Antioxidant-Induced Stress

Abstract
:
1. Introduction
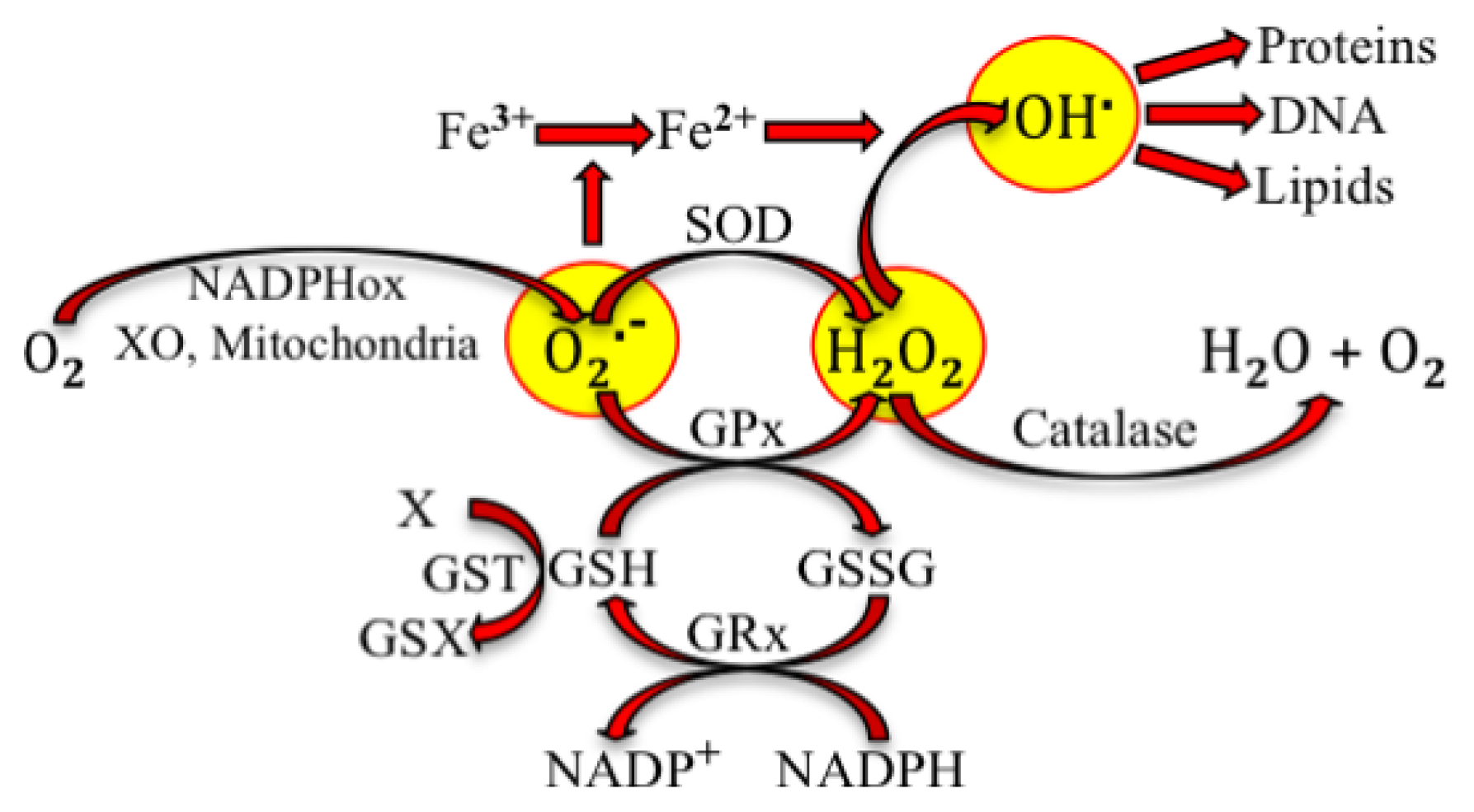
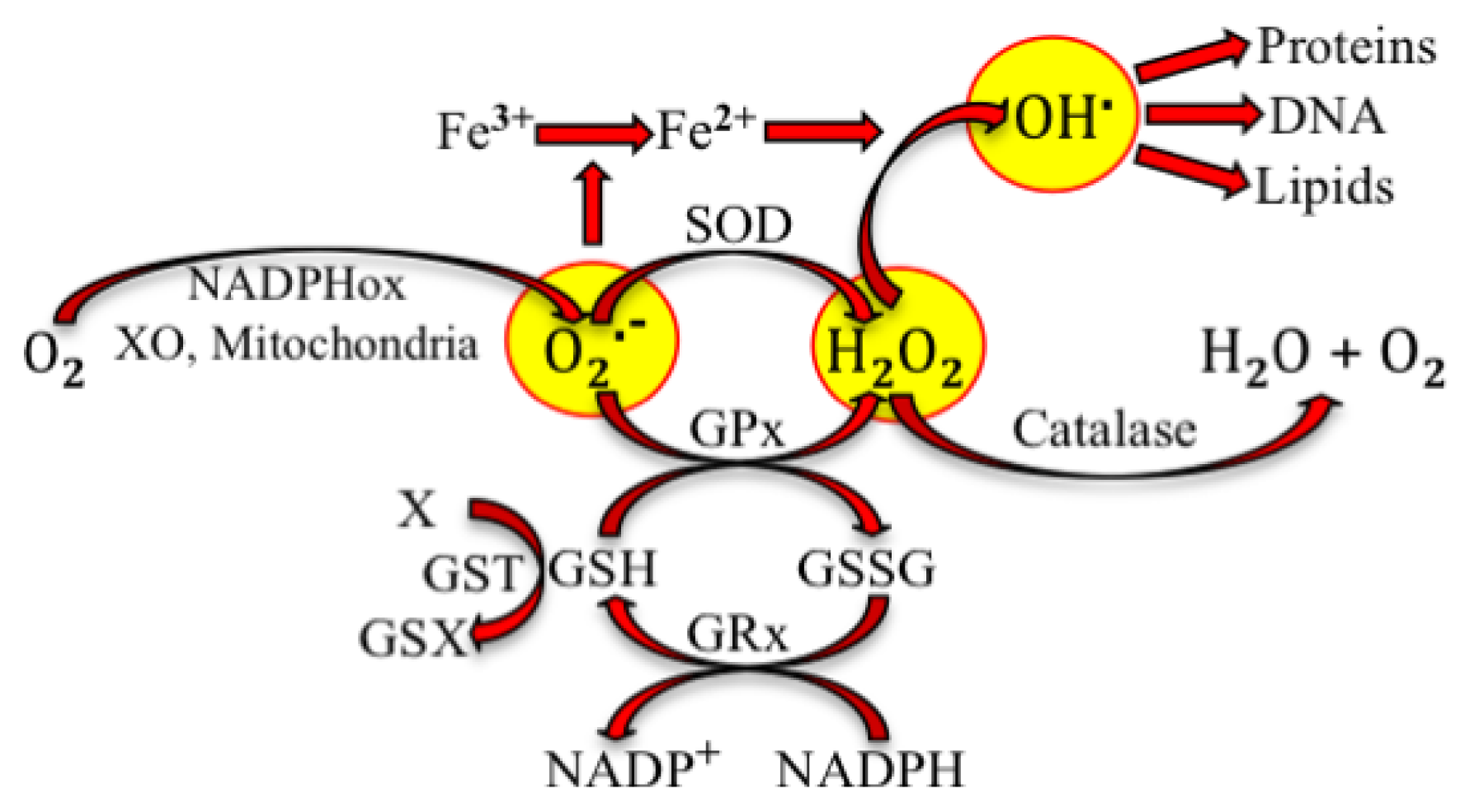
2. Physiological Effects of Reactive Species
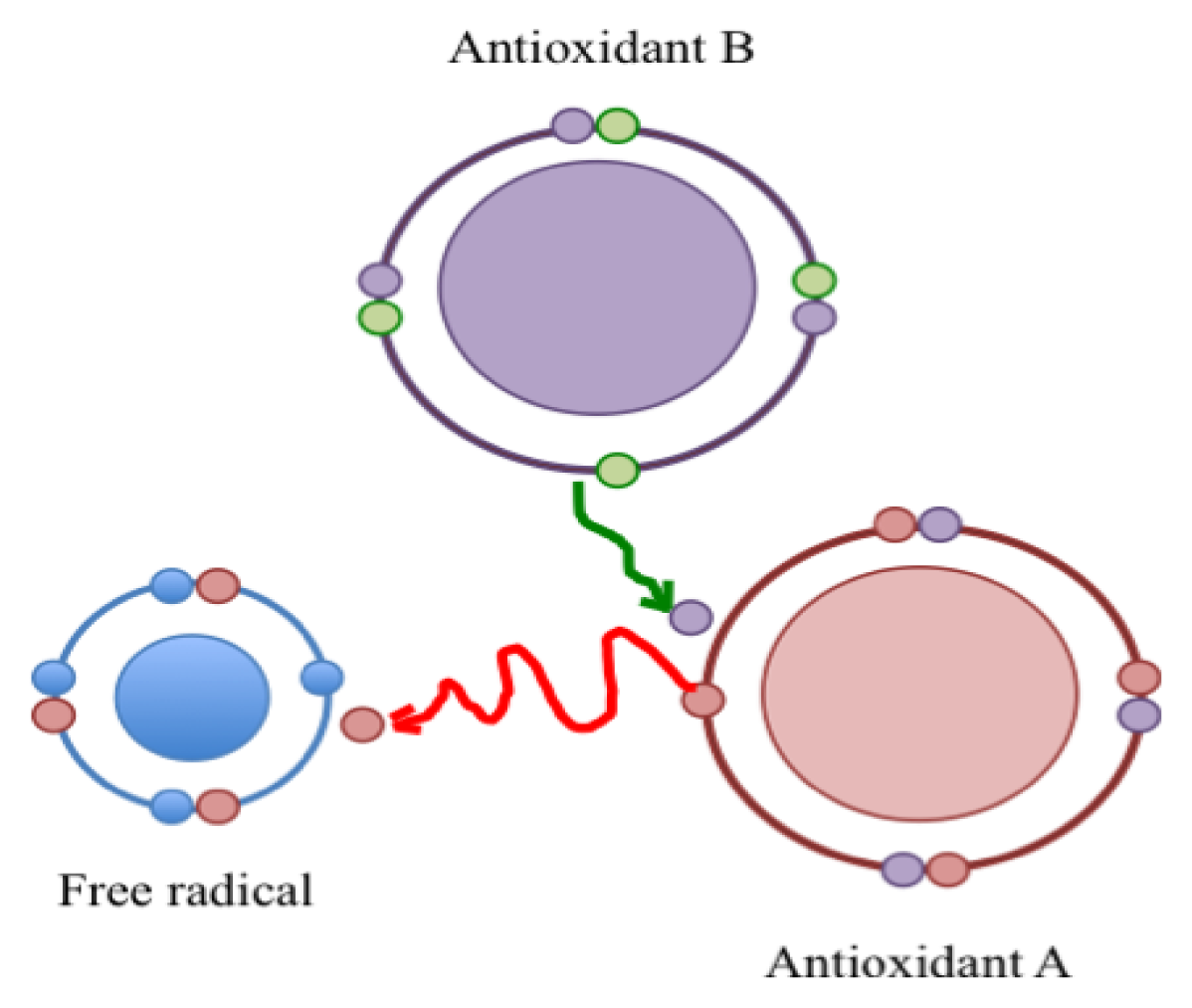
3. Pro-Oxidant Effects of Antioxidants
4. Clinical Trials with Antioxidants
4.1. Beneficial Effects of Antioxidants
4.2. Antioxidants Do not Change the Evolution of Diseases Related to Oxidative Stress
4.3. Harmful Effects of Antioxidants
5. Conclusions
Acknowledgments
References
- McCord, J.M.; Keele, B.B., Jr; Fridovich, I. An enzyme-based theory of obligate anaerobiosis: The physiological function of superoxide dismutase. Proc. Natl. Acad. Sci. USA 1971, 68, 1024–1027. [Google Scholar]
- Laranjinha, J. Oxidative Stress: From 1980’s to Recent Update. In Oxidative Stress, Inflammation and Angiogenesis in the Metabolic Syndrome; Soares, R., Costa, C., Eds.; Springer, Science + business Media: New York, NY, USA, 2009; pp. 21–32. [Google Scholar]
- Gutierrez, J.; Ballinger, S.W.; Darley-Usmar, V.M.; Landar, A. Free radicals, mitochondria, and oxidized lipids: The emerging role in signal transduction in vascular cells. Circ. Res 2006, 99, 924–932. [Google Scholar]
- Durackova, Z. Some current insights into oxidative stress. Physiol. Res 2010, 59, 459–469. [Google Scholar]
- Halliwell, B. The wanderings of a free radical. Free Radic. Biol. Med 2009, 46, 531–542. [Google Scholar]
- Davies, M.J.; Fu, S.; Dean, R.T. Protein hydroperoxides can give rise to reactive free radicals. Biochem. J 1995, 305, 643–649. [Google Scholar]
- Narwaley, M.; Michail, K.; Arvadia, P.; Siraki, A.G. Drug-induced protein free radical formation is attenuated by unsaturated fatty acids by scavenging drug-derived phenyl radical metabolites. Chem. Res. Toxicol 2011, 24, 1031–1039. [Google Scholar]
- Rahmanto, A.S.; Morgan, P.E.; Hawkins, C.L.; Davies, M.J. Cellular effects of peptide and protein hydroperoxides. Free Radic. Biol. Med 2010, 48, 1071–1078. [Google Scholar]
- Yamada, K.; Yamamiya, I.; Utsumi, H. In vivo detection of free radicals induced by diethylnitrosamine in rat liver tissue. Free Radic. Biol. Med 2006, 40, 2040–2046. [Google Scholar]
- North, J.A.; Spector, A.A.; Buettner, G.R. Detection of lipid radicals by electron paramagnetic resonance spin trapping using intact cells enriched with polyunsaturated fatty acid. J. Biol. Chem 1992, 267, 5743–5746. [Google Scholar]
- Leonarduzzi, G.; Gamba, P.; Gargiulo, S.; Biasi, F.; Poli, G. Inflammation-related gene expression by lipid oxidation-derived products in the progression of atherosclerosis. Free Radic. Biol. Med 2011, 52, 19–34. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M. The definition and measurement of antioxidants in biological systems. Free Radic. Biol. Med 1995, 18, 125–126. [Google Scholar]
- Dündar, Y.; Aslan, R. Antioxidative stress. Eastern J. Med 2000, 5, 45–47. [Google Scholar]
- Pechanova, O.; Simko, F. Chronic antioxidant therapy fails to ameliorate hypertension: Potential mechanisms behind. J. Hypertens Suppl 2009, 27, S32–S36. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol 2007, 39, 44–84. [Google Scholar]
- Nemoto, M.; Nishimura, R.; Sasaki, T.; Hiki, Y.; Miyashita, Y.; Nishioka, M.; Fujimoto, K.; Sakuma, T.; Ohashi, T.; Fukuda, K.; et al. Genetic association of glutathione peroxidase-1 with coronary artery calcification in type 2 diabetes: A case control study with multi-slice computed tomography. Cardiovasc. Diabetol 2007, 6, 23:1–23:7. [Google Scholar]
- Voetsch, B.; Jin, R.C.; Bierl, C.; Benke, K.S.; Kenet, G.; Simioni, P.; Ottaviano, F.; Damasceno, B.P.; Annichino-Bizacchi, J.M.; Handy, D.E.; et al. Promoter polymorphisms in the plasma glutathione peroxidase (gpx-3) gene: A novel risk factor for arterial ischemic stroke among young adults and children. Stroke 2007, 38, 41–49. [Google Scholar]
- Naganuma, T.; Nakayama, T.; Sato, N.; Fu, Z.; Soma, M.; Aoi, N.; Hinohara, S.; Doba, N.; Usami, R. Association of extracellular superoxide dismutase gene with cerebral infarction in women: A haplotype-based case-control study. Hereditas 2008, 145, 283–292. [Google Scholar]
- Samoila, O.C.; Carter, A.M.; Futers, S.T.; Otiman, G.; Anghel, A.; Tamas, L.; Seclaman, E. Polymorphic variants of extracellular superoxide dismutase gene in a romanian population with atheroma. Biochem. Genet 2008, 46, 634–643. [Google Scholar]
- Manfredi, S.; Federici, C.; Picano, E.; Botto, N.; Rizza, A.; Andreassi, M.G. Gstm1, gstt1 and cyp1a1 detoxification gene polymorphisms and susceptibility to smoking-related coronary artery disease: A case-only study. Mutat. Res 2007, 621, 106–112. [Google Scholar]
- Afanas’ev, I.B. Free radical mechanisms of aging processes under physiological conditions. Biogerontology 2005, 6, 283–290. [Google Scholar]
- Afanas’ev, I.B. Signaling functions of free radicals superoxide & nitric oxide under physiological & pathological conditions. Mol. Biotechnol 2007, 37, 2–4. [Google Scholar]
- Liaudet, L.; Vassalli, G.; Pacher, P. Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front. Biosci 2009, 14, 4809–4814. [Google Scholar]
- Cadenas, E. Mitochondrial free radical production and cell signaling. Mol. Aspects Med 2004, 25, 17–26. [Google Scholar]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Gu, G.J.; Li, Y.P.; Peng, Z.Y.; Xu, J.J.; Kang, Z.M.; Xu, W.G.; Tao, H.Y.; Ostrowski, R.P.; Zhang, J.H.; Sun, X.J. Mechanism of ischemic tolerance induced by hyperbaric oxygen preconditioning involves upregulation of hypoxia-inducible factor-1alpha and erythropoietin in rats. J. Appl. Physiol 2008, 104, 1185–1191. [Google Scholar]
- Glantz, L.; Avramovich, A.; Trembovler, V.; Gurvitz, V.; Kohen, R.; Eidelman, L.A.; Shohami, E. Ischemic preconditioning increases antioxidants in the brain and peripheral organs after cerebral ischemia. Exp. Neurol 2005, 192, 117–124. [Google Scholar]
- Obrenovitch, T.P. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol. Rev 2008, 88, 211–247. [Google Scholar]
- Pradillo, J.M.; Romera, C.; Hurtado, O.; Cardenas, A.; Moro, M.A.; Leza, J.C.; Davalos, A.; Castillo, J.; Lorenzo, P.; Lizasoain, I. Tnfr1 upregulation mediates tolerance after brain ischemic preconditioning. J. Cereb. Blood Flow Metab 2005, 25, 193–203. [Google Scholar]
- Vartanian, K.B.; Stevens, S.L.; Marsh, B.J.; Williams-Karnesky, R.; Lessov, N.S.; Stenzel-Poore, M.P. Lps preconditioning redirects tlr signaling following stroke: Trif-irf3 plays a seminal role in mediating tolerance to ischemic injury. J. Neuroinflamm 2011, 8, 140:1–140:12. [Google Scholar]
- Kaur, R.; Jaggi, A.S.; Singh, N. Studies on effect of stress preconditioning in restrain stress-induced behavioral alterations. Yakugaku Zasshi 2010, 130, 215–221. [Google Scholar]
- Gidday, J.M. Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci 2006, 7, 437–448. [Google Scholar]
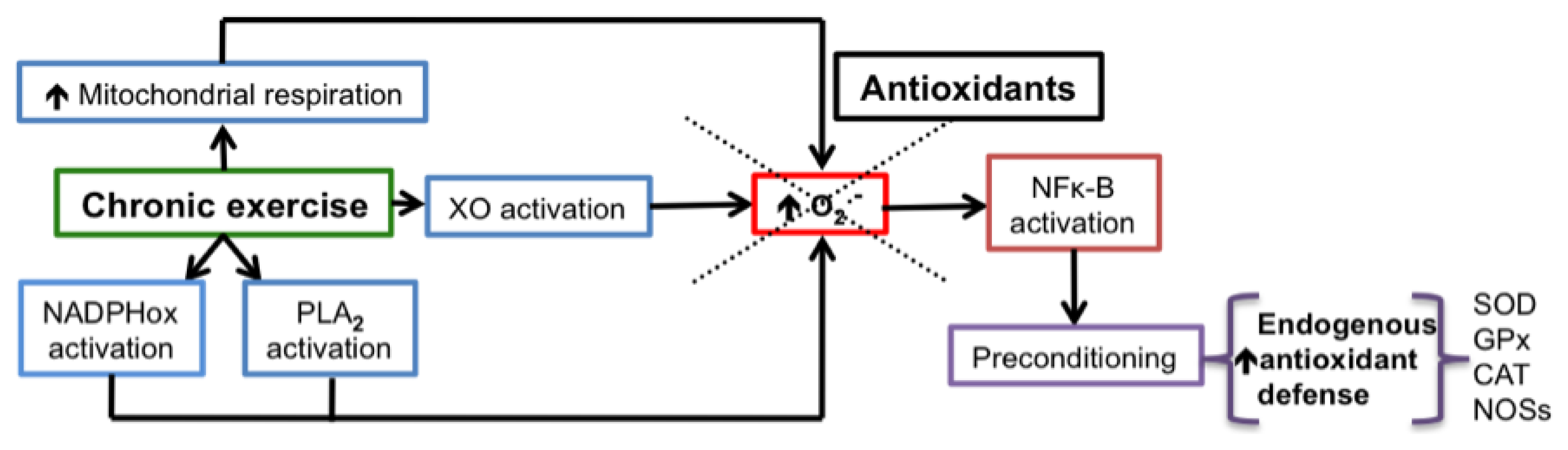
- Boveris, A.; Navarro, A. Systemic and mitochondrial adaptive responses to moderate exercise in rodents. Free Radic. Biol. Med 2008, 44, 224–229. [Google Scholar]
- Gomez-Cabrera, M.C.; Domenech, E.; Vina, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med 2008, 44, 126–131. [Google Scholar]
- Syu, G.D.; Chen, H.I.; Jen, C.J. Severe exercise and exercise training exert opposite effects on human neutrophil apoptosis via altering the redox status. PLoS One 2011, 6, e24385. [Google Scholar]
- Fisher-Wellman, K.; Bell, H.K.; Bloomer, R.J. Oxidative stress and antioxidant defense mechanisms linked to exercise during cardiopulmonary and metabolic disorders. Oxid. Med. Cell. Longev 2009, 2, 43–51. [Google Scholar]
- Lima-Cabello, E.; Cuevas, M.J.; Garatachea, N.; Baldini, M.; Almar, M.; Gonzalez-Gallego, J. Eccentric exercise induces nitric oxide synthase expression through nuclear factor-kappab modulation in rat skeletal muscle. J. Appl. Physiol 2010, 108, 575–583. [Google Scholar]
- George, L.; Lokhandwala, M.F.; Asghar, M. Exercise activates redox-sensitive transcription factors and restores renal d1 receptor function in old rats. Am. J. Physiol. Renal Physiol 2009, 297, F1174–F1180. [Google Scholar]
- Ji, L.L. Modulation of skeletal muscle antioxidant defense by exercise: Role of redox signaling. Free Radic. Biol. Med 2008, 44, 142–152. [Google Scholar]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar]
- Childs, A.; Jacobs, C.; Kaminski, T.; Halliwell, B.; Leeuwenburgh, C. Supplementation with vitamin c and n-acetyl-cysteine increases oxidative stress in humans after an acute muscle injury induced by eccentric exercise. Free Radic. Biol. Med 2001, 31, 745–753. [Google Scholar]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar]
- Peternelj, T.T.; Coombes, J.S. Antioxidant supplementation during exercise training: Beneficial or detrimental? Sports Med 2011, 41, 1043–1069. [Google Scholar]
- Sevanian, A.; Davies, K.J.; Hochstein, P. Serum urate as an antioxidant for ascorbic acid. Am. J. Clin. Nutr 1991, 54, 1129S–1134S. [Google Scholar]
- Pietta, P.G. Flavonoids as antioxidants. J. Nat. Prod 2000, 63, 1035–1042. [Google Scholar]
- Martin, H.D.; Ruck, C.; Schmidt, M.; Sell, S.; Beutner, S.; Mayer, B.; Walsh, R. Chemistry of carotenoid oxidation and free radical reactions. Pure Appl. Chem 1999, 71, 2253–2262. [Google Scholar]
- Halliwell, B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch. Biochem. Biophys 2008, 476, 107–112. [Google Scholar]
- Damiani, E.; Astolfi, P.; Carloni, P.; Stipa, P.; Greci, L. Antioxidants: How They Work. In Oxidants in Biology; Valacchi, G., Davis, P.A., Eds.; Springer Science + Buisness Media: New York, NY, USA, 2008; pp. 251–266. [Google Scholar]
- Botti, H.; Batthyany, C.; Trostchansky, A.; Radi, R.; Freeman, B.A.; Rubbo, H. Peroxynitrite-mediated alpha-tocopherol oxidation in low-density lipoprotein: A mechanistic approach. Free Radic. Biol. Med 2004, 36, 152–162. [Google Scholar]
- Maguire, J.J.; Wilson, D.S.; Packer, L. Mitochondrial electron transport-linked tocopheroxyl radical reduction. J. Biol. Chem 1989, 264, 21462–21465. [Google Scholar]
- Duracková, Z. Oxidants, Antioxidants and Oxidative Stress. In Mitochondrial Medicine; Gvozdjáková, A., Ed.; Springer Science + Business Media: New York, NY, USA, 2008; pp. 19–54. [Google Scholar]
- Liu, C.; Russell, R.M.; Wang, X.D. Alpha-tocopherol and ascorbic acid decrease the production of beta-apo-carotenals and increase the formation of retinoids from beta-carotene in the lung tissues of cigarette smoke-exposed ferrets in vitro. J. Nutr 2004, 134, 426–430. [Google Scholar]
- Yeum, K.J.; Aldini, G.; Russell, R.M.; Krinsky, N.I. Antioxidant/pro-oxidant Actions of Carotenoids. In Carotenoids; Birkhäuser Verlag: Basel, Switzerland, 2009; Volume 5, pp. 235–268. [Google Scholar]
- Moini, H.; Packer, L.; Saris, N.E. Antioxidant and prooxidant activities of alpha-lipoic acid and dihydrolipoic acid. Toxicol. Appl. Pharmacol 2002, 182, 84–90. [Google Scholar]
- Poljsak, B.; Gazdag, Z.; Jenko-Brinovec, S.; Fujs, S.; Pesti, M.; Belagyi, J.; Plesnicar, S.; Raspor, P. Pro-oxidative vs antioxidative properties of ascorbic acid in chromium(vi)-induced damage: An in vivo and in vitro approach. J. Appl. Toxicol 2005, 25, 535–548. [Google Scholar]
- Maurya, D.K.; Devasagayam, T.P. Antioxidant and prooxidant nature of hydroxycinnamic acid derivatives ferulic and caffeic acids. Food Chem. Toxicol 2010, 48, 3369–3373. [Google Scholar]
- Lakshman, M.R. Alpha and omega of carotenoid cleavage. J. Nutr 2004, 134, 241S–245S. [Google Scholar]
- Polyakov, N.E.; Leshina, T.V.; Konovalova, T.A.; Kispert, L.D. Carotenoids as scavengers of free radicals in a fenton reaction: Antioxidants or pro-oxidants? Free Radic. Biol. Med 2001, 31, 398–404. [Google Scholar]
- Siems, W.; Wiswedel, I.; Salerno, C.; Crifo, C.; Augustin, W.; Schild, L.; Langhans, C.D.; Sommerburg, O. Beta-carotene breakdown products may impair mitochondrial functions--potential side effects of high-dose beta-carotene supplementation. J. Nutr. Biochem 2005, 16, 385–397. [Google Scholar]
- Siems, W.; Salerno, C.; Crifo, C.; Sommerburg, O.; Wiswedel, I. Beta-carotene degradation products - formation, toxicity and prevention of toxicity. Forum Nutr 2009, 61, 75–86. [Google Scholar]
- Long, L.H.; Hoi, A.; Halliwell, B. Instability of, and generation of hydrogen peroxide by, phenolic compounds in cell culture media. Arch. Biochem. Biophys 2010, 501, 162–169. [Google Scholar]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans 2007, 35, 1147–1150. [Google Scholar]
- Halliwell, B.; Lee, C.Y. Using isoprostanes as biomarkers of oxidative stress: Some rarely considered issues. Antioxid. Redox Signal 2010, 13, 145–156. [Google Scholar]
- Gutteridge, J.M.; Halliwell, B. Antioxidants: Molecules, medicines, and myths. Biochem. Biophys. Res Commun 2010, 393, 561–564. [Google Scholar]
- Fortes, C.; Virgili, F. Antioxidant vitamins are not “Just antioxidants”: Not necessarily harmful when targeted to the right population. Biofactors 2008, 33, 177–180. [Google Scholar]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med 2011, 51, 327–336. [Google Scholar]
- Bowman, T.S.; Bassuk, S.S.; Gaziano, M. Interventional Trials of Antioxidants. In Atherosclerosis and Oxidant Stress: A New Perspective; Hotzman, J.L., Ed.; Springer: New York, NY, USA, 2007; pp. 25–50. [Google Scholar]
- Stampfer, M.J.; Hennekens, C.H.; Manson, J.E.; Colditz, G.A.; Rosner, B.; Willett, W.C. Vitamin e consumption and the risk of coronary disease in women. N. Engl. J. Med 1993, 328, 1444–1449. [Google Scholar]
- Rimm, E.B.; Stampfer, M.J.; Ascherio, A.; Giovannucci, E.; Colditz, G.A.; Willett, W.C. Vitamin e consumption and the risk of coronary heart disease in men. N. Engl. J. Med 1993, 328, 1450–1456. [Google Scholar]
- Losonczy, K.G.; Harris, T.B.; Havlik, R.J. Vitamin e and vitamin c supplement use and risk of all-cause and coronary heart disease mortality in older persons: The established populations for epidemiologic studies of the elderly. Am. J. Clin. Nutr 1996, 64, 190–196. [Google Scholar]
- Hasnain, B.I.; Mooradian, A.D. Recent trials of antioxidant therapy: What should we be telling our patients? Cleve. Clin. J. Med 2004, 71, 327–334. [Google Scholar]
- Todd, S.; Woodward, M.; Tunstall-Pedoe, H.; Bolton-Smith, C. Dietary antioxidant vitamins and fiber in the etiology of cardiovascular disease and all-causes mortality: Results from the scottish heart health study. Am. J. Epidemiol 1999, 150, 1073–1080. [Google Scholar]
- Myung, S.K.; Ju, W.; Kim, S.C.; Kim, H. Vitamin or antioxidant intake (or serum level) and risk of cervical neoplasm: A meta-analysis. BJOG: Int. J. Obstet. Gynaecol 2011, 118, 1285–1291. [Google Scholar]
- Stolzenberg-Solomon, R.Z.; Sheffler-Collins, S.; Weinstein, S.; Garabrant, D.H.; Mannisto, S.; Taylor, P.; Virtamo, J.; Albanes, D. Vitamin e intake, alpha-tocopherol status, and pancreatic cancer in a cohort of male smokers. Am. J. Clin. Nutr 2009, 89, 584–591. [Google Scholar]
- Weinstein, S.J.; Wright, M.E.; Lawson, K.A.; Snyder, K.; Mannisto, S.; Taylor, P.R.; Virtamo, J.; Albanes, D. Serum and dietary vitamin e in relation to prostate cancer risk. Cancer Epidemiol. Biomark. Prev 2007, 16, 1253–1259. [Google Scholar]
- Bobe, G.; Weinstein, S.J.; Albanes, D.; Hirvonen, T.; Ashby, J.; Taylor, P.R.; Virtamo, J.; Stolzenberg-Solomon, R.Z. Flavonoid intake and risk of pancreatic cancer in male smokers (finland). Cancer Epidemiol. Biomark. Prev 2008, 17, 553–562. [Google Scholar]
- Heart Protection Study Collaborative Group. Mrc/bhf heart protection study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 23–33.
- Klipstein-Grobusch, K.; Geleijnse, J.M.; den Breeijen, J.H.; Boeing, H.; Hofman, A.; Grobbee, D.E.; Witteman, J.C. Dietary antioxidants and risk of myocardial infarction in the elderly: The rotterdam study. Am. J. Clin. Nutr 1999, 69, 261–266. [Google Scholar]
- Yusuf, S.; Dagenais, G.; Pogue, J.; Bosch, J.; Sleight, P. Vitamin e supplementation and cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study investigators. N. Engl. J. Med 2000, 342, 154–160. [Google Scholar]
- GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin e after myocardial infarction: Results of the gissi-prevenzione trial. Gruppo italiano per lo studio della sopravvivenza nell’infarto miocardico. Lancet 1999, 354, 447–455.
- Rytter, E.; Vessby, B.; Asgard, R.; Ersson, C.; Moussavian, S.; Sjodin, A.; Abramsson-Zetterberg, L.; Moller, L.; Basu, S. Supplementation with a combination of antioxidants does not affect glycaemic control, oxidative stress or inflammation in type 2 diabetes subjects. Free Radic. Res 2010, 44, 1445–1453. [Google Scholar]
- Suksomboon, N.; Poolsup, N.; Sinprasert, S. Effects of vitamin e supplementation on glycaemic control in type 2 diabetes: Systematic review of randomized controlled trials. J. Clin. Pharm. Ther 2011, 36, 53–63. [Google Scholar]
- Arain, M.A.; Abdul Qadeer, A. Systematic review on “Vitamin e and prevention of colorectal cancer”. Pak. J. Pharm. Sci 2010, 23, 125–130. [Google Scholar]
- Ward, N.C.; Hodgson, J.M.; Croft, K.D.; Burke, V.; Beilin, L.J.; Puddey, I.B. The combination of vitamin c and grape-seed polyphenols increases blood pressure: A randomized, double-blind, placebo-controlled trial. J. Hypertens 2005, 23, 427–434. [Google Scholar]
- Cheung, M.C.; Zhao, X.Q.; Chait, A.; Albers, J.J.; Brown, B.G. Antioxidant supplements block the response of hdl to simvastatin-niacin therapy in patients with coronary artery disease and low hdl. Arterioscler. Thromb. Vasc. Biol 2001, 21, 1320–1326. [Google Scholar]
- Kim, Y.I. Does a high folate intake increase the risk of breast cancer? Nutr. Rev 2006, 64, 468–475. [Google Scholar]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L., Jr; Valanis, B.; Williams, J.H., Jr; et al. Risk factors for lung cancer and for intervention effects in caret, the beta-carotene and retinol efficacy trial. J. Natl. Cancer Inst. 1996, 88, 1550–1559. [Google Scholar]
- Albanes, D.; Heinonen, O.P.; Taylor, P.R.; Virtamo, J.; Edwards, B.K.; Rautalahti, M.; Hartman, A.M.; Palmgren, J.; Freedman, L.S.; Haapakoski, J.; et al. Alpha-tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: Effects of base-line characteristics and study compliance. J. Natl. Cancer Inst 1996, 88, 1560–1570. [Google Scholar]
- The Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. The effect of vitamin e and beta carotene on the incidence of lung cancer and other cancers in male smokers. The alpha-tocopherol, beta carotene cancer prevention study group. N. Engl. J. Med. 1994, 330, 1029–1035.
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004, 364, 1219–1228. [Google Scholar]
- Myung, S.K.; Kim, Y.; Ju, W.; Choi, H.J.; Bae, W.K. Effects of antioxidant supplements on cancer prevention: Meta-analysis of randomized controlled trials. Ann. Oncol 2010, 21, 166–179. [Google Scholar]
- Mursu, J.; Robien, K.; Harnack, L.J.; Park, K.; Jacobs, D.R., Jr. Dietary supplements and mortality rate in older women: The iowa women’s health study. Arch. Intern. Med. 2011, 171, 1625–1633. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studies reporting beneficial effects of antioxidants | ||||||
|---|---|---|---|---|---|---|
| Study | N | Age (years) | Follow up period | Antioxidant | Main Outcome | References |
| Nurses’ Health Study (NHS) | 87,245 | 34–59 | 8 years | Vitamin E | Significant reduction in CHD risk | [67,68] |
| Health Professional Follow up Study (HPFS) | 39,910 | 40–75 | 4 years | Vitamin E | Significant reduction in CHD risk | [67,69] |
| Established Populations for Epidemiological Studies of the Elderly | 11,178 | 67–105 | 6 years | Vitamin E with or without other Vitamins | Vitamin E associated with a significant reduction CHD risk | [67,70] |
| First National Health and Nutrition Examination Survey (NHANES I) | 11,348 | 25–74 | 10 years | Vitamin C | Inverse correlation between Vitamin C and all-cause CVD death in men; not women | [71] |
| Scottish Heart Health Study | 7869 | 40–59 | 10 years | Vitamins C, E and β-carotene | Significant reduction of CHD; only in men | [71,72] |
| Meta-analysis | 10,073 | 18–90 | NR | Vitamins C, E and B12, and β-carotene | Preventive effects on cervical neoplasms | [73] |
| Alpha-Tocopherol, Beta carotene Cancer Prevention Study (ATBC) | 27,111 | 50–69 | 16–19.4 years | Alphatocopherol, β-carotene and flavonoids | Alpha-tocopherol was associated with reduced risk of pancreatic and prostate cancer. Flavonoids were associated with decreased risk of pancreatic cancer | [74–76] |
| Heart Protection Study | 20,536 | 40–80 | 5 years | Vitamins C and E and β-carotene | No reductions in blood pressure, morbidity or mortality | [14,77] |
| Rotterdam Study | 4802 | 55–95 | 4 years | Vitamins C and E and β carotene | No effects of Vitamin E on the risk of myocardial infarction | [71,78] |
| Scottish Heart Health Study | 7869 | 40–59 | 10 years | Vitamins C and E and β-carotene | No effects on all-cause mortality | [71,72] |
| Primary Prevention Project | 4495 | 64 (average) | 3.6 years | Vitamin E and low-dose aspirin | Vitamin E had no beneficial effect. Trial terminated because other studies demonstrated the beneficial effect of aspirin on cardiovascular mortality | [71] |
| Heart Outcomes Prevention Evaluation Study (HOPE) | 9544 | >55 | 4.5 years | Vitamin E | No effect of Vitamin E | [71,79] |
| Gruppo Italiano per lo Studio della Supravvivenza nell’ Infarto Miocardico (GISSI) | 11,324 | 59.3 (average) | 3.5 years | Vitamin E and omega-3 oils | No effect of Vitamin E | [71,80] |
| Study on well controlled diabetic patients | 40 | 61.9 | 12 weeks | Extracts of fruits and vegetables | No effect of the extracts | [81] |
| Meta-analysis including studies performed in Type 2 diabetic patients | 418 | 20–80 | 8 weeks | Vitamin E | No effects on metabolic control | [82] |
| Meta-analysis | 94,069 | >49 | 7–10 years | Vitamin E | No effect on colorectal cancer | [83] |
| Randomized, double blind, placebo controlled study in hypertensive patients | 69 | 62 (average) | 6 weeks | Vitamin C and grape seed polyphenols | Increase of blood pressure and no effect on either endothelium dependent vasodilation or oxidative stress | [84] |
| Randomized study in patients with CAD | 169 | 52 (average) | 3 years | One statin, Vitamin C, vitamin E, β-carotene and selenium | Antioxidants blunted the effect of statins on HDL | [71,85] |
| Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial (PLCO) | 25,400 | 55–74 | 10 years | Folic acid | Folic acid supplementation significantly increased breast cancer | [86] |
| Beta Carotene and Retinal Efficacy Trial (CARET) | 18,314 | 58 (average) | Stopped after 2 years | Vitamin A and β-carotene | Antioxidant treatment was associated with an increased incidence of lung cancer and mortality | [71,87] |
| Alpha- Tocopherol Beta-Carotene Cancer Prevention Study (ATBC) | 29,133 | 50–69 | 8 years | α-tocopherol, β-carotene | Antioxidants increased the incidence and mortality of lung cancer | [88,89] |
| Meta-analysis | 131,727 | 55 (average) | 1–12 years | β-carotene, Vitamin A, Vitamin E, selenium | The antioxidant treatment did not prevent gastrointestinal cancer but significantly increased mortality | [90] |
| Meta-analysis | 161,045 | 58.4 (average) | 5.3–5.8 years | β-carotene, Vitamin C, Vitamin E, selenium | Increased risk of bladder cancer in 4 of 22 of the trials included in the analysis | [91] |
| Study in healthy men | 14 | 24.4 (average) | 7 days | Vitamin C and N-Acetylcysteine | Increase of oxidative stress produced by exercise | [41] |
| Study in healthy men | 39 | 25–35 | 4 weeks | Vitamin C and Vitamin E | Antioxidants blocked the increase of insulin sensitivity and expression produced by exercise | [42] |
| Meta-analysis | 338 * | NR | 1 day– 6 weeks | Allopurinol, Coenzyme Q, Vitamins (C, E, B6, B12), α-lipoic acid, β-carotene, lutein, Nacetylcysteine, Selenium and/or Zinc | Any of these effects (compared to groups treated with placebo): ↓ training induced improvement in physical performance, ↓ exercise-induced oxidative stress preconditioning, ↑ CK, ↑ inflammatory biomarkers, prevented beneficial effects on insulin sensitivity and expression | [43] |
| Iowa Woman’s Health Study | 38,772 | >60 | 14 years | Dietary vitamins and mineral supplements | May be associated with increased total mortality risk | [92] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Villanueva, C.; Kross, R.D. Antioxidant-Induced Stress. Int. J. Mol. Sci. 2012, 13, 2091-2109. https://doi.org/10.3390/ijms13022091
Villanueva C, Kross RD. Antioxidant-Induced Stress. International Journal of Molecular Sciences. 2012; 13(2):2091-2109. https://doi.org/10.3390/ijms13022091
Chicago/Turabian StyleVillanueva, Cleva, and Robert D. Kross. 2012. "Antioxidant-Induced Stress" International Journal of Molecular Sciences 13, no. 2: 2091-2109. https://doi.org/10.3390/ijms13022091
APA StyleVillanueva, C., & Kross, R. D. (2012). Antioxidant-Induced Stress. International Journal of Molecular Sciences, 13(2), 2091-2109. https://doi.org/10.3390/ijms13022091