A Molecular Dynamics (MD) and Quantum Mechanics/Molecular Mechanics (QM/MM) Study on Ornithine Cyclodeaminase (OCD): A Tale of Two Iminiums


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Methods
2.1. Molecular Dynamics (MD) Equilibration
2.2. QM/MM Computations
3. Results and Discussion
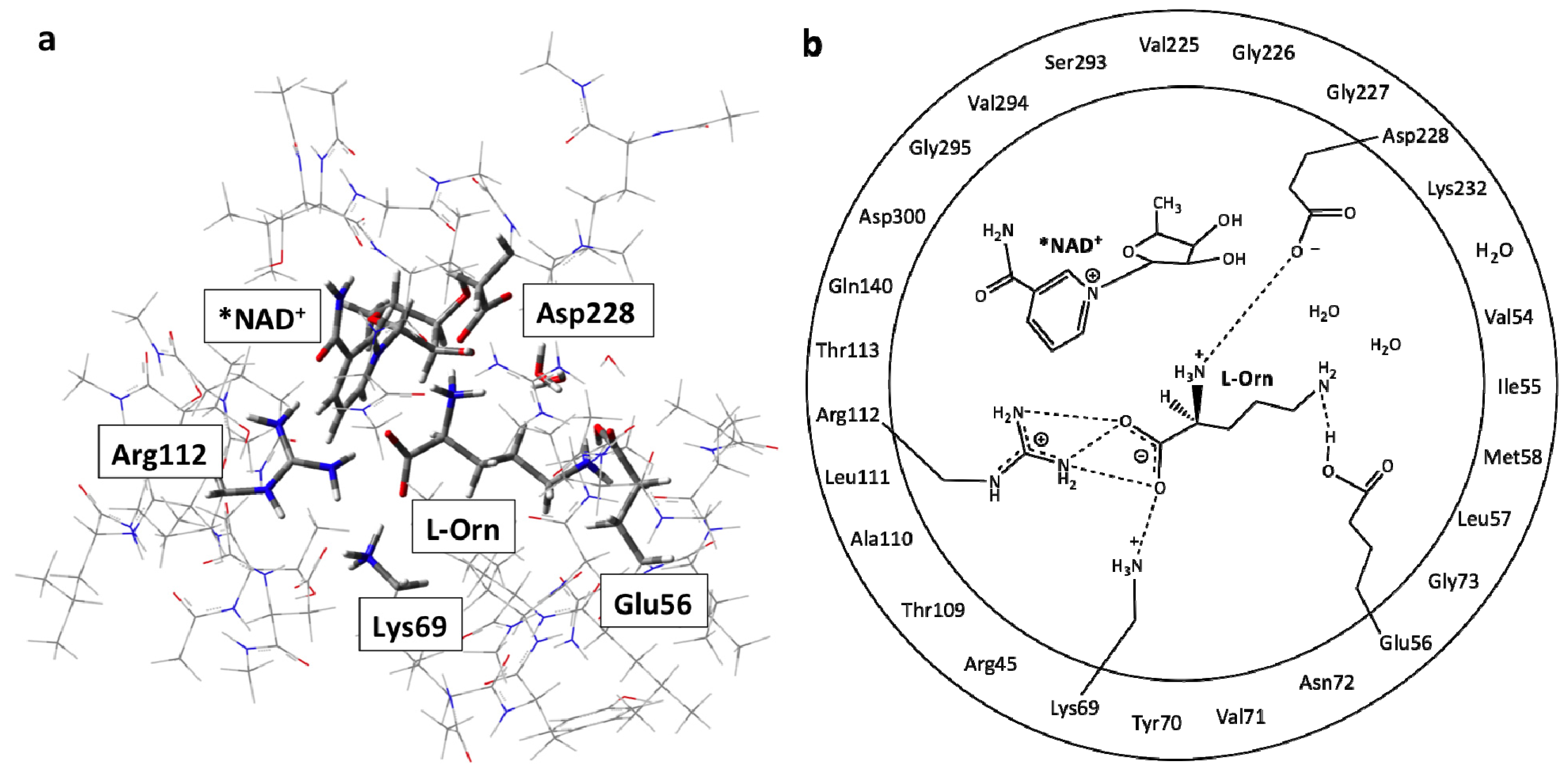
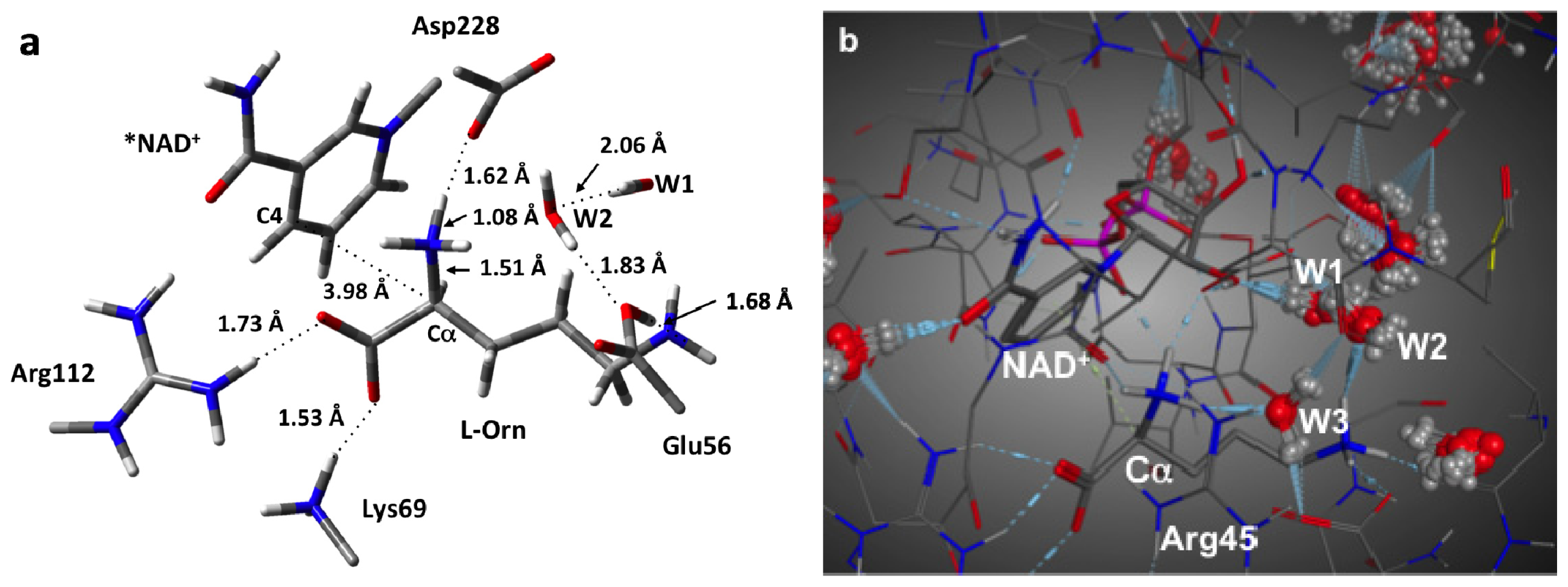
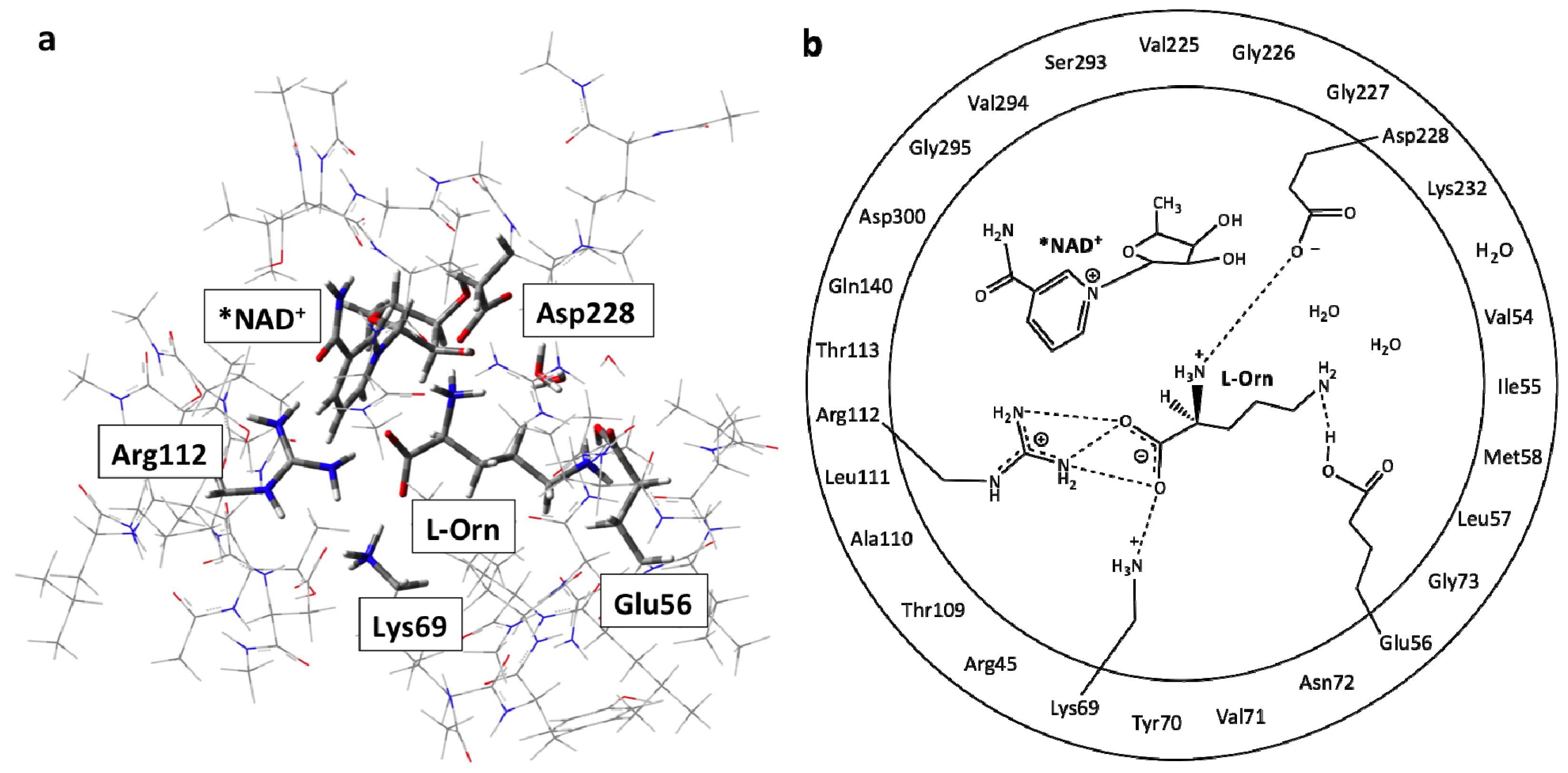
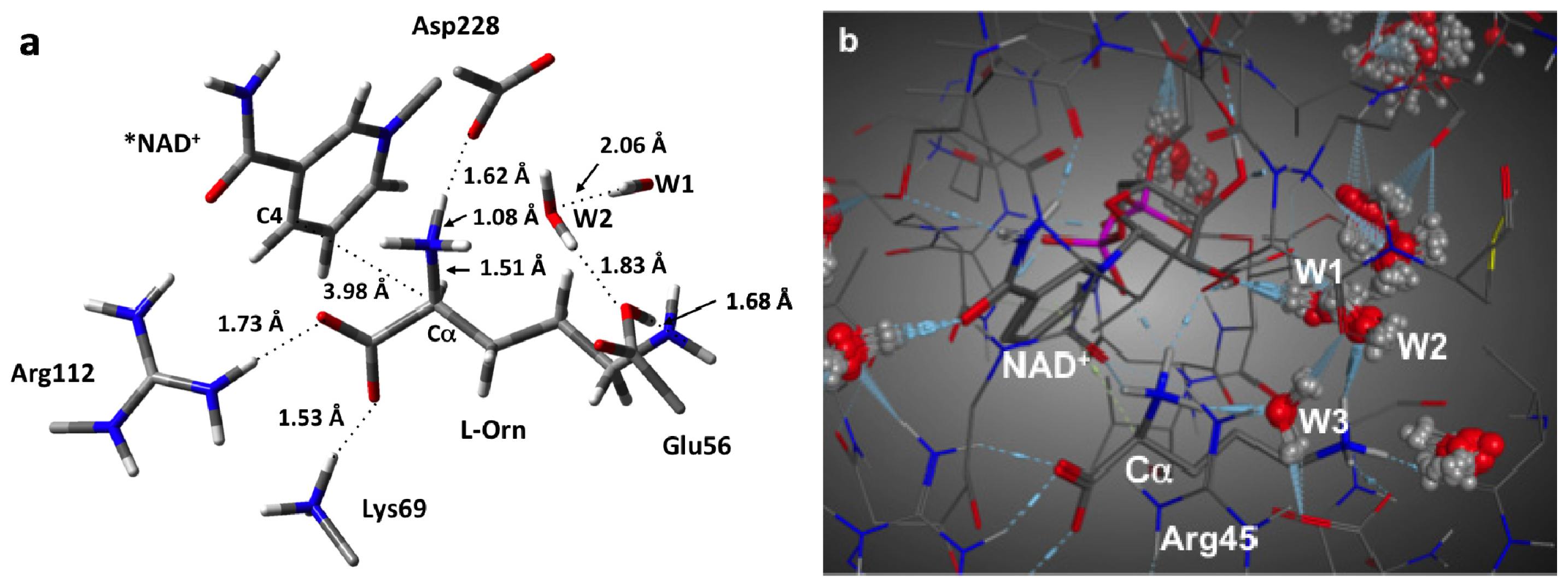
3.1. Structure of the Active Site with the Substrate L-Ornithine and NAD+ Cofactor Bound
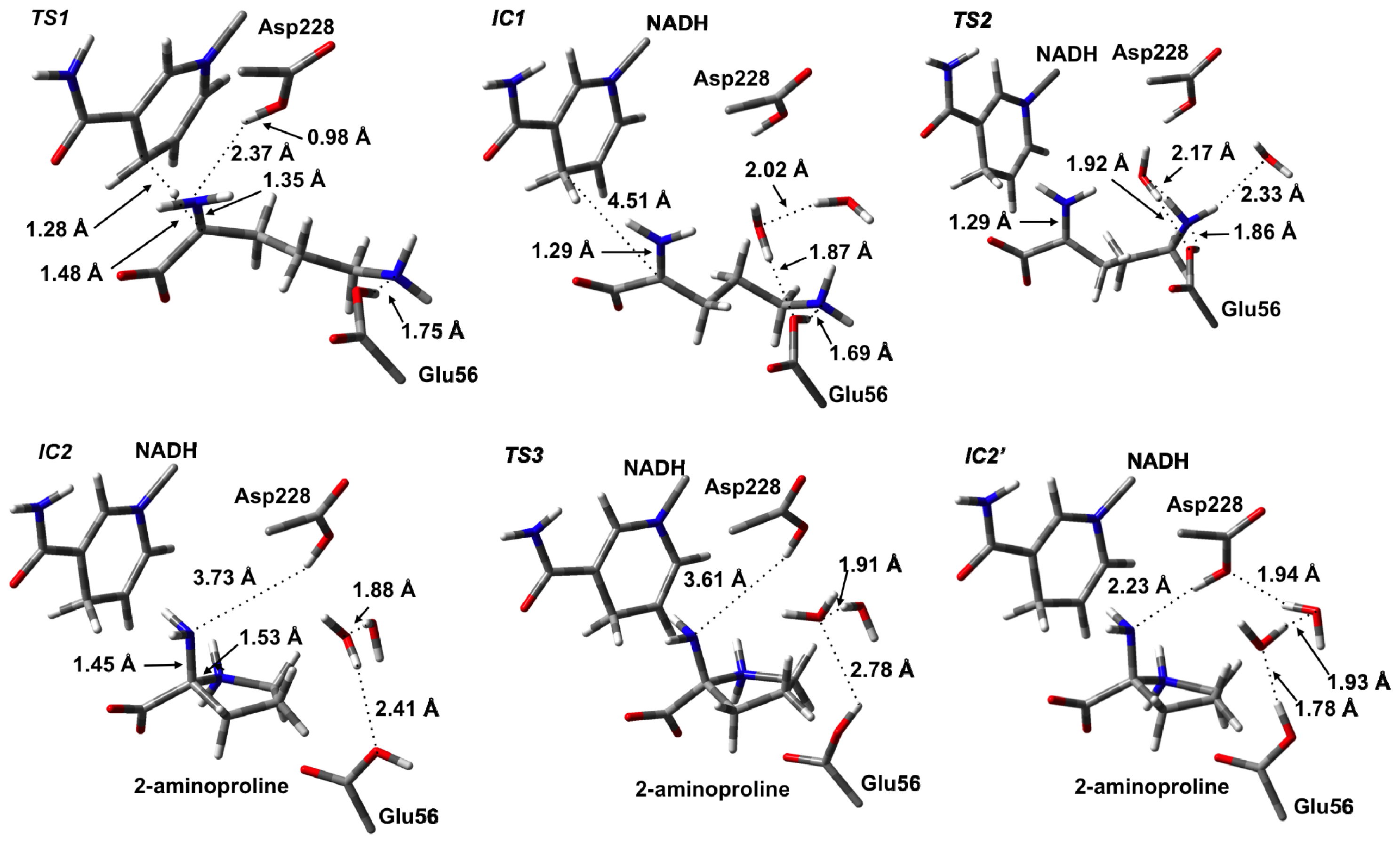
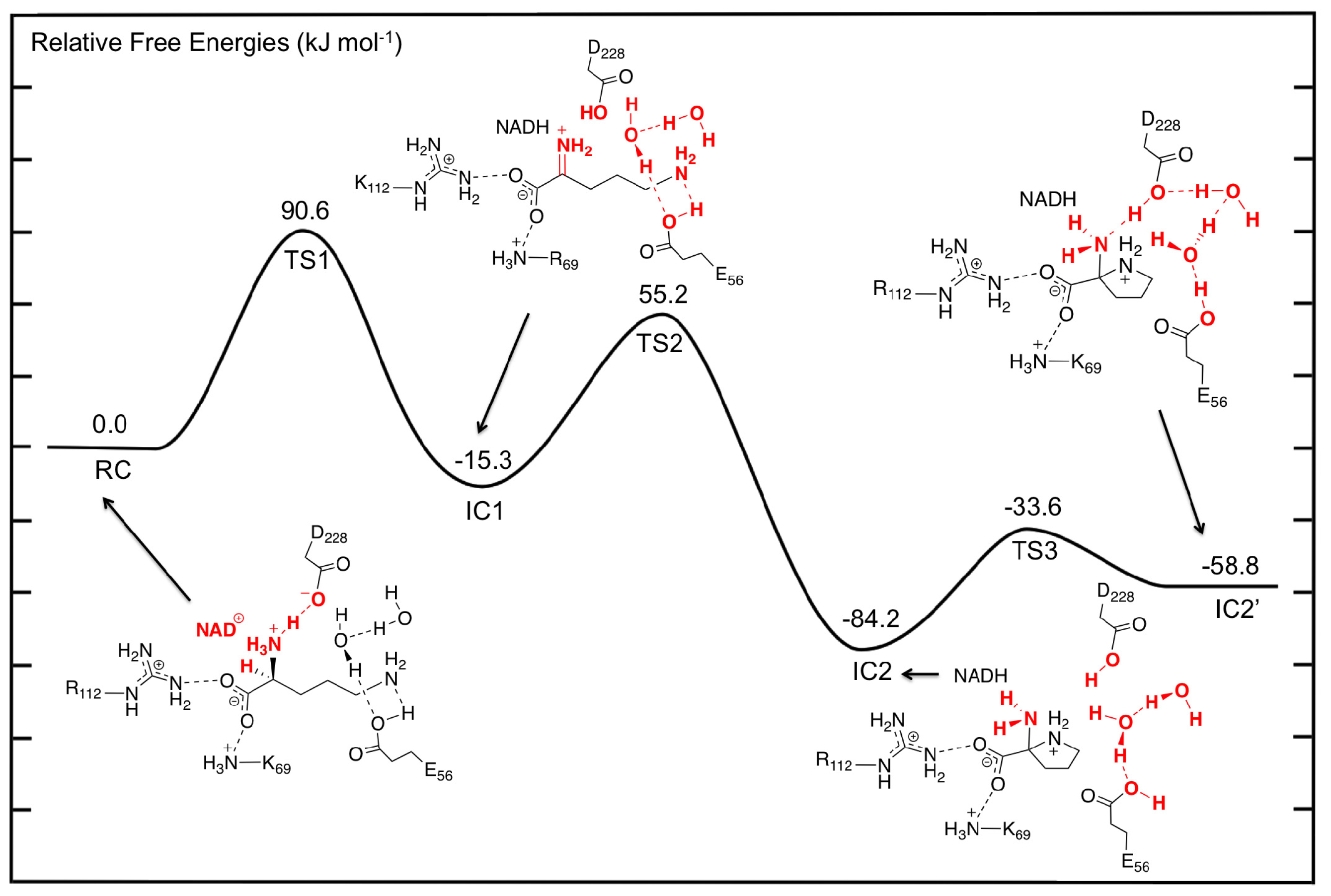
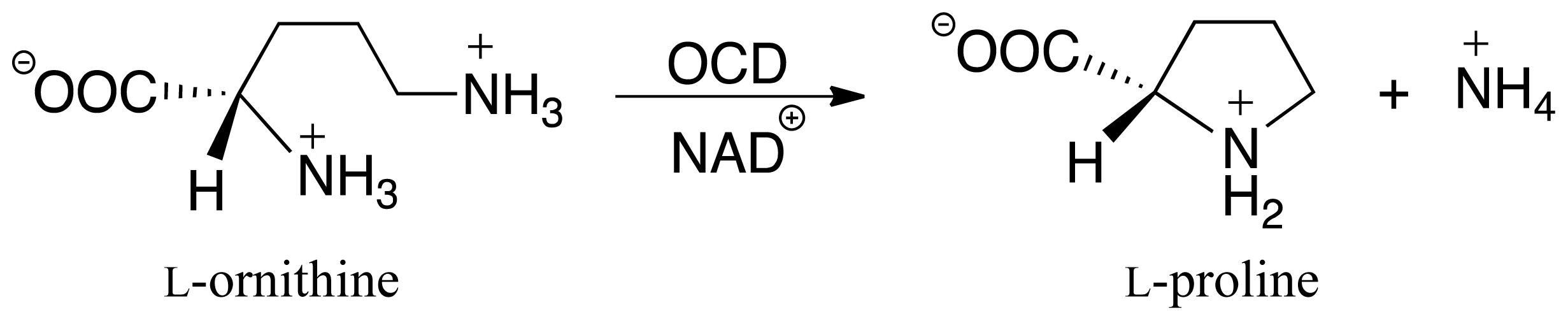
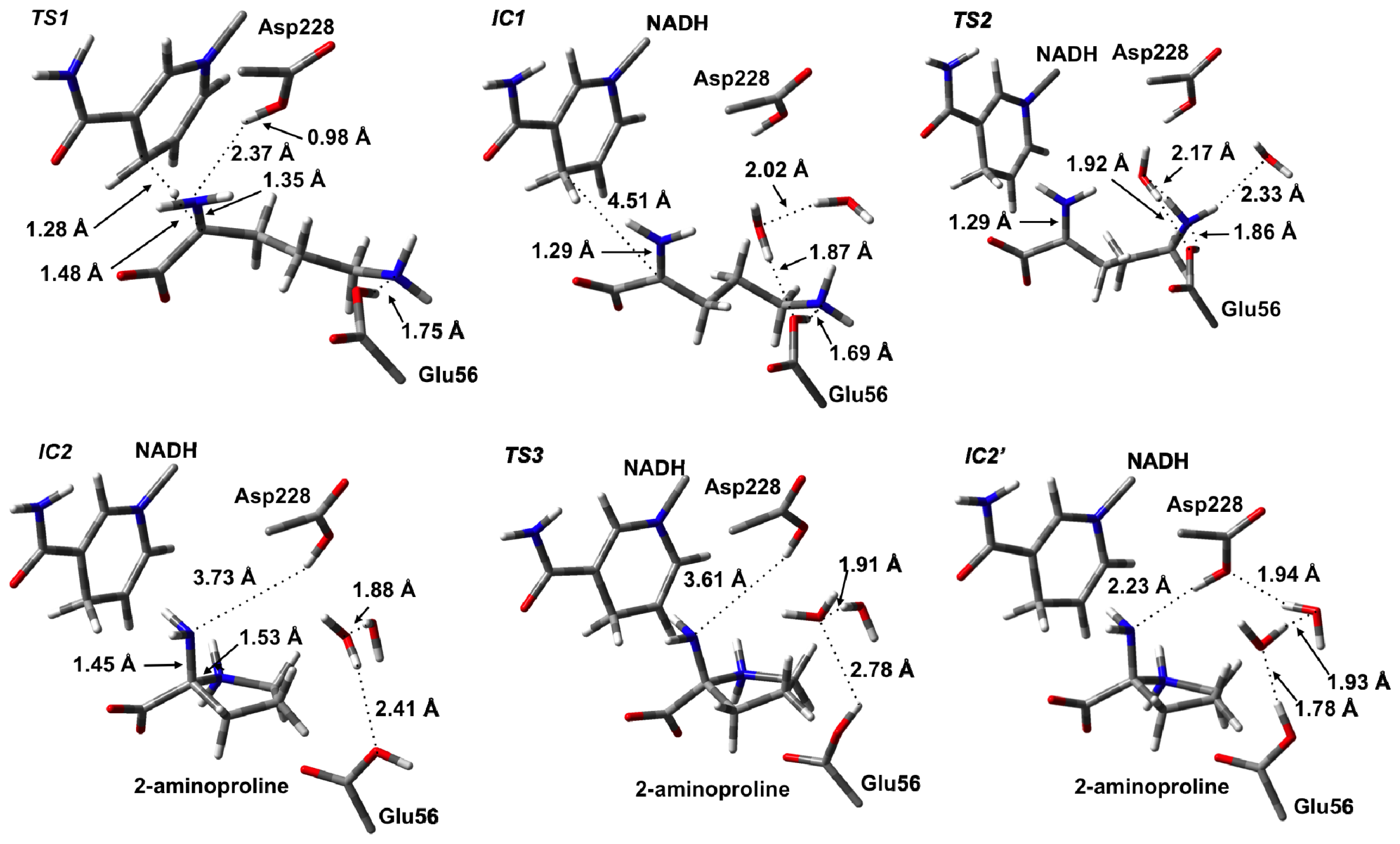
3.2. L-Orn Oxidation and Formation of 2-Aminoproline (AP)
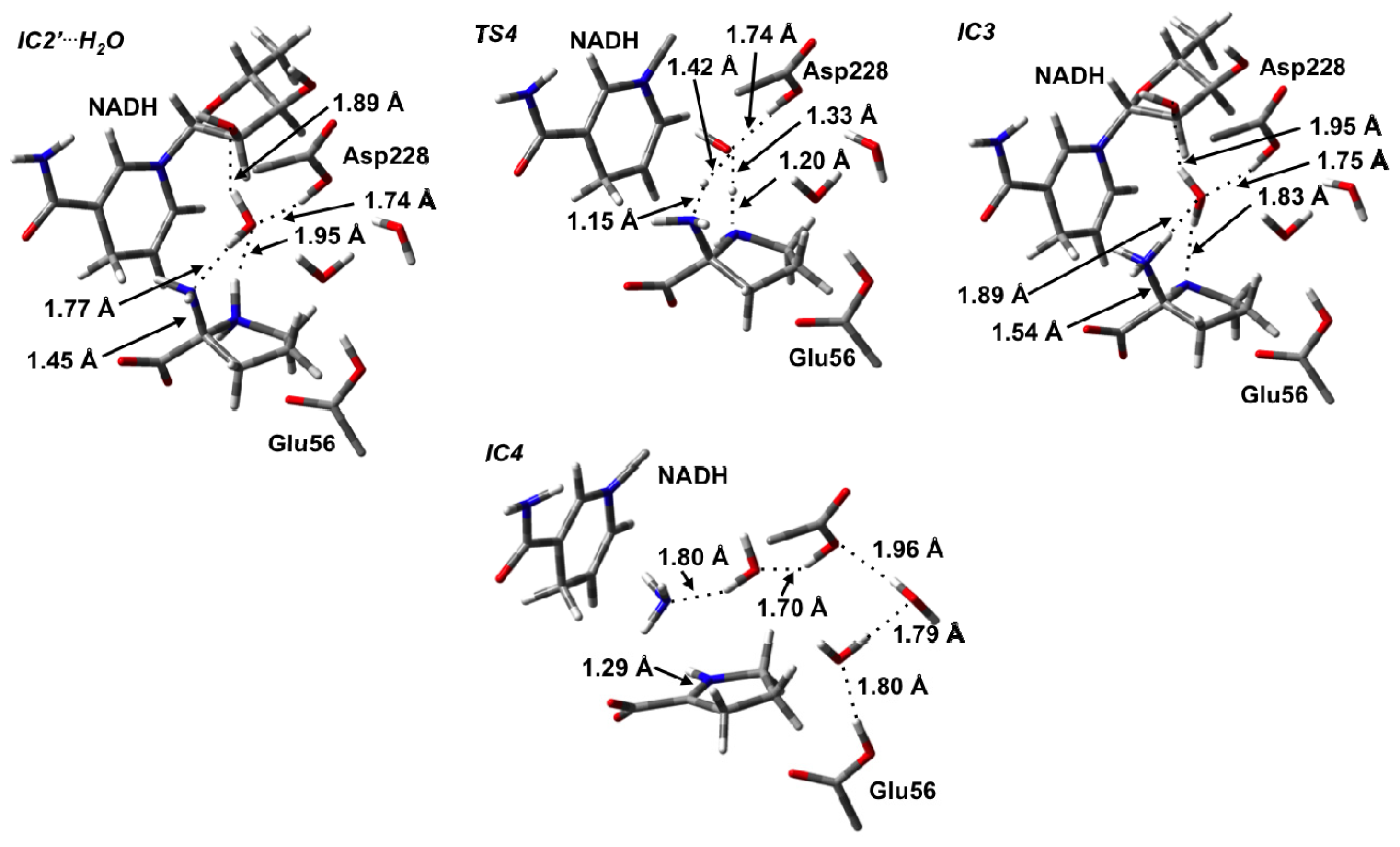
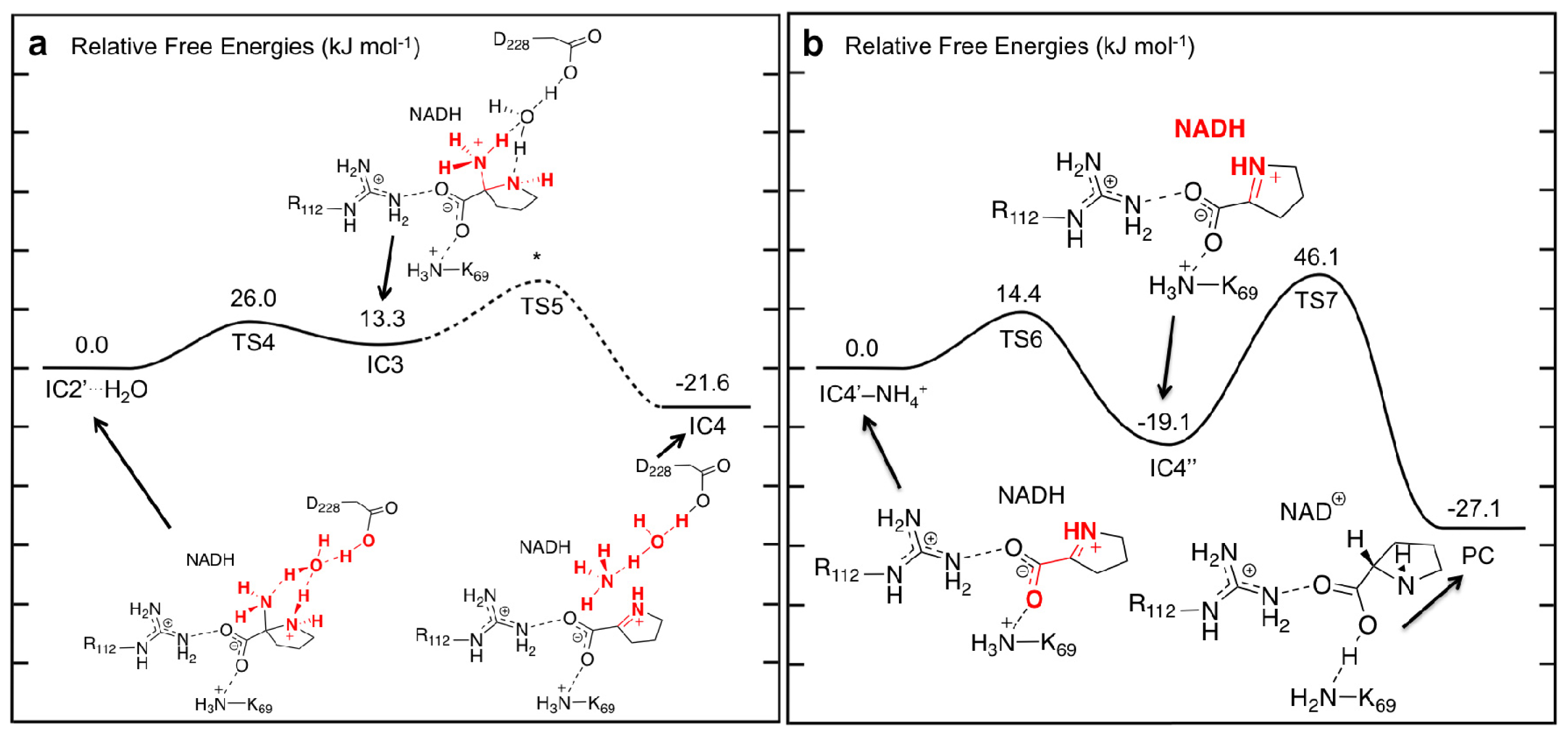
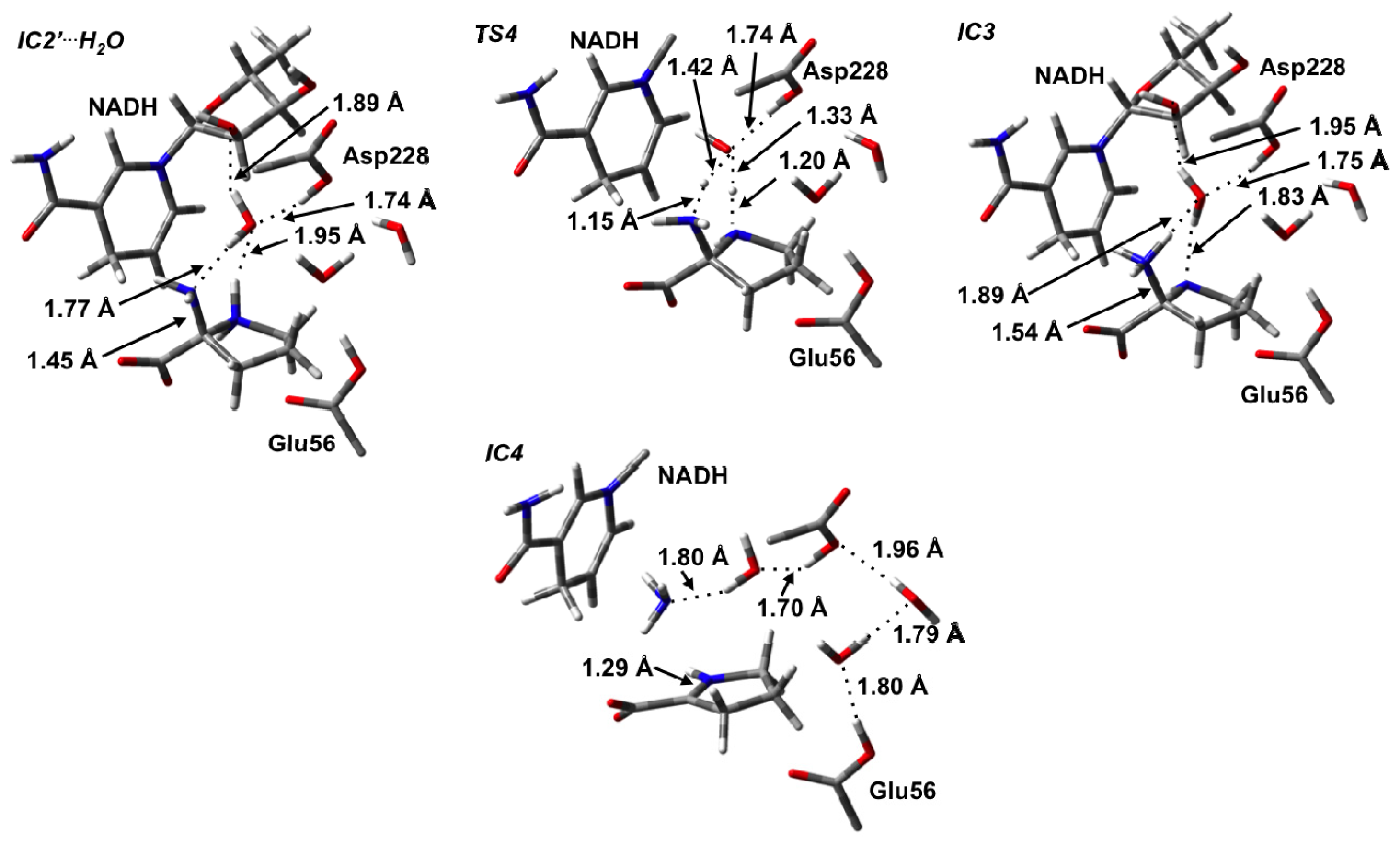
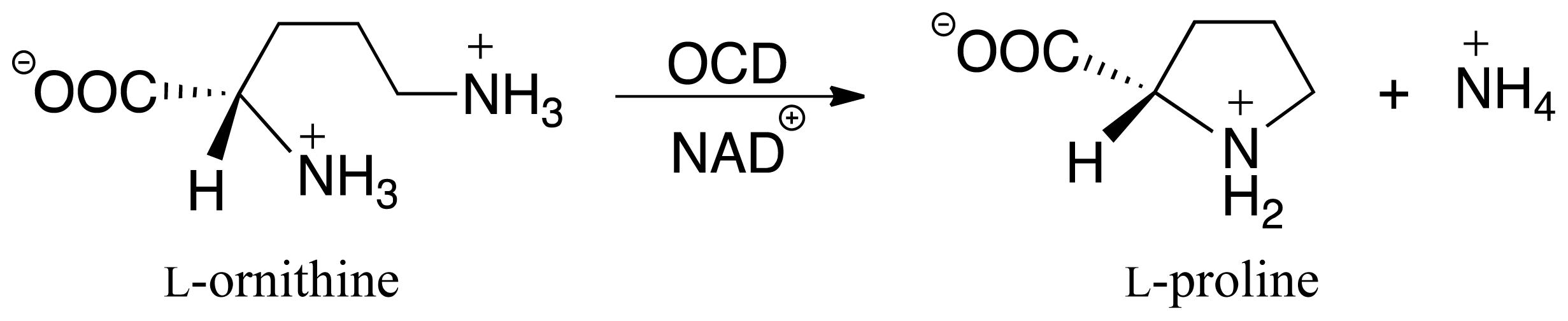
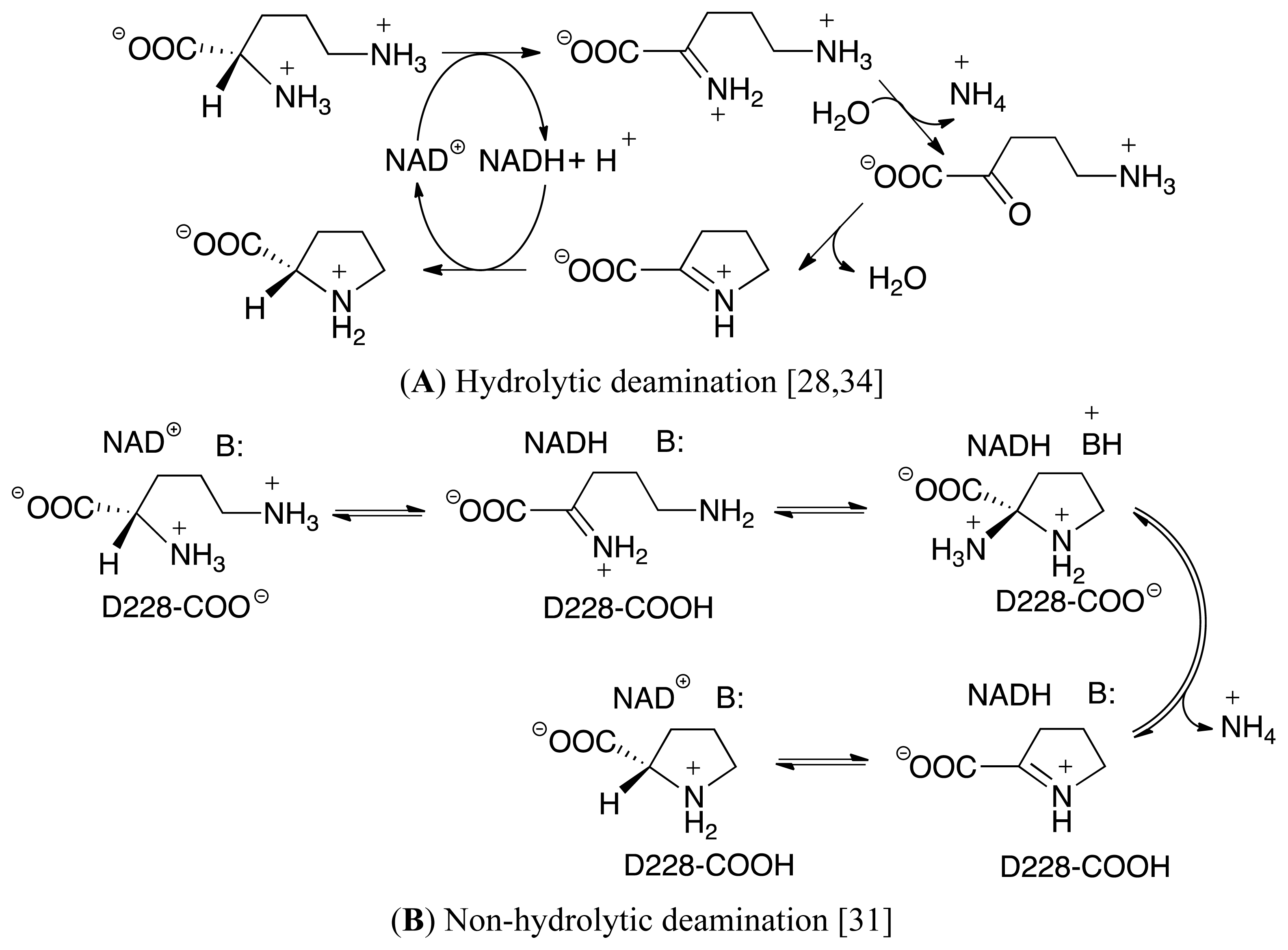
3.3. Deamination of 2-Aminoproline (AP)
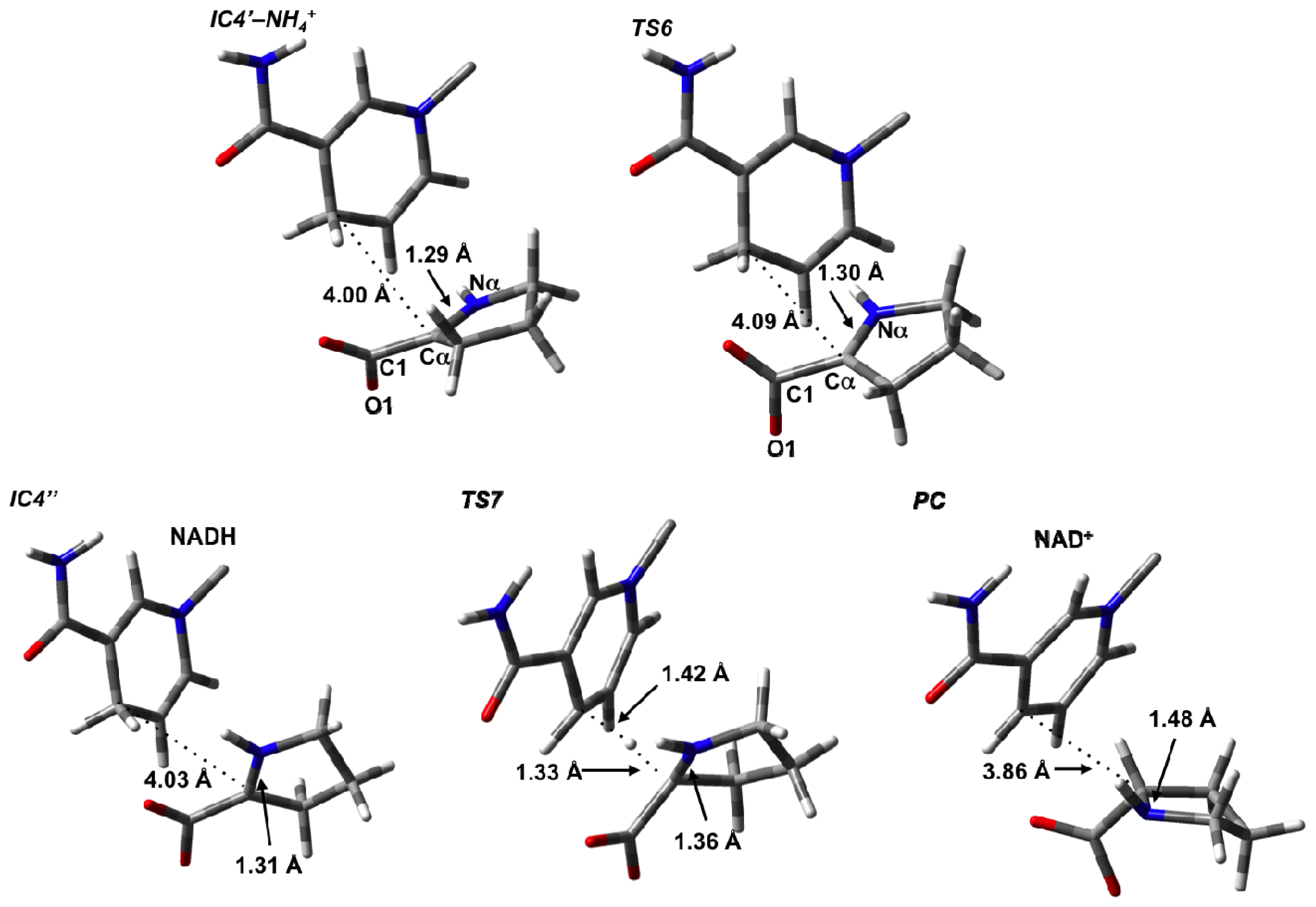
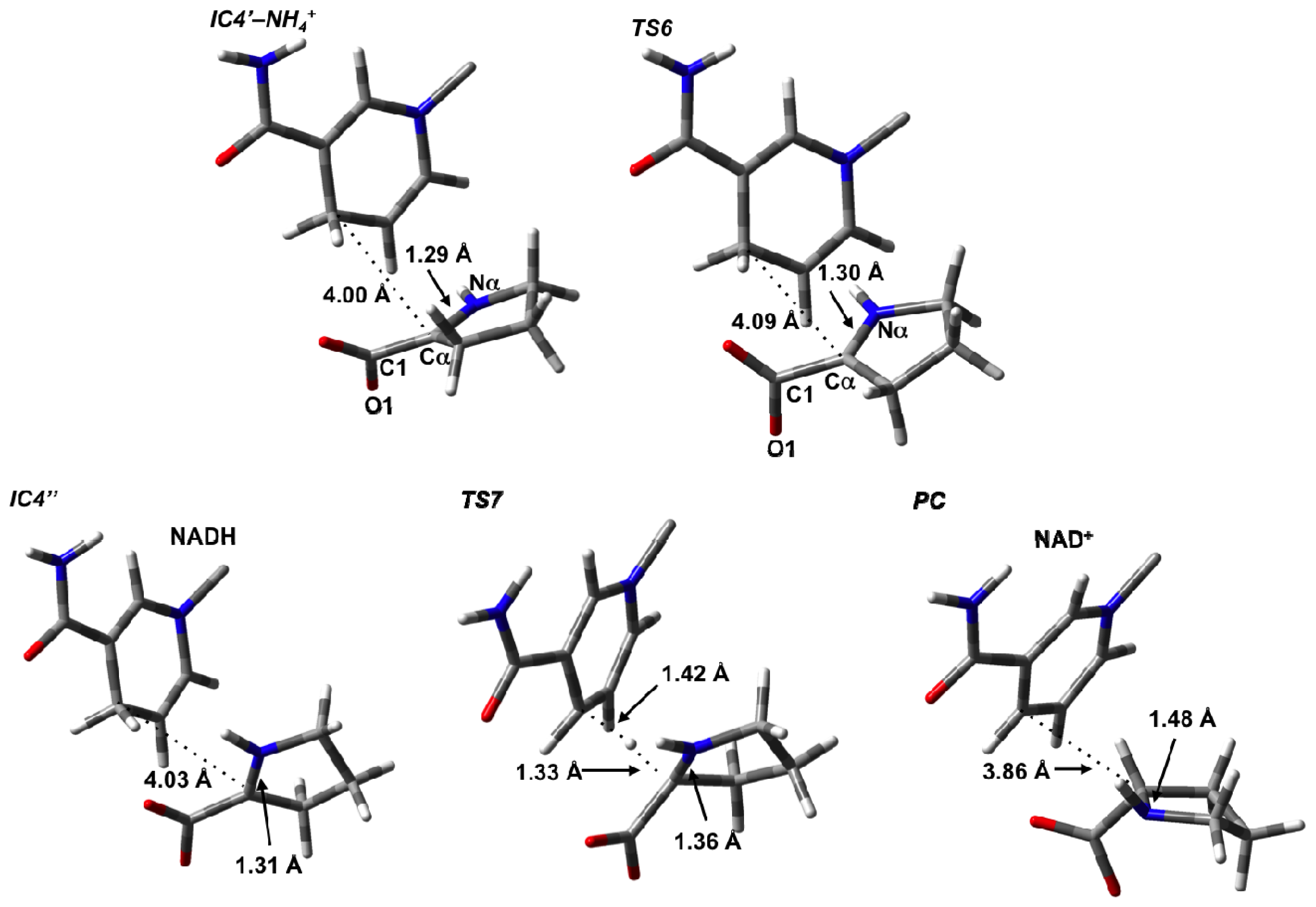
3.4. Hydride Transfer from NADH onto P2C to Give the Final Product L-Proline
4. Conclusions
Acknowledgments
References
- Meng, Z.H.; Lou, Z.Y.; Liu, Z.; Li, M.; Zhao, X.D.; Bartlam, M.; Rao, Z.H. Crystal structure of human pyrroline-5-carboxylate reductase. J. Mol. Biol 2006, 359, 1364–1377. [Google Scholar]
- Yura, T.; Vogel, H.J. Pyrroline-5-carboxylate reductase of Neurospora crassa; partial purification and some properties. J. Biol. Chem 1959, 234, 335–338. [Google Scholar]
- Valle, D.; Blaese, R.M.; Phang, J.M. Increased sensitivity of lymphocyte Δ1-pyrroline-5-carboxylate reductase to inhibition by proline with transformation. Nature 1975, 253, 214–216. [Google Scholar]
- Shiono, T.; Kador, P.F.; Kinoshita, J.H. Stimulation of the hexose-monophosphate pathway by pyrroline-5-carboxylate reductase in the lens. Exp. Eye Res 1985, 41, 767–775. [Google Scholar]
- Yeh, G.C.; Harris, S.C.; Phang, J.M. Pyrroline-5-carboxylate reductase in human-erythrocytes—A comparison of differential regulation. J. Clin. Invest 1981, 67, 1042–1046. [Google Scholar]
- Murahama, M.; Yoshida, T.; Hayashi, F.; Ichino, T.; Sanada, Y.; Wada, K. Purification and characterization of Δ1-pyrroline-5-carboxylate reductase isoenzymes, indicating differential distribution in spinach (Spinacia oleracea L.) leaves. Plant Cell Physiol 2001, 42, 742–750. [Google Scholar]
- Deutch, C.E.; Klarstrom, J.L.; Link, C.L.; Ricciardi, D.L. Oxidation of L-thiazolidine-4-carboxylate by Δ1-pyrroline-5-carboxylate reductase in Escherichia coli. Curr. Microbiol 2001, 42, 442–446. [Google Scholar]
- Basch, J.J.; Wickham, E.D.; Farrell, H.M. Pyrroline-5-carboxylate reductase in lactating bovine mammary glands. J. Dairy Sci 1996, 79, 1361–1368. [Google Scholar]
- Forlani, G.; Petrollino, D.; Fusetti, M.; Romanini, L.; Nocek, B.; Joachimiak, A.; Berlicki, L.; Kafarski, P. Δ1-pyrroline-5-carboxylate reductase as a new target for therapeutics: Inhibition of the enzyme from Streptococcus pyogenes and effects in vivo. Amino Acids 2012, 42, 2283–2291. [Google Scholar]
- Nakajima, K.; Natsu, S.; Mizote, T.; Nagata, Y.; Aoyania, K.; Fukuda, Y.; Nagata, K. Possible involvement of put A gene in Helicobacter pylori colonization in the stomach and motility. Biomed. Res 2008, 29, 9–18. [Google Scholar]
- Hoper, D.; Volker, U.; Hecker, M. Comprehensive characterization of the contribution of individual SigB-dependent general stress genes to stress resistance of Bacillus subtilis. J. Bacteriol 2005, 187, 2810–2826. [Google Scholar]
- Forlani, G.; Giberti, S.; Berlicki, L.; Petrollino, D.; Kafarski, P. Plant P5C reductase as a new target for aminomethylenebisphosphonates. J. Agric. Food Chem 2007, 55, 4340–4347. [Google Scholar]
- Hare, P.D.; Cress, W.A. Metabolic implications of stress-induced proline accumulation in plants. Plant Growth Regul 1997, 21, 79–102. [Google Scholar]
- Nocek, B.; Chang, C.; Li, H.; Lezondra, L.; Holzle, D.; Collart, F.; Joachimiak, A. Crystal structures of Δ1-pyrroline-5-carboxylate reductase from human pathogens Neisseria meningitides and Streptococcus pyogenes. J. Mol. Biol 2005, 354, 91–106. [Google Scholar]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar]
- Maxwell, S.A.; Davis, G.E. Differential gene expression in p53-mediated apoptosis-resistant vs. apoptosis-sensitive tumor cell lines. Proc. Natl. Acad. Sci. USA 2000, 97, 13009–13014. [Google Scholar]
- Donald, S.P.; Sun, X.Y.; Hu, C.A.A.; Yu, J.; Mei, J.M.; Valle, D.; Phang, J.M. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res 2001, 61, 1810–1815. [Google Scholar]
- Chen, C.B.; Dickman, M.B. Proline suppresses apoptosis in the fungal pathogen Colletotrichum trifolii. Proc. Natl. Acad. Sci. USA 2005, 102, 3459–3464. [Google Scholar]
- Hagedorn, C.H.; Phang, J.M. Catalytic transfer of hydride ions from NADPH to oxygen by the interconversions of proline and Δ1-pyrroline-5-carboxylate. Arch. Biochem. Biophys 1986, 248, 166–174. [Google Scholar]
- Hagedorn, C.H. Demonstration of a NADPH-linked Δ1-pyrroline-5-carboxylate proline shuttle in a cell-free rat-liver system. Biochim. Biophys. Acta 1986, 884, 11–17. [Google Scholar]
- Smith, R.J.; Downing, S.J.; Phang, J.M.; Lodato, R.F.; Aoki, T.T. Biosynthesis and metabolism of arginine in bacteria. Proc. Natl. Acad. Sci. USA 1980, 77, 5221–5225. [Google Scholar]
- Deutch, A.H.; Smith, C.J.; Rushlow, K.E.; Kretschmer, P.J. Escherichia-coli Δ1-pyrroline-5-carboxylate reductase—Gene sequence, protein overproduction and purification. Nucleic Acids Res 1982, 10, 7701–7714. [Google Scholar]
- Cunin, R.; Glansdorff, N.; Pierard, A.; Stalon, V. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev 1986, 50, 314–352. [Google Scholar]
- Aral, B.; Kamoun, P. The proline biosynthesis in living organisms. Amino Acids 1997, 13, 189–217. [Google Scholar]
- Tanner, J.J. Structural biology of proline catabolism. Amino Acids 2008, 35, 719–730. [Google Scholar]
- Petrollino, D.; Forlani, G. Coenzyme preference of Streptococcus pyogenes Δ1-pyrroline-5-carboxylate reductase: Evidence supporting NADPH as the physiological electron donor. Amino Acids 2012, 43, 493–497. [Google Scholar]
- Soto, M.J.; Vandillewijn, P.; Olivares, J.; Toro, N. Ornithine cyclodeaminase activity in Rhizobium-meliloti. FEMS Microbiol. Lett 1994, 119, 209–213. [Google Scholar]
- Graupner, M.; White, R.H. Methanococcus jannaschii generates L-proline by cyclization of L-ornithine. J. Bacteriol 2001, 183, 5203–5205. [Google Scholar]
- Alam, S.; Wang, S.C.; Ruzicka, F.J.; Frey, P.A.; Wedekind, J.E. Crystallization and X-ray diffraction analysis of ornithine cyclodeaminase from Pseudomonas putida. Acta Crystallogr. Sect. D Biol. Crystallogr 2004, 60, 941–944. [Google Scholar]
- Gallagher, D.T.; Monbouquette, H.G.; Schroder, I.; Robinson, H.; Holden, M.J.; Smith, N.N. Structure of alanine dehydrogenase from Archaeoglobus: Active site analysis and relation to bacterial cyclodeaminases and mammalian μ-crystallin. J. Mol. Biol 2004, 342, 119–130. [Google Scholar]
- Goodman, J.L.; Wang, S.; Alam, S.; Ruzicka, F.J.; Frey, P.A.; Wedekind, J.E. Ornithine cyclodeaminase: Structure, mechanism of action, and implications for the μ-crystallin family. Biochemistry 2004, 43, 13883–13891. [Google Scholar]
- Carugo, O.; Argos, P. NADP-dependent enzymes. I. Conserved stereochemistry of cofactor binding. Proteins 1997, 28, 10–28. [Google Scholar]
- Andres, J.; Moliner, V.; Safont, V.S.; Domingo, L.R.; Picher, M.T. On transition structures for hydride transfer step in enzyme catalysis. A comparative study on models of glutathione reductase derived from semiempirical, HF, and DFT methods. J. Org. Chem 1996, 61, 7777–7783. [Google Scholar]
- Walsh, C. Enzymatic Reaction Mechanisms; W. H. Freeman: San Francisco, CA, USA, 1979. [Google Scholar]
- Klinman, J.P. Isotope-effects and structure-reactivity correlations in yeast alcohol-dehydrogenase reaction—Study of enzyme-catalyzed oxidation of aromatic alcohols. Biochemistry 1976, 15, 2018–2026. [Google Scholar]
- Lipscomb, W.N. Structure and catalysis of enzymes. Annu. Rev. Biochem 1983, 52, 17–34. [Google Scholar]
- Sans, N.; Schroder, G.; Schroder, J. The Noc region of Ti plasmid C58 codes for arginase and ornithine cyclodeaminase. Eur. J. Biochem 1987, 167, 81–87. [Google Scholar]
- Erdtman, E.; Bushnell, E.A.C.; Gauld, J.W.; Eriksson, L.A. Computational studies on Schiff-base formation: Implications for the catalytic mechanism of porphobilinogen synthase. Comput. Theor. Chem 2011, 963, 479–489. [Google Scholar]
- Almasi, J.N.; Bushnell, E.A.C.; Gauld, J.W. A QM/MM-based computational investigation on the catalytic mechanism of saccharopine reductase. Molecules 2011, 16, 8569–8589. [Google Scholar]
- Godoy-Alcantar, C.; Yatsimirsky, A.K.; Lehn, J.M. Structure-stability correlations for imine formation in aqueous solution. J. Phys. Org. Chem 2005, 18, 979–985. [Google Scholar]
- Hall, N.E.; Smith, B.J. High-level ab initio molecular orbital calculations of imine formation. J. Phys. Chem. A 1998, 102, 4930–4938. [Google Scholar]
- Szefczyk, B.; Kedzierski, P.; Sokalski, W.A.; Leszczynski, J. Theoretical insights into catalysis by phosphonoacetaldehyde hydrolase. Mol. Phys 2006, 104, 2203–2211. [Google Scholar]
- Conant, J.B.; Bartlett, P.D. A quantitative study of semicarbazone formation. J. Am. Chem. Soc 1932, 54, 2881–2899. [Google Scholar]
- Hill, R.L.; Crowell, T.I. Structural effects in the reactivity of primary amines with piperonal. J. Am. Chem. Soc 1956, 78, 2284–2286. [Google Scholar]
- Jencks, W.P. Reactions of nucleophilic reagents with phosphoramidate. J. Am. Chem. Soc 1959, 81, 475–481. [Google Scholar]
- Lowry, T.H.; Richardson, K.S. Mechanism and Theory in Organic Chemistry; Harper and Row Publishers: New York, NY, USA, 1976. [Google Scholar]
- Santerre, G.M.; Hansrote, C.J., Jr; Crowell, T.I. The reaction of aromatic aldehydes with n-butylamine. acid catalysis and substituent effects. J. Am. Chem. Soc. 1958, 80, 1254–1257. [Google Scholar]
- Hallen, A.; Cooper, A.J.L.; Jamie, J.F.; Haynes, P.A.; Willows, R.D. Mammalian forebrain ketimine reductase identified as μ-crystallin; potential regulation by thyroid hormones. J. Neurochem 2011, 118, 379–387. [Google Scholar]
- Kathiresan, T.; Krishnan, K.; Krishnakumar, V.; Agrawal, R.; Anand, A.; Muralidhar, D.; Mishra, A.K.; Dhople, V.M.; Aggrawal, R.K.; Sharma, Y. Triose phosphate isomerase, a novel enzyme-crystallin, and tau-crystallin in crocodile cornea—High accumulation of both proteins during late embryonic development. FEBS J 2006, 273, 3370–3380. [Google Scholar]
- Kim, R.Y.; Gasser, R.; Wistow, G.J. μ-crystallin is a mammalian homolog of Agrobacterium ornithine cyclodeaminase and is expressed in human retina. Proc. Natl. Acad. Sci. USA 1992, 89, 9292–9296. [Google Scholar]
- Jaenicke, R.; Slingsby, C. Lens crystallins and their microbial homologs: Structure, stability, and function. Crit. Rev. Biochem. Mol. Biol 2001, 36, 435–499. [Google Scholar]
- Llano, J.; Gauld, J.W. Mechanistics of Enzyme Catalysis: From Small to Large Active-Site Models. In Quantum Biochemistry: Electronic Structure and Biological Activity; Matta, C.F., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Volume 2, pp. 643–666. [Google Scholar]
- MOE, version 2010.10; Chemical Computing Group: Montreal, QC, Canada, 2010.
- Bond, S.D.; Leimkuhler, B.J.; Laird, B.B. The Nosé-Poincaré method for constant temperature molecular dynamics. J. Comput. Phys 1999, 151, 114–134. [Google Scholar]
- Bushnell, E.A.C.; Erdtman, E.; Llano, J.; Eriksson, L.A.; Gauld, J.W. The first branching point in porphyrin biosynthesis: A systematic docking, molecular dynamics and quantum mechanical/molecular mechanical study of substrate binding and mechanism of uroporphyrinogen-III decarboxylase. J. Comput. Chem 2011, 32, 822–834. [Google Scholar]
- Gaussian 09, Revision B.01; Gaussian Inc: Wallingford, CT, USA, 2010.
- Bearpark, M.J.; Ogliaro, F.; Vreven, T.; Boggio-Pasqua, M.; Frisch, M.J.; Larkin, S.M.; Robb, M.A. CASSCF calculations for excited states of large molecules: Choosing when to use the RASSCF, ONIOM and MMVB approximations. Aip. Conf. Proc 2007, 2, 583–585. [Google Scholar]
- Dapprich, S.; Komaromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struc. Theochem 1999, 461, 1–21. [Google Scholar]
- Humbel, S.; Sieber, S.; Morokuma, K. The IMOMO method: Integration of different levels of molecular orbital approximations for geometry optimization of large systems: Test for n-butane conformation and SN2 reaction: RCl + Cl−. J. Chem. Phys 1996, 105, 1959–1967. [Google Scholar]
- Maseras, F.; Morokuma, K. IMOMM—A new integrated ab-initio plus molecular mechanics geometry optimization scheme of equilibrium structures and transition-states. J. Comput. Chem 1995, 16, 1170–1179. [Google Scholar]
- Morokuma, K.; Musaev, D.G.; Vreven, T.; Basch, H.; Torrent, M.; Khoroshun, D.V. Model studies of the structures, reactivities, and reaction mechanisms of metalloenzymes. IBM J. Res. Dev 2001, 45, 367–395. [Google Scholar]
- Svensson, M.; Humbel, S.; Froese, R.D.J.; Matsubara, T.; Sieber, S.; Morokuma, K. ONIOM: A multilayered integrated MO + MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)3)2 + H2 oxidative addition. J. Phys. Chem 1996, 100, 19357–19363. [Google Scholar]
- Vreven, T.; Byun, K.S.; Komaromi, I.; Dapprich, S.; Montgomery, J.A.; Morokuma, K.; Frisch, M.J. Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J. Chem. Theory Comput 2006, 2, 815–826. [Google Scholar]
- Vreven, T.; Morokuma, K. On the application of the IMOMO (integrated molecular orbital plus molecular orbital) method. J. Comput. Chem 2000, 21, 1419–1432. [Google Scholar]
- Vreven, T.; Morokuma, K.; Farkas, O.; Schlegel, H.B.; Frisch, M.J. Geometry optimization with QM/MM, ONIOM, and other combined methods. I. Microiterations and constraints. J. Comput. Chem 2003, 24, 760–769. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys 1993, 98, 1372–1377. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem 2005, 26, 1668–1688. [Google Scholar]
- Hu, L.H.; Soderhjelm, P.; Ryde, U. On the convergence of QM/MM energies. J. Chem. Theory Comput 2011, 7, 761–777. [Google Scholar]
- Brazier, J.B.; Tomkinson, N.C.O. Secondary and primary amine catalysts for iminium catalysis. Top. Curr. Chem 2009, 291, 281–347. [Google Scholar]
- Roy, D.; von Ragué Schleyer, P. Chemical Origin of Life: How do Five HCN Molecules Combine to form Adenine under Prebiotic and Interstellar Conditions. In Quantum Biochemistry: Electronic Structure and Biological Activity; Matta, C.F., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; Volume 1, pp. 199–217. [Google Scholar]


© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ion, B.F.; Bushnell, E.A.C.; Luna, P.D.; Gauld, J.W. A Molecular Dynamics (MD) and Quantum Mechanics/Molecular Mechanics (QM/MM) Study on Ornithine Cyclodeaminase (OCD): A Tale of Two Iminiums. Int. J. Mol. Sci. 2012, 13, 12994-13011. https://doi.org/10.3390/ijms131012994
Ion BF, Bushnell EAC, Luna PD, Gauld JW. A Molecular Dynamics (MD) and Quantum Mechanics/Molecular Mechanics (QM/MM) Study on Ornithine Cyclodeaminase (OCD): A Tale of Two Iminiums. International Journal of Molecular Sciences. 2012; 13(10):12994-13011. https://doi.org/10.3390/ijms131012994
Chicago/Turabian StyleIon, Bogdan F., Eric A. C. Bushnell, Phil De Luna, and James W. Gauld. 2012. "A Molecular Dynamics (MD) and Quantum Mechanics/Molecular Mechanics (QM/MM) Study on Ornithine Cyclodeaminase (OCD): A Tale of Two Iminiums" International Journal of Molecular Sciences 13, no. 10: 12994-13011. https://doi.org/10.3390/ijms131012994
APA StyleIon, B. F., Bushnell, E. A. C., Luna, P. D., & Gauld, J. W. (2012). A Molecular Dynamics (MD) and Quantum Mechanics/Molecular Mechanics (QM/MM) Study on Ornithine Cyclodeaminase (OCD): A Tale of Two Iminiums. International Journal of Molecular Sciences, 13(10), 12994-13011. https://doi.org/10.3390/ijms131012994