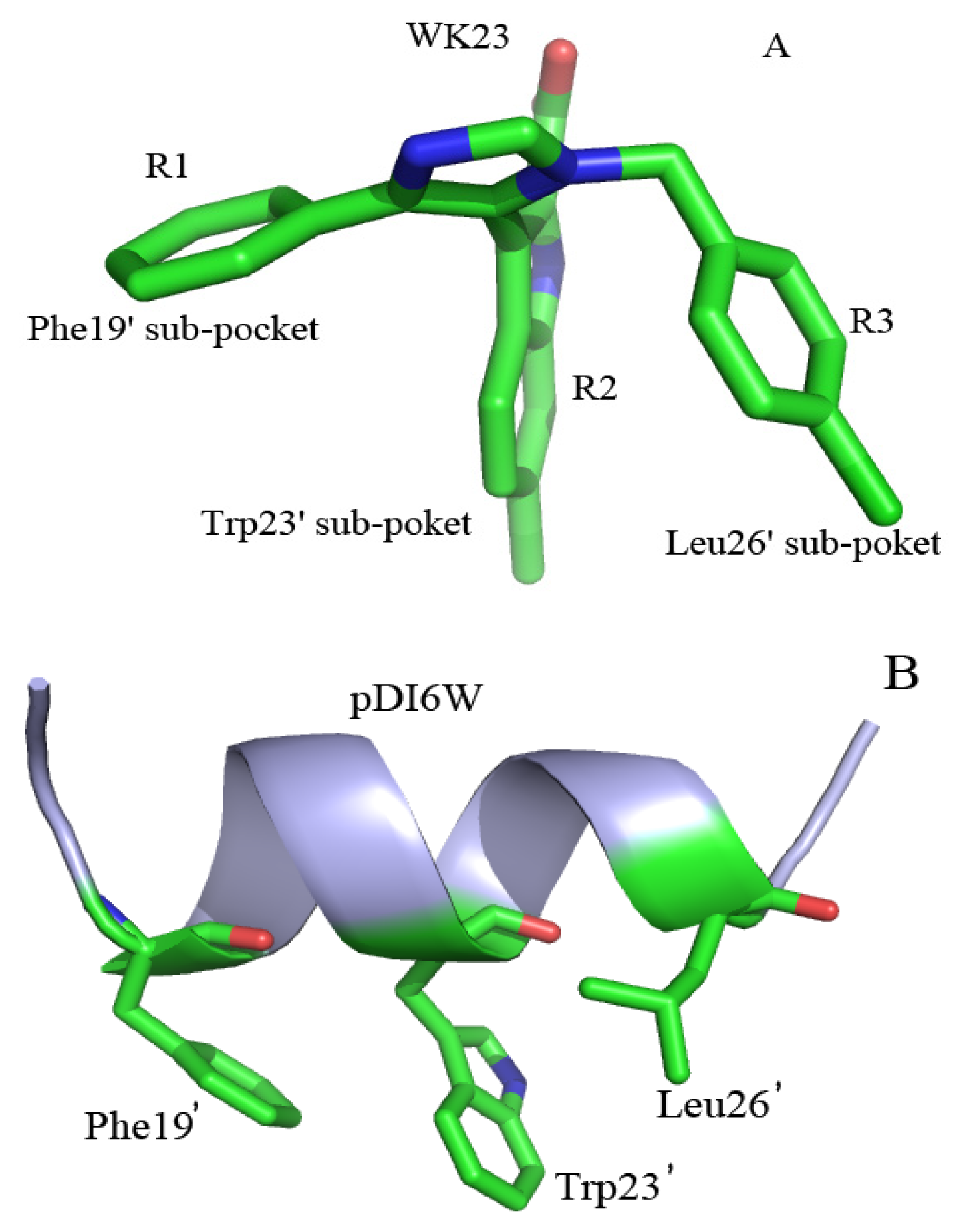
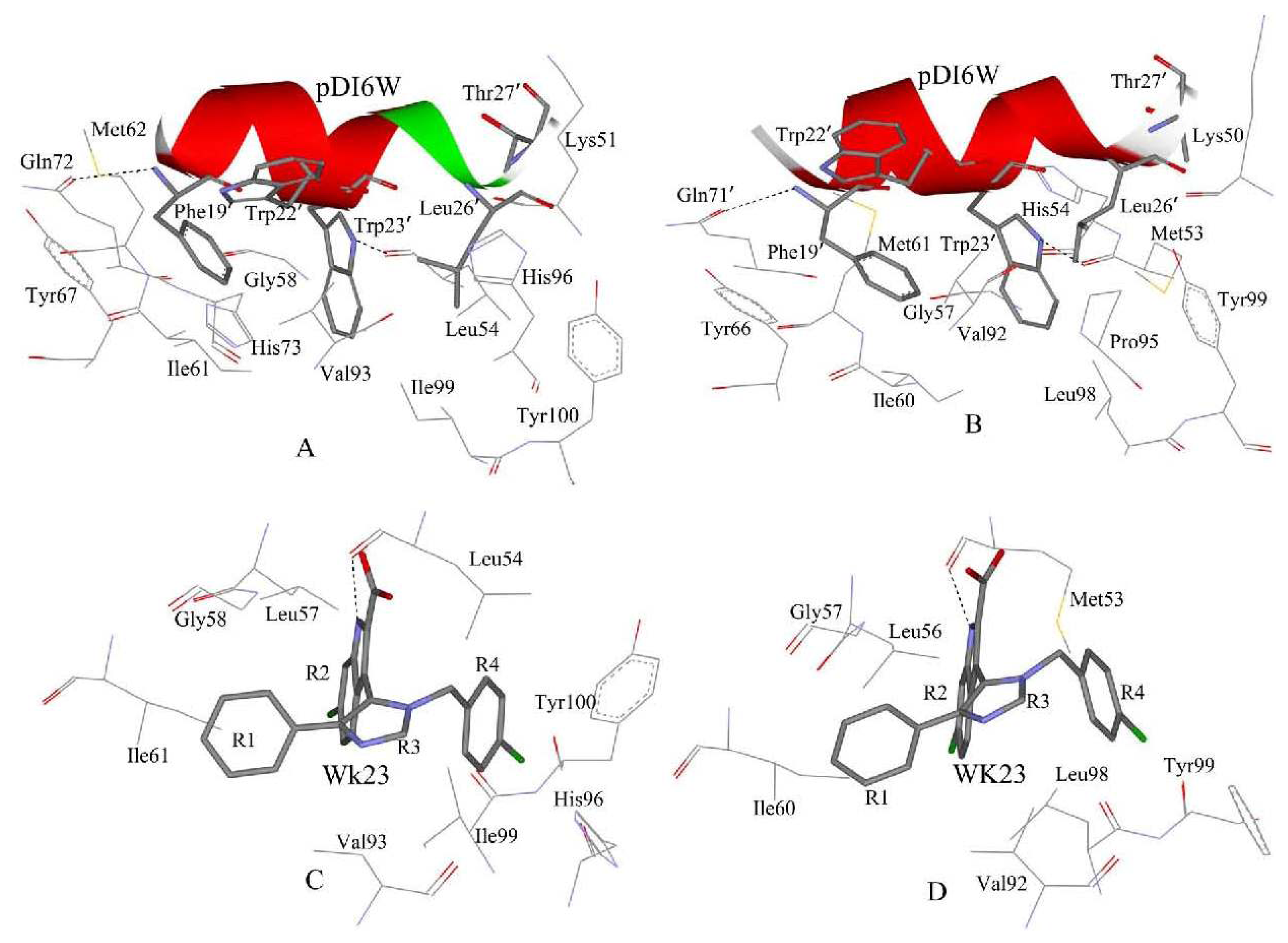
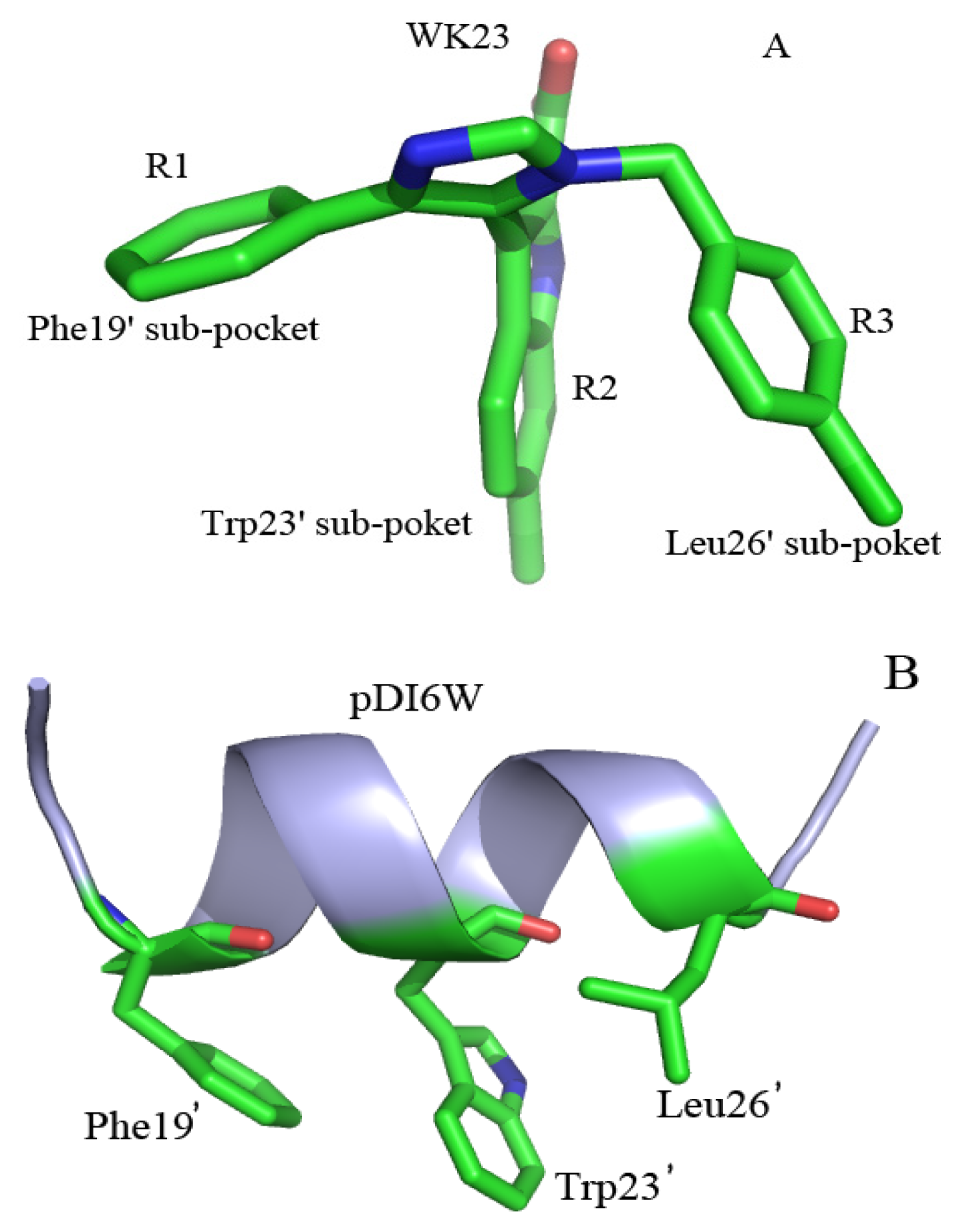
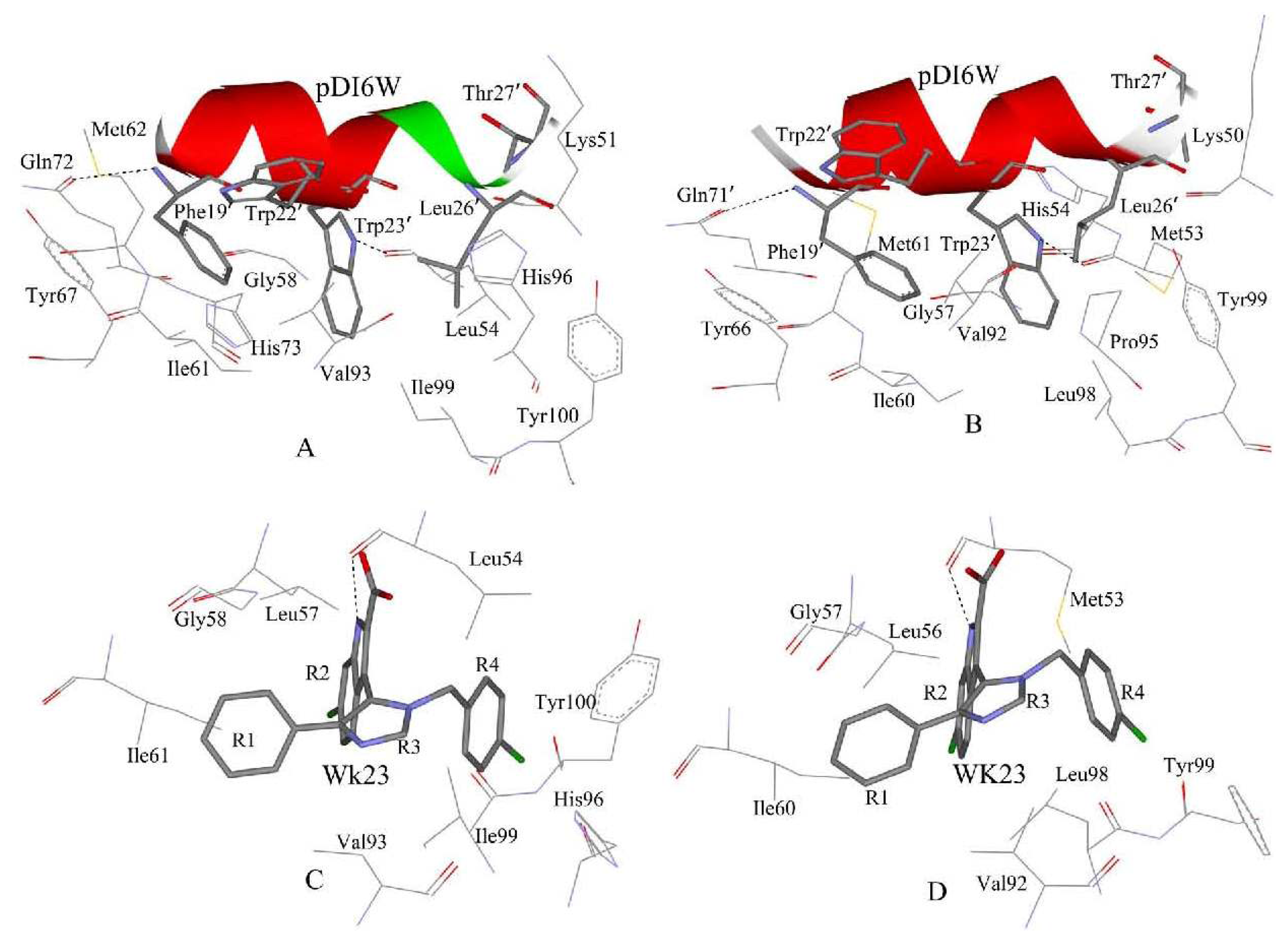
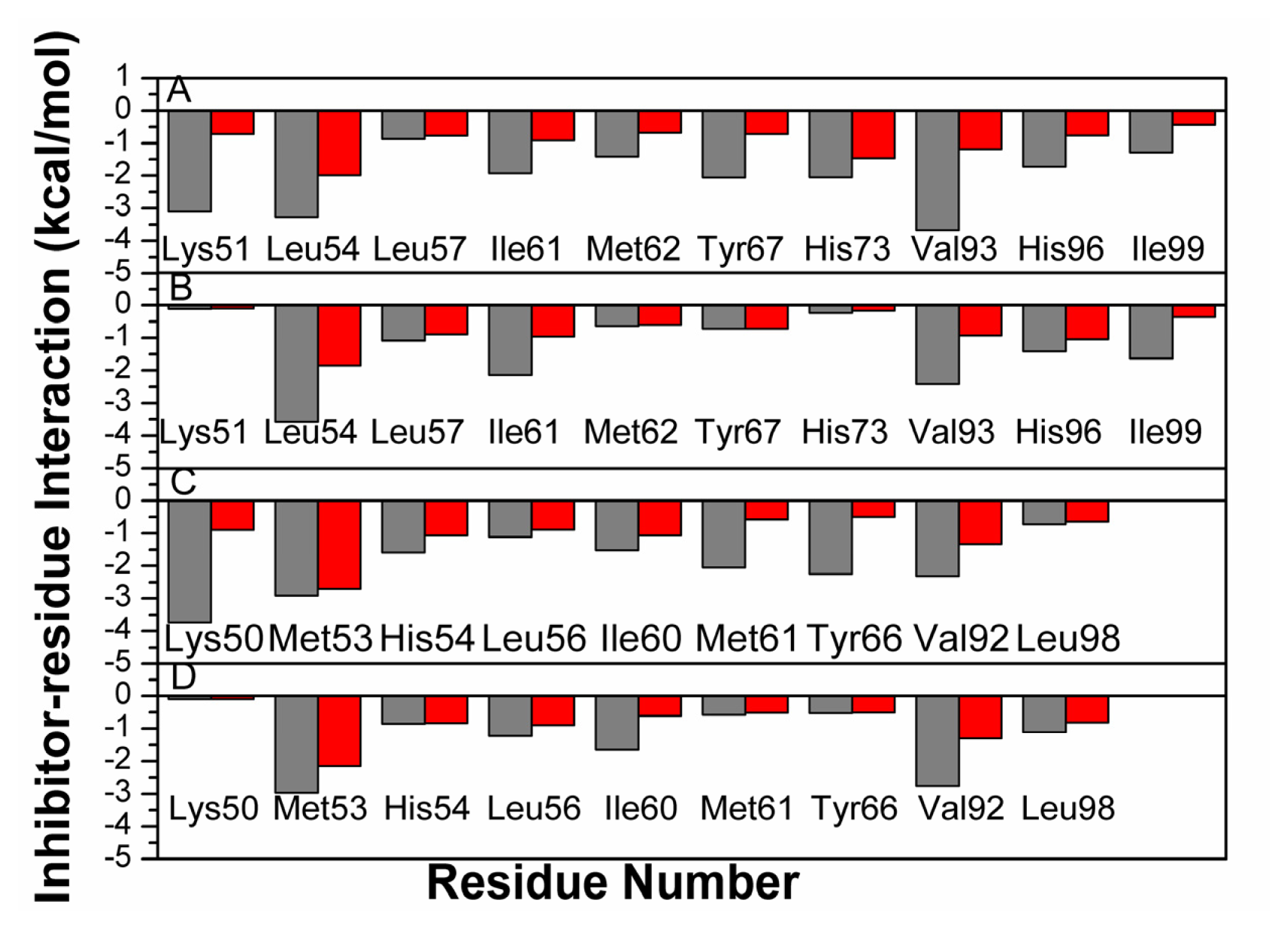
2.4. Binding Mode Predictions of Inhibitors to MDM2/MDMX
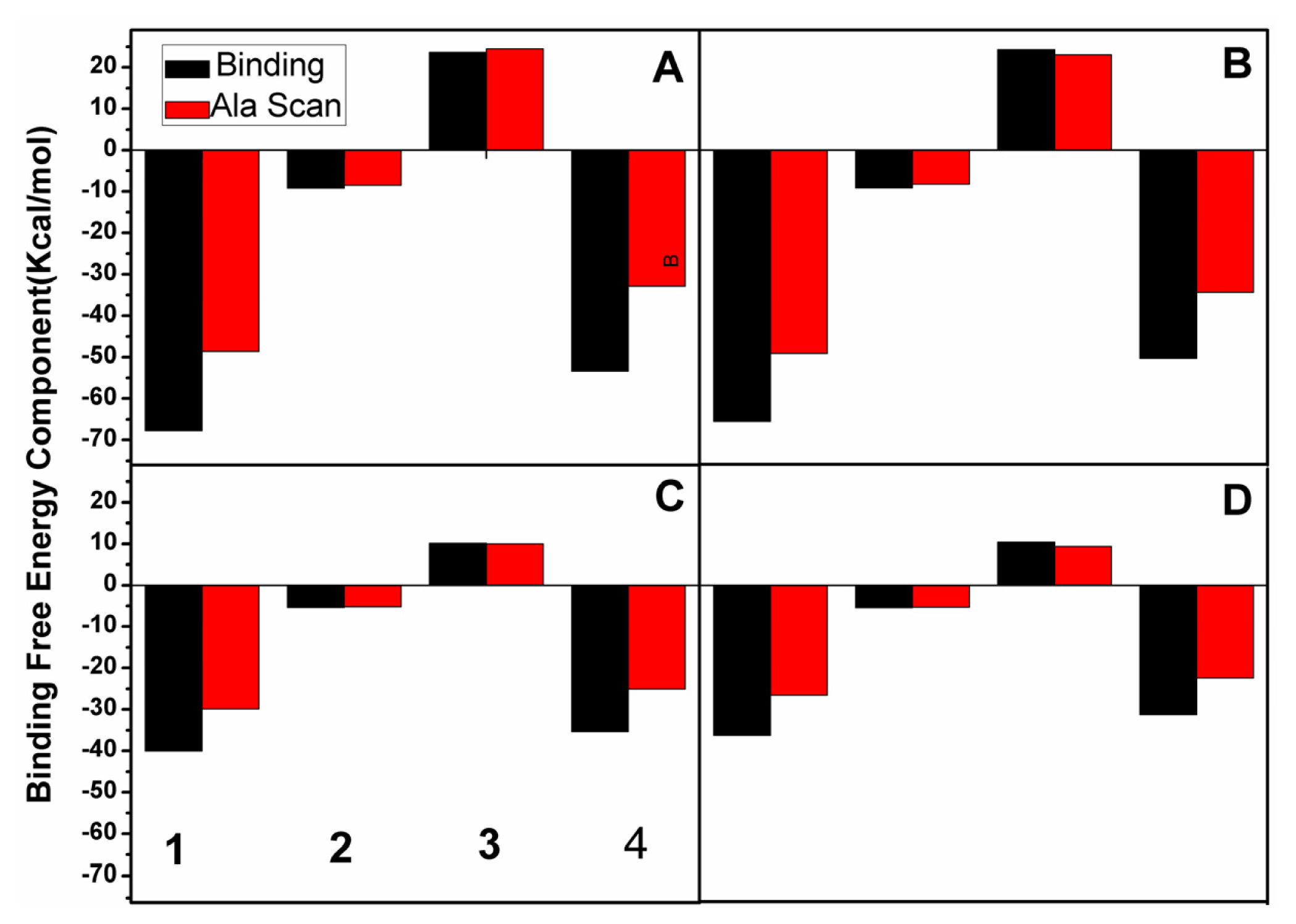
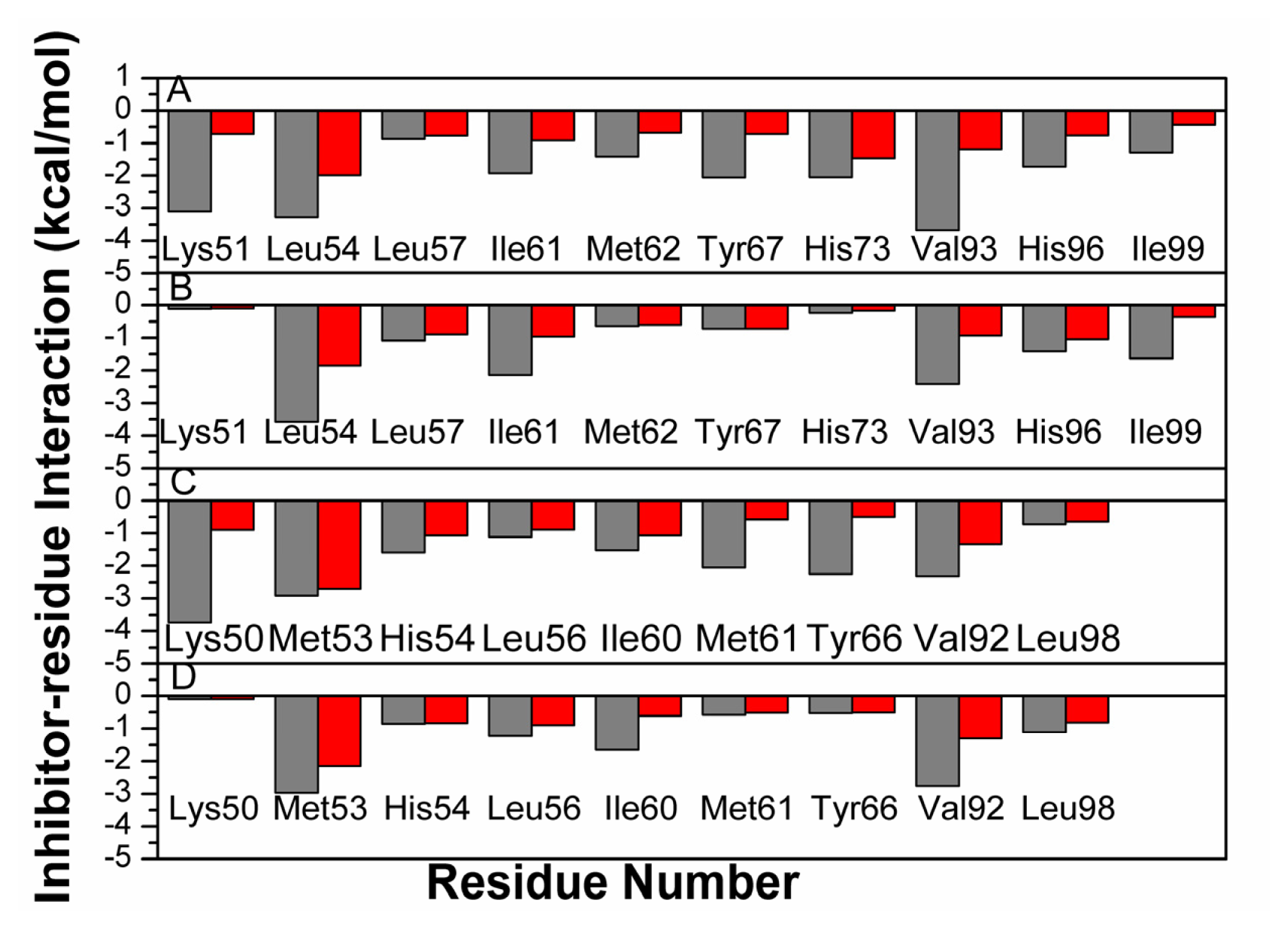
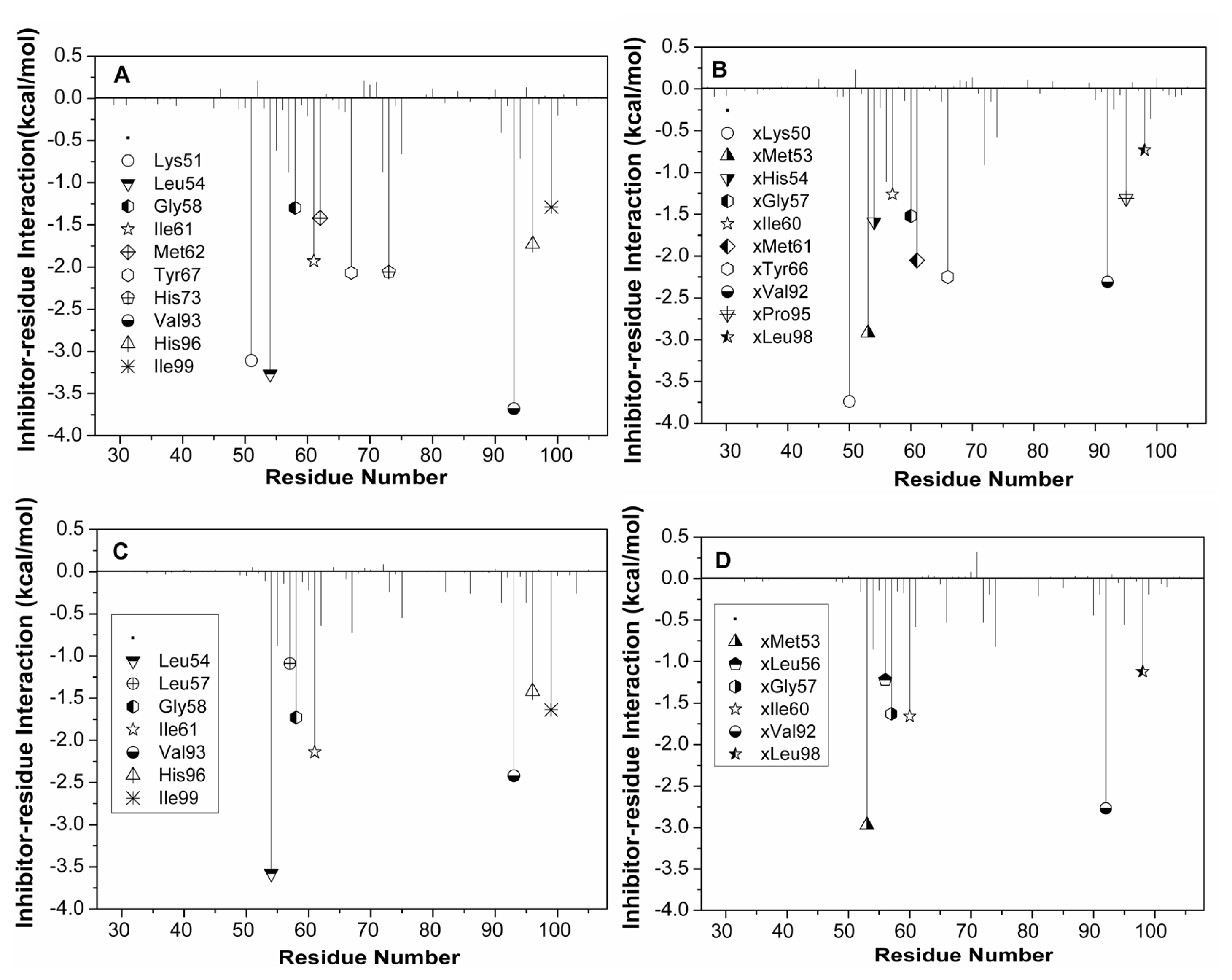
2.5. Computational Alanine Scanning
3. Experimental Section
3.1. System Preparation
3.2. Molecular Dynamics Simulations
3.3. MM-GBSA Calculations
3.4. Inhibitor-Residue Interaction Decomposition
3.5. Computational Alanine Scanning Mutagensis
4. Conclusions
Acknowledgment
References
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol 2007, 8, 275–283. [Google Scholar]
- Shmueli, A.; Oren, M. Regulation of p53 by Mdm2: Fate Is in the Numbers. Mol. Cell 2004, 13, 4–5. [Google Scholar]
- de Graaf, P.; Little, N.A.; Ramos, Y.F.M.; Meulmeester, E.; Letteboer, S.J.F.; Jochemsen, A.G. Hdmx protein stability is regulated by the ubiquitin ligase activity of Mdm2. J. Biol. Chem 2003, 278, 38315–38324. [Google Scholar]
- Uldrijan, S.; Pannekoek, W.J.; Vousden, K.H. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J 2006, 26, 102–112. [Google Scholar]
- Veprintsev, D.B.; Freund, S.; Andreeva, A.; Rutledge, S.E.; Tidow, H.; Cañadillas, J.M.P.; Blair, C.M.; Fersht, A.R. Core domain interactions in full-length p53 in solution. Proc. Natl. Acad. Sci. USA 2006, 103, 2115–2118. [Google Scholar]
- Popowicz, G.M.; Czarna, A.; Wolf, S. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53-MDMX/MDM2 antagonist drug discovery. Cell Cycle 2010, 9, 1104–1111. [Google Scholar]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar]
- Hu, B.; Gilkes, D.M.; Farooqi, B.; Sebti, S.M.; Chen, J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J. Biol. Chem 2006, 281, 33030–33035. [Google Scholar]
- Wade, M.; Wang, Y.V.; Wahl, G.M. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 2010, 20, 299–309. [Google Scholar]
- Czarna, A.; Popowicz, G.M.; Pecak, A.; Wolf, S.; Dubin, G.; Holak, T.A. High affinity interaction of the p53 peptide-analogue with human Mdm2 and Mdmx. Cell Cycle 2009, 8, 1176–1184. [Google Scholar]
- Marine, J.C.W.; Dyer, M.A.; Jochemsen, A.G. MDMX: From bench to bedside. J. Cell Sci 2007, 120, 371–378. [Google Scholar]
- Boettger, V.; Boettger, A.; Garcia-Echeverria, C.; Ramos, Y.; van der Eb, A.J.; Jochemsen, A.G.; Lane, D.P. Comparative study of the p53-mdm2 and p53-MDMX interfaces. Oncogene 1999, 18, 189–199. [Google Scholar]
- Murray, J.K.; Gellman, S.H. Targeting protein–protein interactions: Lessons from p53/MDM2. Biopolymers 2007, 88, 657–686. [Google Scholar]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar]
- Vassilev, L.T. MDM2 inhibitors for cancer therapy. Trends. Mol. Med 2007, 13, 23–31. [Google Scholar]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar]
- Bixby, D.; Kujawski, L.; Wang, S.; Malek, S.N. The pre-clinical development of MDM2 inhibitors in chronic lymphocytic leukemia uncovers a central role for p53 status in sensitivity to Mdm2 inhibitor-mediated apoptosis. Cell Cycle 2008, 7, 971–979. [Google Scholar]
- Cheok, C.F.; Lane, D.P. New developments in small molecules targeting p53 pathways in anticancer therapy. Drug Dev. Res 2008, 69, 289–296. [Google Scholar]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol 2009, 49, 223–241. [Google Scholar]
- Tanimura, S.; Ohtsuka, S.; Mitsui, K.; Shirouzu, K.; Yoshimura, A.; Ohtsubo, M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett 1999, 447, 5–9. [Google Scholar]
- Jackson, M.W.; Berberich, S.J. MdmX protects p53 from Mdm2-mediated degradation. Mol. Cell. Biol 2000, 20, 1001–1007. [Google Scholar]
- Pirolli, D.; Carelli Alinovi, C.; Capoluongo, E.; Satta, M.A.; Concolino, P.; Giardina, B.; De Rosa, M.C. Insight into a novel p53 single point mutation (G389E) by molecular dynamics simulations. Int. J. Mol. Sci 2010, 12, 128–140. [Google Scholar]
- Popowicz, G.M.; Czarna, A.; Rothweiler, U.; Szwagierczak, A.; Krajewski, M.; Weber, L.; Holak, T.A. Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle 2007, 6, 2386–2392. [Google Scholar]
- Joseph, T.L.; Madhumalar, A.; Brown, C.J.; Lane, D.P.; Verma, C. Differential binding of p53 and nutlin to MDM2 and MDMX: Computational studies. Cell Cycle 2010, 9, 1167–1181. [Google Scholar]
- Li, C.; Pazgier, M.; Yuan, W.; Liu, M.; Wei, G.; Lu, W.Y.; Lu, W. Systematic mutational analysis of peptide inhibition of the p53-MDM2/MDMX interactions. J. Mol. Biol 2010, 398, 200–213. [Google Scholar]
- Lu, S.Y.; Jiang, Y.J.; Zou, J.W.; Wu, T.X. Molecular modeling and molecular dynamics simulation studies on pyrrolopyrimidine-based [alpha]-helix mimetic as dual inhibitors of MDM2 and MDMX. J. Mol. Graph. Mod 2011, 30, 167–178. [Google Scholar]
- Reed, D.; Shen, Y.; Shelat, A.A.; Arnold, L.A.; Ferreira, A.M.; Zhu, F.; Mills, N.; Smithson, D.C.; Regni, C.A.; Bashford, D. Identification and characterization of the first small molecule inhibitor of MDMX. J. Biol. Chem 2010, 285, 10786–10796. [Google Scholar]
- Dezi, C.; Carotti, A.; Magnani, M.; Baroni, M.; Padova, A.; Cruciani, G.; Macchiarulo, A.; Pellicciari, R. Molecular interaction fields and 3D-QSAR studies of p53-MDM2 inhibitors suggest additional features of ligand-target interaction. J. Chem. Inf. Model 2010, 50, 1451–1465. [Google Scholar]
- Phan, J.; Li, Z.; Kasprzak, A.; Li, B.; Sebti, S.; Guida, W.; Schönbrunn, E.; Chen, J. Structure-based design of high affinity peptides inhibiting the interaction of p53 with MDM2 and MDMX. J. Biol. Chem 2010, 285, 2174–2183. [Google Scholar]
- Kallen, J.; Goepfert, A.; Blechschmidt, A.; Izaac, A.; Geiser, M.; Tavares, G.; Ramage, P.; Furet, P.; Masuya, K.; Lisztwan, J. Crystal structures of human MdmX (HdmX) in complex with p53 peptide analogues reveal surprising conformational changes. J. Biol. Chem 2009, 284, 8812–8821. [Google Scholar]
- Hu, G.; Wang, D.; Liu, X.; Zhang, Q. A computational analysis of the binding model of MDM2 with inhibitors. J. Comput. Aided Mol. Des 2010, 24, 687–697. [Google Scholar]
- Zhong, H.; Carlson, H.A. Computational studies and peptidomimetic design for the human p53–MDM2 complex. Proteins 2005, 58, 222–234. [Google Scholar]
- Cummins, P.L.; Gready, J.E. Computer-aided drug design: A free energy perturbation study on the binding of methyl-substituted pterins and N5-deazapterins to dihydrofolate reductase. J. Comput. Aided Mol. Des 1993, 7, 535–555. [Google Scholar]
- Kuhn, B.; Kollman, P.A.; Stahl, M. Prediction of pKa shifts in proteins using a combination of molecular mechanical and continuum solvent calculations. J. Comput. Chem 2004, 25, 1865–1872. [Google Scholar]
- Jiao, D.; Zhang, J.; Duke, R.E.; Li, G.; Schnieders, M.J.; Ren, P. Trypsin ligand binding free energies from explicit and implicit solvent simulations with polarizable potential. J. Comput. Chem 2009, 30, 1701–1711. [Google Scholar]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc 2001, 123, 5221–5230. [Google Scholar]
- Wang, W.; Kollman, P.A. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model1. J. Mol. Biol 2000, 303, 567–582. [Google Scholar]
- Wang, W.; Lim, W.A.; Jakalian, A.; Wang, J.; Luo, R.; Bayly, C.I.; Kollman, P.A. An analysis of the interactions between the Sem-5 SH3 domain and its ligands using molecular dynamics, free energy calculations, and sequence analysis. J. Am. Chem. Soc 2001, 123, 3986–3994. [Google Scholar]
- Swanson, J.M.J.; Henchman, R.H.; McCammon, J.A. Revisiting free energy calculations: A theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys. J 2004, 86, 67–74. [Google Scholar]
- Tong, Y.; Mei, Y.; Li, Y.L.; Ji, C.G.; Zhang, J.Z.H. Electrostatic polarization makes a substantial contribution to the free energy of avidin-biotin binding. J. Am. Chem. Soc 2010, 132, 5137–5142. [Google Scholar]
- Chen, J.; Wang, J.; Xu, B.; Zhu, W.; Li, G. Insight into Mechanism of small molecule inhibitors of the MDM2-p53 interaction: Molecular dynamics simulation and free energy analysis. J. Mol. Graph. Model 2011, 30, 46–53. [Google Scholar]
- Chen, J.; Zhang, S.; Liu, X.; Zhang, Q. Insights into drug resistance of mutations D30N and I50V to HIV-1 protease inhibitor TMC-114: Free energy calculation and molecular dynamic simulation. J. Mol. Model 2010, 16, 459–468. [Google Scholar]
- Wu, E.L.; Han, K.L.; Zhang, J.Z.H. Selectivity of neutral/weakly basic P1 group inhibitors of thrombin and trypsin by a molecular dynamics study. Chem. Eur. J 2008, 14, 8704–8714. [Google Scholar]
- Hou, T.; Yu, R. Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: Mechanism for binding and drug resistance. J. Med. Chem 2007, 50, 1177–1188. [Google Scholar]
- Xu, Y.; Wang, R. A computational analysis of the binding affinities of FKBP12 inhibitors using the MM PB/SA method. Proteins 2006, 64, 1058–1068. [Google Scholar]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem 1996, 100, 19824–19839. [Google Scholar]
- Mongan, J.; Simmerling, C.; McCammon, J.A.; David, A.; Onufriev, A. Generalized Born model with a simple, robust molecular volume correction. J. Chem. Theory. Comput 2007, 3, 156–169. [Google Scholar]
- Riley, K.E.; Merz, K.M., Jr. Insights into the strength and origin of halogen bonding: The halobenzene-formaldehyde dimer. J. Phys.Chem. A 2007, 111, 1688–1694. [Google Scholar]
- Ding, Y.; Mei, Y.; Zhang, J.Z.H.; Tao, F.M. Efficient bond function basis set for interaction energies. J. Comput. Chem 2008, 29, 275–279. [Google Scholar]
- Kovalevsky, A.Y.; Liu, F.; Leshchenko, S.; Ghosh, A.K.; Louis, J.M.; Harrison, R.W.; Weber, I.T. Ultra-high resolution crystal structure of HIV-1 protease mutant reveals two binding sites for clinical inhibitor TMC114. J. Mol. Biol 2006, 363, 161–173. [Google Scholar]
- Kovalevsky, A.Y.; Tie, Y.; Liu, F.; Boross, P.I.; Wang, Y.F.; Leshchenko, S.; Ghosh, A.K.; Harrison, R.W.; Weber, I.T. Effectiveness of nonpeptide clinical inhibitor TMC-114 on HIV-1 protease with highly drug resistant mutations D30N, I50V, and L90M. J. Med. Chem 2006, 49, 1379–1387. [Google Scholar]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, I.C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.C.; Zhang, W.; et al. AMBER 10; University of California, San Francisco: San Francisco, CA, USA, 2008. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 98; Gaussian, Inc: Pittsburgh, PA, USA, 1998. [Google Scholar]
- Cieplak, P.; Cornell, W.D.; Bayly, C.; Kollman, P.A. Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem 1995, 16, 1357–1377. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- Coleman, T.G.; Mesick, H.C.; Darby, R.L. Numerical integration. Ann. Biomed. Eng 1977, 5, 322–328. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- Connolly, M.L. Analytical molecular surface calculation. J. Appl. Cryst 1983, 16, 548–558. [Google Scholar]
- Hao, G.F.; Yang, G.F. The role of Phe82 and Phe351 in auxin-induced substrate perception by TIR1 ubiquitin ligase: A novel insight from molecular dynamics simulations. PLoS One 2010, 5, e10742. [Google Scholar]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol 2003, 330, 891–913. [Google Scholar]
- Massova, I.; Kollman, P.A. Computational alanine scanning to probe protein-protein interactions: a novel approach to evaluate binding free energies. J. Am. Chem. Soc 1999, 121, 8133–8143. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components b | pDI6W + MDM2 | pDI6W + MDMX | WK23 + MDM2 | WK23 + MDMX | ||||
|---|---|---|---|---|---|---|---|---|
| mean | std c | Mean | std c | mean | std c | mean | std c | |
| ΔGele | −135.20 | 0.41 | −144.89 | 0.32 | −0.95 | 0.01 | −1.78 | 0.05 |
| ΔGvdw | −67.76 | 0.16 | −65.49 | 0.17 | −40.05 | 0.21 | −36.26 | 0.31 |
| ΔGpol | 149.67 | 0.31 | 169.18 | 0.36 | 11.05 | 0.08 | 12.18 | 0.10 |
| ΔGnopol | −9.18 | 0.05 | −9.11 | 0.02 | −5.36 | 0.21 | −5.41 | 0.02 |
| ΔGele + pol | 23.65 | 0.20 | 24.29 | 0.20 | 10.09 | 0.06 | 10.39 | 0.07 |
| ΔGgb | −53.29 | 0.23 | −50.31 | 0.24 | −35.31 | 0.14 | −31.28 | 0.14 |
| −TΔS | 31.36 | 0.12 | 30.57 | 0.21 | 18.50 | 0.11 | 15.61 | 0.18 |
| ΔGbind | −21.93 | −19.74 | −16.81 | −14.89 | ||||
| ΔGexpd | −10.5 | −9.73 | −8.26 | −6.08 | ||||
| Protein | Donor a | Acceptor | Distance(Å) b | Angle(°) b | Occupancy(%) c |
|---|---|---|---|---|---|
| MDM2 | Trp23′-NE1-HE1 | Leu54-O | 2.92 | 146.79 | 97.20 |
| Phe19′-N-H | Gln72-OE1 | 3.01 | 151.71 | 71.34 | |
| WK23-N8-H75 | Leu54-O | 2.90 | 154.12 | 98.95 | |
| MDMX | Trp23′-NE1-HE1 | Met53-O | 2.89 | 140.29 | 98.76 |
| Phe19′-N-H | Gln71-OE1 | 3.01 | 150.94 | 69.66 | |
| WK23-N8-H103 | Met53-O | 2.92 | 153.75 | 99.29 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, J.; Zhang, D.; Zhang, Y.; Li, G. Computational Studies of Difference in Binding Modes of Peptide and Non-Peptide Inhibitors to MDM2/MDMX Based on Molecular Dynamics Simulations. Int. J. Mol. Sci. 2012, 13, 2176-2195. https://doi.org/10.3390/ijms13022176
Chen J, Zhang D, Zhang Y, Li G. Computational Studies of Difference in Binding Modes of Peptide and Non-Peptide Inhibitors to MDM2/MDMX Based on Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2012; 13(2):2176-2195. https://doi.org/10.3390/ijms13022176
Chicago/Turabian StyleChen, Jianzhong, Dinglin Zhang, Yuxin Zhang, and Guohui Li. 2012. "Computational Studies of Difference in Binding Modes of Peptide and Non-Peptide Inhibitors to MDM2/MDMX Based on Molecular Dynamics Simulations" International Journal of Molecular Sciences 13, no. 2: 2176-2195. https://doi.org/10.3390/ijms13022176
APA StyleChen, J., Zhang, D., Zhang, Y., & Li, G. (2012). Computational Studies of Difference in Binding Modes of Peptide and Non-Peptide Inhibitors to MDM2/MDMX Based on Molecular Dynamics Simulations. International Journal of Molecular Sciences, 13(2), 2176-2195. https://doi.org/10.3390/ijms13022176