UBE4B: A Promising Regulatory Molecule in Neuronal Death and Survival



{kind=link}
Abstract
:1. Introduction
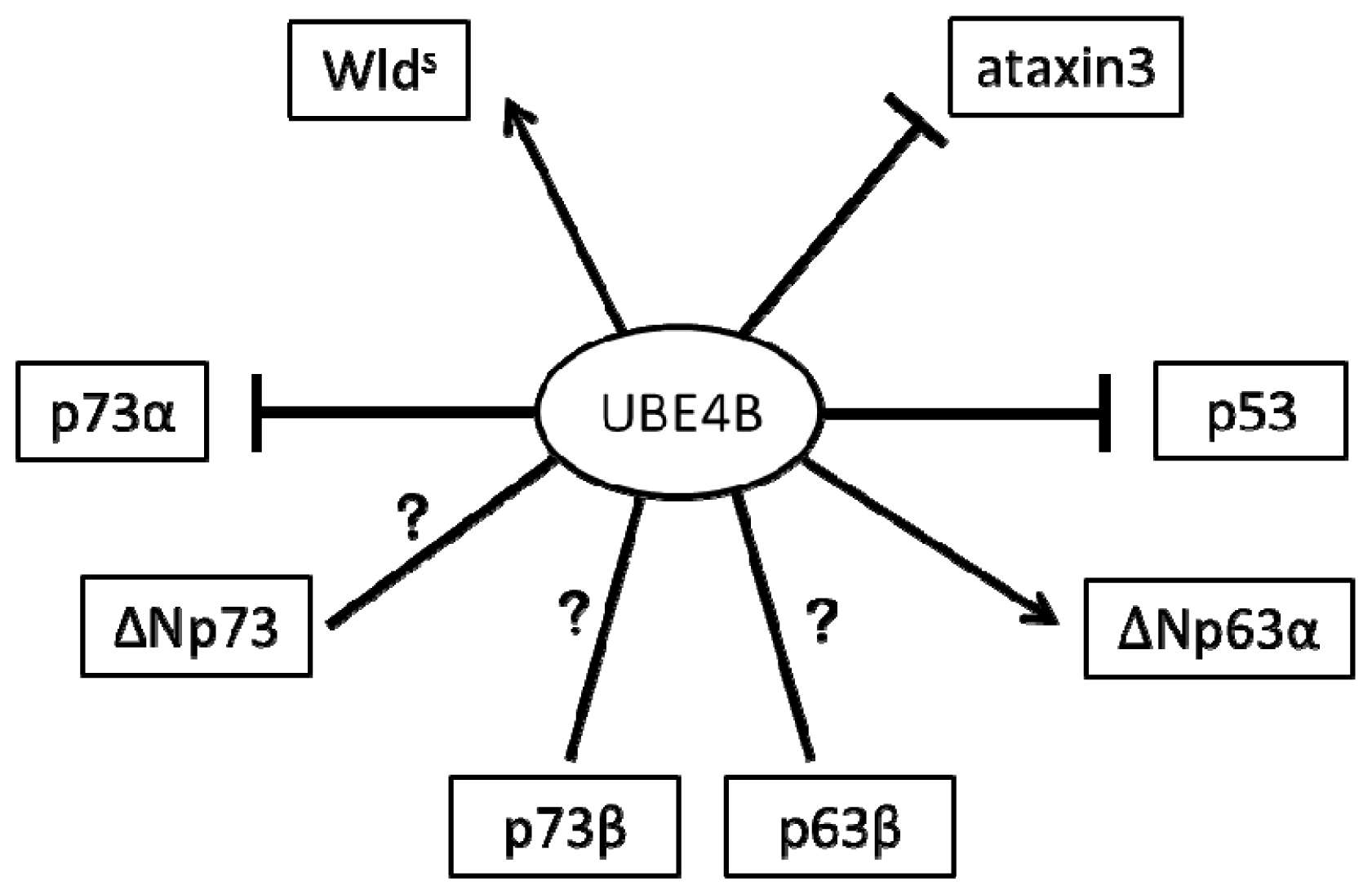
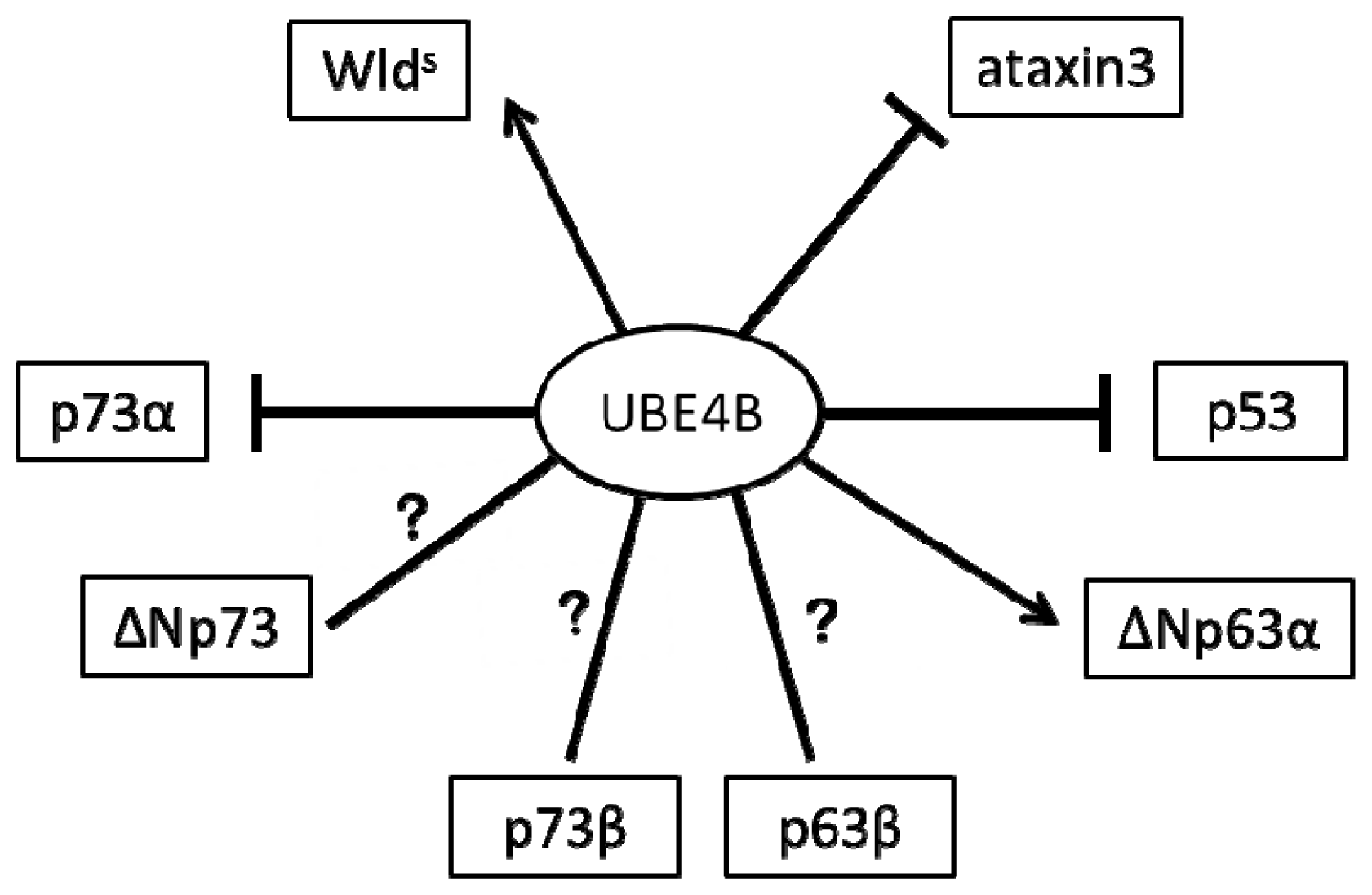
2. The Role of UBE4B in the Nervous System via p53 Family Regulation
2.1. UBE4B and p53 in the Nervous System
2.2. UBE4B and p73 in the Nervous System
2.3. UBE4B and p63 in the Nervous System
3. The Role of UBE4B in the Nervous System Is Independent of p53 Family Regulation
4. Conclusions
Acknowledgments
References
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar]
- Jacobs, W.B.; Kaplan, D.R.; Miller, F.D. The p53 family in nervous system development and disease. J. Neurochem 2006, 97, 1571–1584. [Google Scholar]
- Blaschke, A.J.; Staley, K.; Chun, J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 1996, 122, 1165–1174. [Google Scholar]
- Gilmore, E.C.; Nowakowski, R.S.; Caviness, V.S., Jr; Herrup, K. Cell birth, cell death, cell diversity and DNA breaks: How do they all fit together? Trends Neurosci. 2000, 23, 100–105. [Google Scholar]
- Jacobs, W.B.; Walsh, G.S.; Miller, F.D. Neuronal survival and p73/p63/p53: A family affair. Neuroscientist 2004, 10, 443–455. [Google Scholar]
- Wood, K.A.; Youle, R.J. The role of free radicals and p53 in neuron apoptosis in vivo. J. Neurosci 1995, 15, 5851–5857. [Google Scholar]
- Graham, S.H.; Chen, J. Programmed cell death in cerebral ischemia. J. Cereb. Blood Flow Metab 2001, 21, 99–109. [Google Scholar]
- Green, D.R. Apoptotic pathways: The roads to ruin. Cell 1998, 94, 695–698. [Google Scholar]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar]
- Soussi, T. The p53 pathway and human cancer. Br. J. Surg 2005, 92, 1331–1332. [Google Scholar]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar]
- Carter, S.; Vousden, K.H. p53-Ubl fusions as models of ubiquitination, sumoylation and neddylation of p53. Cell Cycle 2008, 7, 2519–2528. [Google Scholar]
- Wu, H.; Pomeroy, S.L.; Ferreira, M.; Teider, N.; Mariani, J.; Nakayama, K.I.; Hatakeyama, S.; Tron, V.A.; Saltibus, L.F.; Spyracopoulos, L.; et al. UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat. Med 2011, 17, 347–355. [Google Scholar]
- Gostissa, M.; Hengstermann, A.; Fogal, V.; Sandy, P.; Schwarz, S.E.; Scheffner, M.; del Sal, G. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J 1999, 18, 6462–6471. [Google Scholar]
- Rodriguez, M.S.; Desterro, J.M.; Lain, S.; Midgley, C.A.; Lane, D.P.; Hay, R.T. SUMO-1 modification activates the transcriptional response of p53. EMBO J 1999, 18, 6455–6461. [Google Scholar]
- Shen, L.N.; Liu, H.; Dong, C.; Xirodimas, D.; Naismith, J.H.; Hay, R.T. Structural basis of NEDD8 ubiquitin discrimination by the deNEDDylating enzyme NEDP1. EMBO J 2005, 24, 1341–1351. [Google Scholar]
- Whitby, F.G.; Xia, G.; Pickart, C.M.; Hill, C.P. Crystal structure of the human ubiquitin-like protein NEDD8 and interactions with ubiquitin pathway enzymes. J. Biol. Chem 1998, 273, 34983–34991. [Google Scholar]
- Feng, L.; Lin, T.; Uranishi, H.; Gu, W.; Xu, Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol. Cell. Biol 2005, 25, 5389–5395. [Google Scholar]
- Ozeki, C.; Sawai, Y.; Shibata, T.; Kohno, T.; Okamoto, K.; Yokota, J.; Tashiro, F.; Tanuma, S.; Sakai, R.; Kawase, T.; et al. Cancer susceptibility polymorphism of p53 at codon 72 affects phosphorylation and degradation of p53 protein. J. Biol. Chem 2011, 286, 18251–18260. [Google Scholar]
- Yang, Y.; Li, C.C.; Weissman, A.M. Regulating the p53 system through ubiquitination. Oncogene 2004, 23, 2096–2106. [Google Scholar]
- Watson, I.R.; Irwin, M.S. Ubiquitin and ubiquitin-like modifications of the p53 family. Neoplasia 2006, 8, 655–666. [Google Scholar]
- Tan, J.M.; Wong, E.S.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.P.; Ho, M.W.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet 2008, 17, 431–439. [Google Scholar]
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G., Jr; Oren, M.; Chen, J.; Lu, H. MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell. Biol. 1999, 19, 3257–3266. [Google Scholar]
- Kaneko, C.; Hatakeyama, S.; Matsumoto, M.; Yada, M.; Nakayama, K.; Nakayama, K.I. Characterization of the mouse gene for the U-box-type ubiquitin ligase UFD2a. Biochem. Biophys. Res. Commun 2003, 300, 297–304. [Google Scholar]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar]
- Armstrong, J.F.; Kaufman, M.H.; Harrison, D.J.; Clarke, A.R. High-frequency developmental abnormalities in p53-deficient mice. Curr. Biol 1995, 5, 931–936. [Google Scholar]
- Sah, V.P.; Attardi, L.D.; Mulligan, G.J.; Williams, B.O.; Bronson, R.T.; Jacks, T. A subset of p53-deficient embryos exhibit exencephaly. Nat. Genet 1995, 10, 175–180. [Google Scholar]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar]
- Cecconi, F.; Alvarez-Bolado, G.; Meyer, B.I.; Roth, K.A.; Gruss, P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 1998, 94, 727–737. [Google Scholar]
- Kuida, K.; Haydar, T.F.; Kuan, C.Y.; Gu, Y.; Taya, C.; Karasuyama, H.; Su, M.S.; Rakic, P.; Flavell, R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998, 94, 325–337. [Google Scholar]
- Yoshida, H.; Kong, Y.Y.; Yoshida, R.; Elia, A.J.; Hakem, A.; Hakem, R.; Penninger, J.M.; Mak, T.W. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998, 94, 739–750. [Google Scholar]
- Kuida, K.; Zheng, T.S.; Na, S.; Kuan, C.; Yang, D.; Karasuyama, H.; Rakic, P.; Flavell, R.A. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 1996, 384, 368–372. [Google Scholar]
- Slack, R.S.; Belliveau, D.J.; Rosenberg, M.; Atwal, J.; Lochmuller, H.; Aloyz, R.; Haghighi, A.; Lach, B.; Seth, P.; Cooper, E.; et al. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J. Cell Biol 1996, 135, 1085–1096. [Google Scholar]
- Miller, F.D.; Kaplan, D.R. On Trk for retrograde signaling. Neuron 2001, 32, 767–770. [Google Scholar]
- Aloyz, R.S.; Bamji, S.X.; Pozniak, C.D.; Toma, J.G.; Atwal, J.; Kaplan, D.R.; Miller, F.D. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell Biol 1998, 143, 1691–1703. [Google Scholar]
- Lee, Y.; McKinnon, P.J. Detection of apoptosis in the central nervous system. Methods Mol. Biol 2009, 559, 273–282. [Google Scholar]
- Chopp, M.; Li, Y.; Zhang, Z.G.; Freytag, S.O. p53 expression in brain after middle cerebral artery occlusion in the rat. Biochem. Biophys. Res. Commun 1992, 182, 1201–1207. [Google Scholar]
- Watanabe, H.; Ohta, S.; Kumon, Y.; Sakaki, S.; Sakanaka, M. Increase in p53 protein expression following cortical infarction in the spontaneously hypertensive rat. Brain Res 1999, 837, 38–45. [Google Scholar]
- Martin, L.J. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis 2000, 7, 613–622. [Google Scholar]
- Levrero, M.; de Laurenzi, V.; Costanzo, A.; Gong, J.; Wang, J.Y.; Melino, G. The p53/p63/p73 family of transcription factors: Overlapping and distinct functions. J. Cell Sci 2000, 113, 1661–1670. [Google Scholar]
- Wu, H.; Leng, R.P. UBE4B, a ubiquitin chain assembly factor, is required for MDM2-mediated p53 polyubiquitination and degradation. Cell Cycle 2011, 10, 1912–1915. [Google Scholar]
- Saeki, Y.; Kudo, T.; Sone, T.; Kikuchi, Y.; Yokosawa, H.; Toh-e, A.; Tanaka, K. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J 2009, 28, 359–371. [Google Scholar]
- Wu, H.; Zeinab, R.A.; Flores, E.R.; Leng, R.P. Pirh2, a ubiquitin E3 ligase, inhibits p73 transcriptional activity by promoting its ubiquitination. Mol. Cancer Res 2011, 9, 1780–1790. [Google Scholar]
- Melino, G.; de Laurenzi, V.; Vousden, K.H. p73: Friend or foe in tumorigenesis. Nat. Rev. Cancer 2002, 2, 605–615. [Google Scholar]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; McKeon, F.; Caput, D. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416, 560–564. [Google Scholar]
- Jost, C.A.; Marin, M.C.; Kaelin, W.G., Jr. p73 is a simian (correction of human) p53-related protein that can induce apoptosis. Nature 1997, 389, 191–194. [Google Scholar]
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000, 289, 304–306. [Google Scholar]
- Walsh, G.S.; Orike, N.; Kaplan, D.R.; Miller, F.D. The invulnerability of adult neurons: A critical role for p73. J. Neurosci 2004, 24, 9638–9647. [Google Scholar]
- Irwin, M.S.; Miller, F.D. p73: Regulator in cancer and neural development. Cell Death Differ 2004, 11, S17–S22. [Google Scholar]
- Wilson, C.; Henry, S.; Smith, M.A.; Bowser, R. The p53 homologue p73 accumulates in the nucleus and localizes to neurites and neurofibrillary tangles in Alzheimer disease brain. Neuropathol. Appl. Neurobiol 2004, 30, 19–29. [Google Scholar]
- Zaika, A.I.; Slade, N.; Erster, S.H.; Sansome, C.; Joseph, T.W.; Pearl, M.; Chalas, E.; Moll, U.M. DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J. Exp. Med 2002, 196, 765–780. [Google Scholar]
- Nakagawa, T.; Takahashi, M.; Ozaki, T.; Watanabe, K.-i.; Todo, S.; Mizuguchi, H.; Hayakawa, T.; Nakagawara, A. Autoinhibitory regulation of p73 by ΔNp73 to modulate cell survival and death through a p73-specific target element within the Delta Np73 promoter. Mol. Cell. Biol. 2002, 22, 2575–2585. [Google Scholar]
- Grob, T.J.; Novak, U.; Maisse, C.; Barcaroli, D.; Luthi, A.U.; Pirnia, F.; Hugli, B.; Graber, H.U.; de Laurenzi, V.; Fey, M.F.; et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ 2001, 8, 1213–1223. [Google Scholar]
- Fillippovich, I.; Sorokina, N.; Gatei, M.; Haupt, Y.; Hobson, K.; Moallem, E.; Spring, K.; Mould, M.; McGuckin, M.A.; Lavin, M.F.; et al. Transactivation-deficient p73α (p73Δexon2) inhibits apoptosis and competes with p53. Oncogene 2001, 20, 514–522. [Google Scholar]
- Irwin, M.S.; Kaelin, W.G. p53 family update: p73 and p63 develop their own identities. Cell Growth Differ 2001, 12, 337–349. [Google Scholar]
- Hosoda, M.; Ozaki, T.; Miyazaki, K.; Hayashi, S.; Furuya, K.; Watanabe, K.; Nakagawa, T.; Hanamoto, T.; Todo, S.; Nakagawara, A. UFD2a mediates the proteasomal turnover of p73 without promoting p73 ubiquitination. Oncogene 2005, 24, 7156–7169. [Google Scholar]
- Ozaki, T.; Hosoda, M.; Miyazaki, K.; Hayashi, S.; Watanabe, K.; Nakagawa, T.; Nakagawara, A. Functional implication of p73 protein stability in neuronal cell survival and death. Cancer Lett 2005, 228, 29–35. [Google Scholar]
- Lee, A.F.; Ho, D.K.; Zanassi, P.; Walsh, G.S.; Kaplan, D.R.; Miller, F.D. Evidence that ΔNp73 promotes neuronal survival by p53-dependent and p53-independent mechanisms. J. Neurosci 2004, 24, 9174–9184. [Google Scholar]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol 2000, 10, 381–391. [Google Scholar]
- Jacobs, W.B.; Govoni, G.; Ho, D.; Atwal, J.K.; Barnabe-Heider, F.; Keyes, W.M.; Mills, A.A.; Miller, F.D.; Kaplan, D.R. p63 is an essential proapoptotic protein during neural development. Neuron 2005, 48, 743–756. [Google Scholar]
- Nicotera, P.; Melino, G. Neurodevelopment on route p63. Neuron 2005, 48, 707–709. [Google Scholar]
- Gressner, O.; Schilling, T.; Lorenz, K.; Schleithoff, E.S.; Koch, A.; Schulze-Bergkamen, H.; Lena, A.M.; Candi, E.; Terrinoni, A.; Catani, M.V.; et al. TAp63α induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J 2005, 24, 2458–2471. [Google Scholar]
- King, K.E.; Ponnamperuma, R.M.; Yamashita, T.; Tokino, T.; Lee, L.A.; Young, M.F.; Weinberg, W.C. ΔNp63α functions as both a positive and a negative transcriptional regulator and blocks in vitro differentiation of murine keratinocytes. Oncogene 2003, 22, 3635–3644. [Google Scholar]
- Yamaguchi, K.; Wu, L.; Caballero, O.L.; Hibi, K.; Trink, B.; Resto, V.; Cairns, P.; Okami, K.; Koch, W.M.; Sidransky, D.; et al. Frequent gain of the p40/p51/p63 gene locus in primary head and neck squamous cell carcinoma. Int. J. Cancer 2000, 86, 684–689. [Google Scholar]
- Pelosi, G.; Pasini, F.; Stenholm, C.S.; Pastorino, U.; Maisonneuve, P.; Sonzogni, A.; Maffini, F.; Pruneri, G.; Fraggetta, F.; Cavallon, A.; et al. p63 immunoreactivity in lung cancer: Yet another player in the development of squamous cell carcinomas? J. Pathol 2002, 198, 100–109. [Google Scholar]
- Crook, T.; Nicholls, J.M.; Brooks, L.; O’Nions, J.; Allday, M.J. High level expression of δN-p63: A mechanism for the inactivation of p53 in undifferentiated nasopharyngeal carcinoma (NPC)? Oncogene 2000, 19, 3439–3444. [Google Scholar]
- Chatterjee, A.; Upadhyay, S.; Chang, X.; Nagpal, J.K.; Trink, B.; Sidransky, D. U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of ΔNp63α. Cell Cycle 2008, 7, 1231–1237. [Google Scholar]
- Westfall, M.D.; Joyner, A.S.; Barbieri, C.E.; Livingstone, M.; Pietenpol, J.A. Ultraviolet radiation induces phosphorylation and ubiquitin-mediated degradation of δNp63α. Cell Cycle 2005, 4, 710–716. [Google Scholar]
- Osada, M.; Inaba, R.; Shinohara, H.; Hagiwara, M.; Nakamura, M.; Ikawa, Y. Regulatory domain of protein stability of human P51/TAP63, a P53 homologue. Biochem. Biophys Res. Commun 2001, 283, 1135–1141. [Google Scholar]
- Koshy, B.T.; Zoghbi, H.Y. The CAG/polyglutamine tract diseases: Gene products and molecular pathogenesis. Brain Pathol 1997, 7, 927–942. [Google Scholar]
- Alves-Rodrigues, A.; Gregori, L.; Figueiredo-Pereira, M.E. Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci 1998, 21, 516–520. [Google Scholar]
- Hayashi, Y.; Kakita, A.; Yamada, M.; Koide, R.; Igarashi, S.; Takano, H.; Ikeuchi, T.; Wakabayashi, K.; Egawa, S.; Tsuji, S.; et al. Hereditary dentatorubral-pallidoluysian atrophy: Detection of widespread ubiquitinated neuronal and glial intranuclear inclusions in the brain. Acta Neuropathol 1998, 96, 547–552. [Google Scholar]
- Matsumoto, M.; Yada, M.; Hatakeyama, S.; Ishimoto, H.; Tanimura, T.; Tsuji, S.; Kakizuka, A.; Kitagawa, M.; Nakayama, K.I. Molecular clearance of ataxin-3 is regulated by a mammalian E4. EMBO J 2004, 23, 659–669. [Google Scholar]
- Lunkes, A.; Mandel, J.L. Polyglutamines, nuclear inclusions and neurodegeneration. Nat. Med 1997, 3, 1201–1202. [Google Scholar]
- Kaneko-Oshikawa, C.; Nakagawa, T.; Yamada, M.; Yoshikawa, H.; Matsumoto, M.; Yada, M.; Hatakeyama, S.; Nakayama, K.; Nakayama, K.I. Mammalian E4 is required for cardiac development and maintenance of the nervous system. Mol. Cell. Biol 2005, 25, 10953–10964. [Google Scholar]
- Morreale, G.; Conforti, L.; Coadwell, J.; Wilbrey, A.L.; Coleman, M.P. Evolutionary divergence of valosin-containing protein/cell division cycle protein 48 binding interactions among endoplasmic reticulum-associated degradation proteins. FEBS J 2009, 276, 1208–1220. [Google Scholar]
- Dai, R.M.; Li, C.C. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat. Cell Biol 2001, 3, 740–744. [Google Scholar]
- Conforti, L.; Wilbrey, A.; Morreale, G.; Janeckova, L.; Beirowski, B.; Adalbert, R.; Mazzola, F.; di Stefano, M.; Hartley, R.; Babetto, E.; et al. Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J. Cell Biol 2009, 184, 491–500. [Google Scholar]
- Ferri, A.; Sanes, J.R.; Coleman, M.P.; Cunningham, J.M.; Kato, A.C. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr. Biol 2003, 13, 669–673. [Google Scholar]
- Samsam, M.; Mi, W.; Wessig, C.; Zielasek, J.; Toyka, K.V.; Coleman, M.P.; Martini, R. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J. Neurosci 2003, 23, 2833–2839. [Google Scholar]
- Wang, M.; Wu, Y.; Culver, D.G.; Glass, J.D. The gene for slow Wallerian degeneration (Wld(s)) is also protective against vincristine neuropathy. Neurobiol. Dis 2001, 8, 155–161. [Google Scholar]
- Coleman, M. Axon degeneration mechanisms: Commonality amid diversity. Nat. Rev. Neurosci 2005, 6, 889–898. [Google Scholar]
- Mack, T.G.; Reiner, M.; Beirowski, B.; Mi, W.; Emanuelli, M.; Wagner, D.; Thomson, D.; Gillingwater, T.; Court, F.; Conforti, L.; et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci 2001, 4, 1199–1206. [Google Scholar]
- Zhang, Z.; Fujiki, M.; Guth, L.; Steward, O. Genetic influences on cellular reactions to spinal cord injury: A wound-healing response present in normal mice is impaired in mice carrying a mutation (WldS) that causes delayed Wallerian degeneration. J. Comp. Neurol 1996, 371, 485–495. [Google Scholar]
- Glass, J.D.; Brushart, T.M.; George, E.B.; Griffin, J.W. Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon. J. Neurocytol 1993, 22, 311–321. [Google Scholar]
- Buckmaster, E.A.; Perry, V.H.; Brown, M.C. The rate of Wallerian degeneration in cultured neurons from wild type and C57BL/WldS mice depends on time in culture and may be extended in the presence of elevated K+ levels. Eur. J. Neurosci 1995, 7, 1596–1602. [Google Scholar]
- Perry, V.H.; Brown, M.C.; Lunn, E.R.; Tree, P.; Gordon, S. Evidence that very slow wallerian degeneration in C57BL/Ola mice is an intrinsic property of the peripheral nerve. Eur. J. Neurosci 1990, 2, 802–808. [Google Scholar]
- Gillingwater, T.H.; Ribchester, R.R. Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J. Physiol 2001, 534, 627–639. [Google Scholar]
- Gillingwater, T.H.; Thomson, D.; Mack, T.G.; Soffin, E.M.; Mattison, R.J.; Coleman, M.P.; Ribchester, R.R. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. J. Physiol 2002, 543, 739–755. [Google Scholar]
- Adalbert, R.; Gillingwater, T.H.; Haley, J.E.; Bridge, K.; Beirowski, B.; Berek, L.; Wagner, D.; Grumme, D.; Thomson, D.; Celik, A.; et al. A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur. J. Neurosci 2005, 21, 271–277. [Google Scholar]
- Deckwerth, T.L.; Johnson, E.M., Jr. Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis). Dev. Biol. 1994, 165, 63–72. [Google Scholar]
- Lunn, E.R.; Perry, V.H.; Brown, M.C.; Rosen, H.; Gordon, S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur. J. Neurosci 1989, 1, 27–33. [Google Scholar]
- Conforti, L.; Fang, G.; Beirowski, B.; Wang, M.S.; Sorci, L.; Asress, S.; Adalbert, R.; Silva, A.; Bridge, K.; Huang, X.P.; et al. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ 2007, 14, 116–127. [Google Scholar]
- Emanuelli, M.; Carnevali, F.; Saccucci, F.; Pierella, F.; Amici, A.; Raffaelli, N.; Magni, G. Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. J. Biol. Chem 2001, 276, 406–412. [Google Scholar]
- Sasaki, Y.; Vohra, B.P.; Lund, F.E.; Milbrandt, J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci 2009, 29, 5525–5535. [Google Scholar]
- Laser, H.; Conforti, L.; Morreale, G.; Mack, T.G.; Heyer, M.; Haley, J.E.; Wishart, T.M.; Beirowski, B.; Walker, S.A.; Haase, G.; et al. The slow Wallerian degeneration protein, WldS, binds directly to VCP/p97 and partially redistributes it within the nucleus. Mol. Biol. Cell 2006, 17, 1075–1084. [Google Scholar]
- Wilbrey, A.L.; Haley, J.E.; Wishart, T.M.; Conforti, L.; Morreale, G.; Beirowski, B.; Babetto, E.; Adalbert, R.; Gillingwater, T.H.; Smith, T.; et al. VCP binding influences intracellular distribution of the slow Wallerian degeneration protein, Wld(S). Mol. Cell. Neurosci 2008, 38, 325–340. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zeinab, R.A.; Wu, H.; Sergi, C.; Leng, R. UBE4B: A Promising Regulatory Molecule in Neuronal Death and Survival. Int. J. Mol. Sci. 2012, 13, 16865-16879. https://doi.org/10.3390/ijms131216865
Zeinab RA, Wu H, Sergi C, Leng R. UBE4B: A Promising Regulatory Molecule in Neuronal Death and Survival. International Journal of Molecular Sciences. 2012; 13(12):16865-16879. https://doi.org/10.3390/ijms131216865
Chicago/Turabian StyleZeinab, Rami Abou, Hong Wu, Consolato Sergi, and Roger Leng. 2012. "UBE4B: A Promising Regulatory Molecule in Neuronal Death and Survival" International Journal of Molecular Sciences 13, no. 12: 16865-16879. https://doi.org/10.3390/ijms131216865