Bioinformatics Tools and Novel Challenges in Long Non-Coding RNAs (lncRNAs) Functional Analysis

Abstract
:1. Introduction
2. The Four Main Roles of Long Non-Coding RNAs
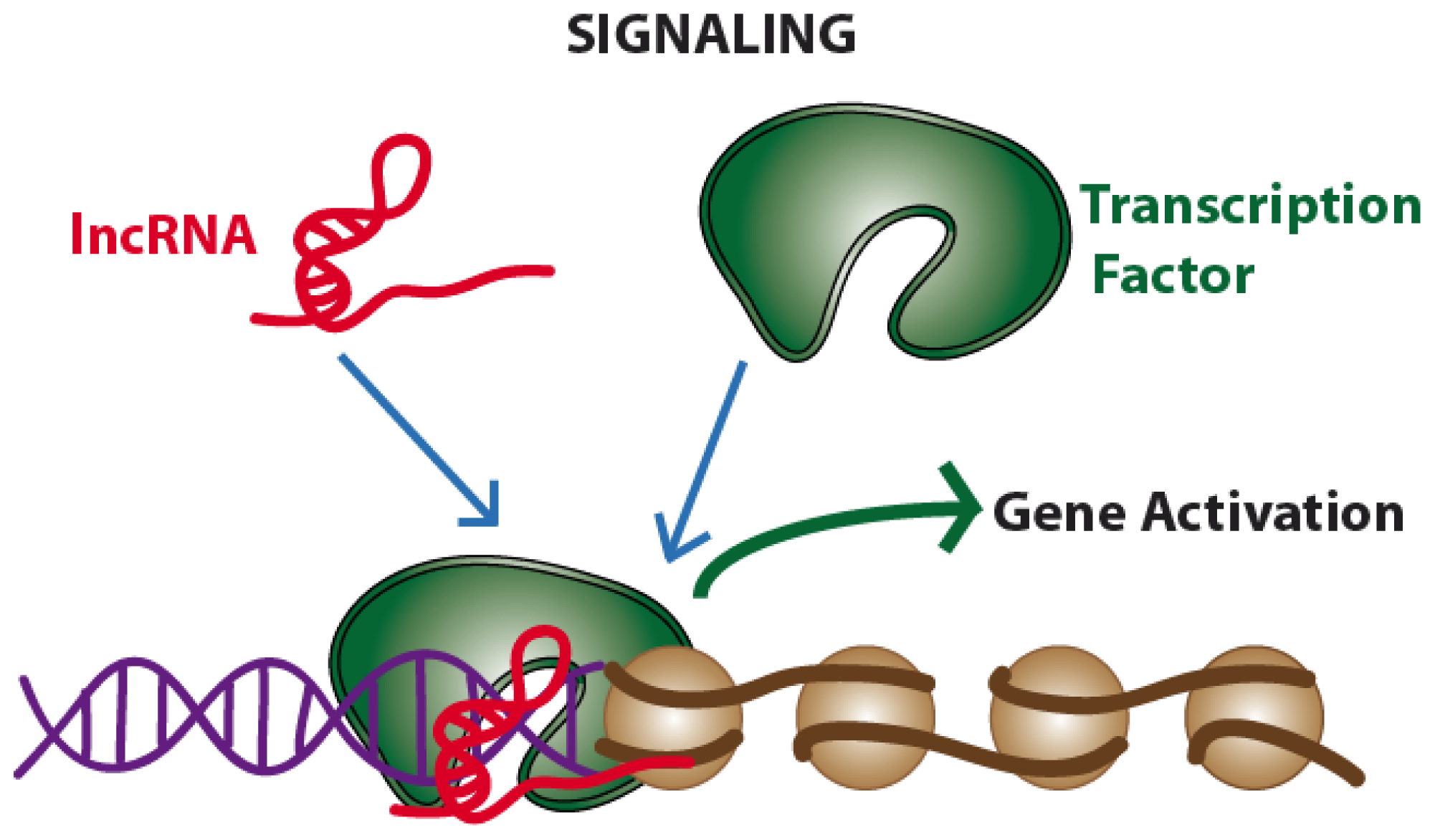
2.1. Signaling. Long Non-Coding RNAs Acting as Gene Expression Enhancers or Repressors
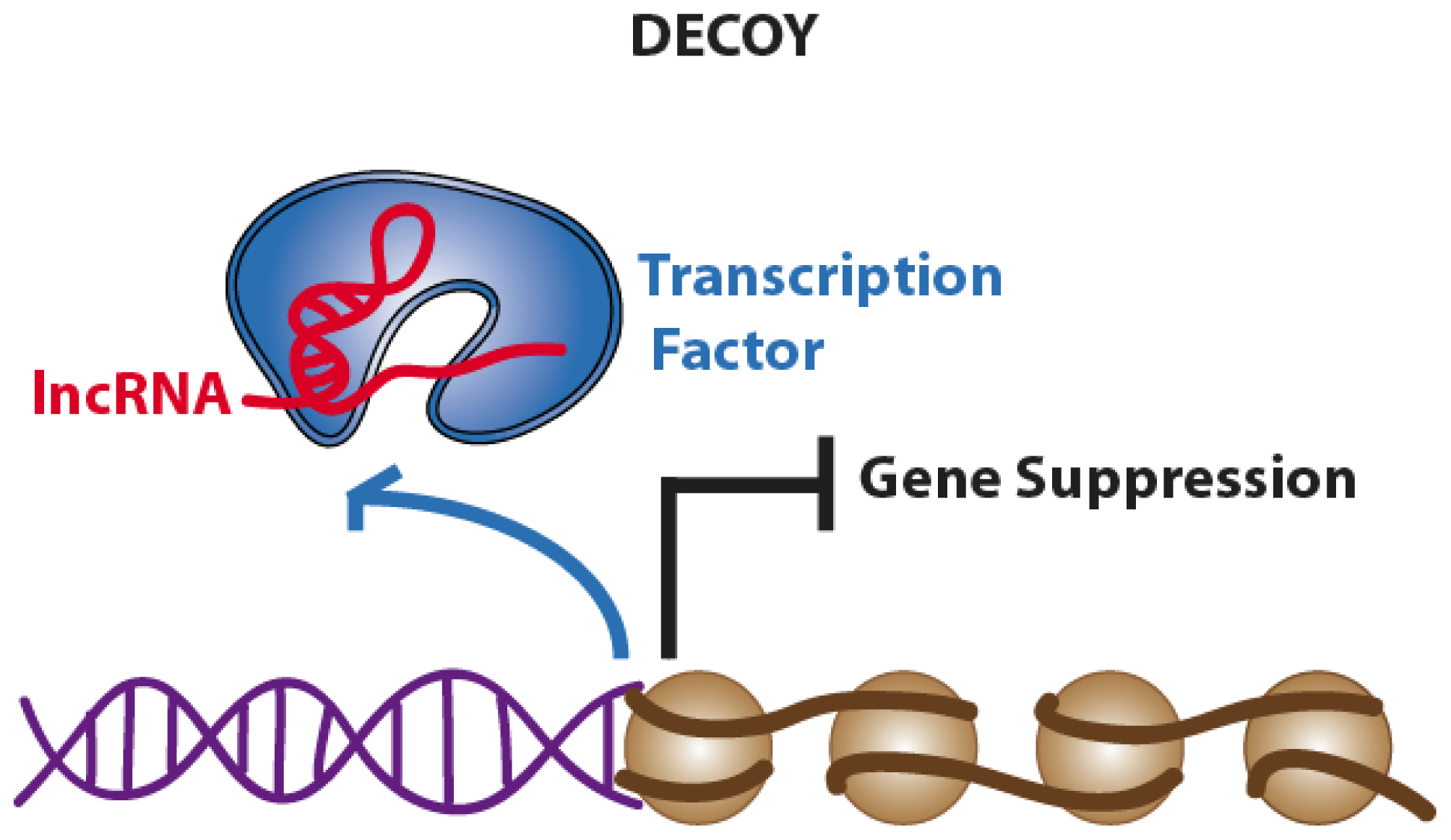
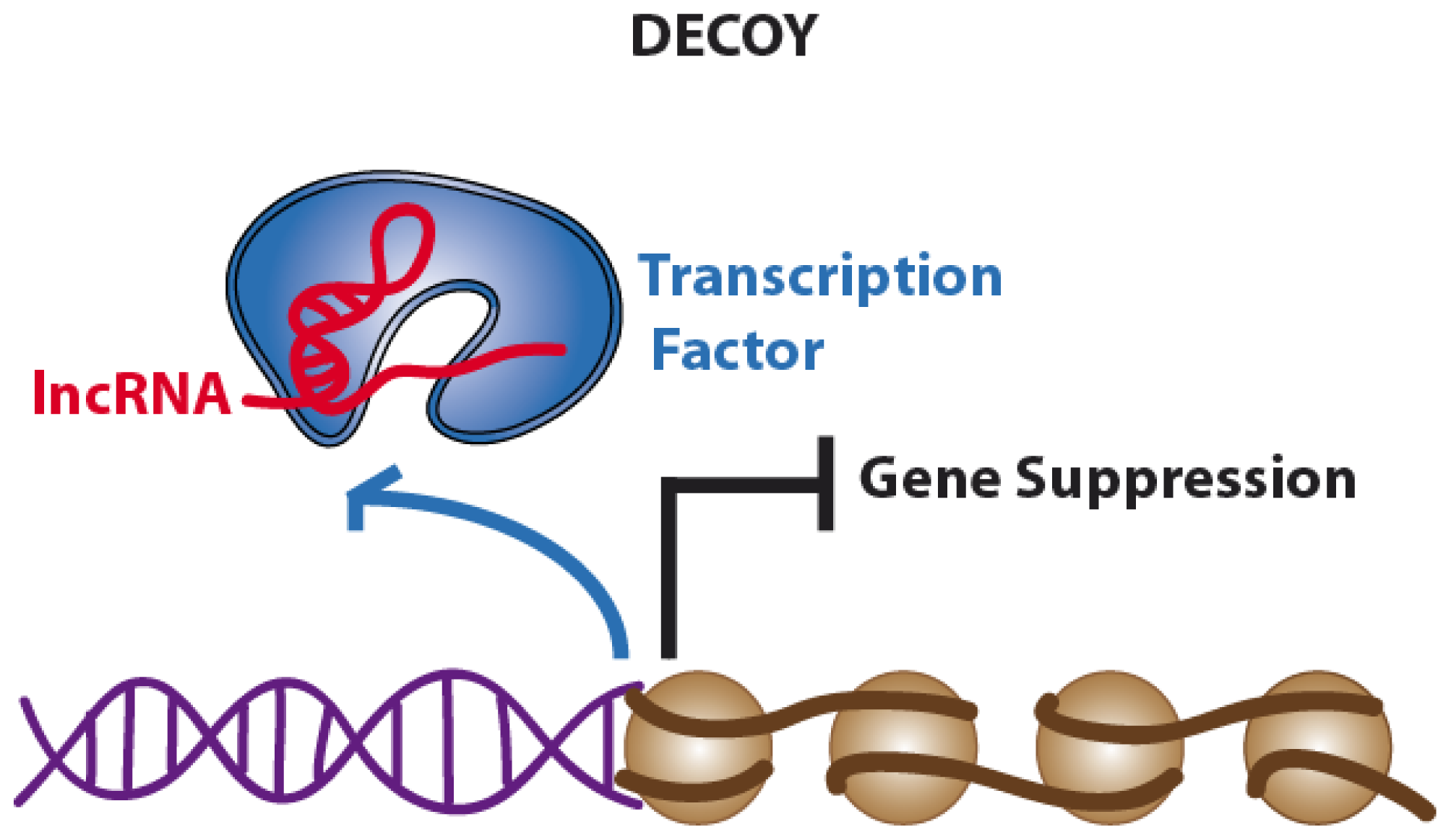
2.2. Decoys. Long Non-Coding RNAs Acting as Molecular Sinks
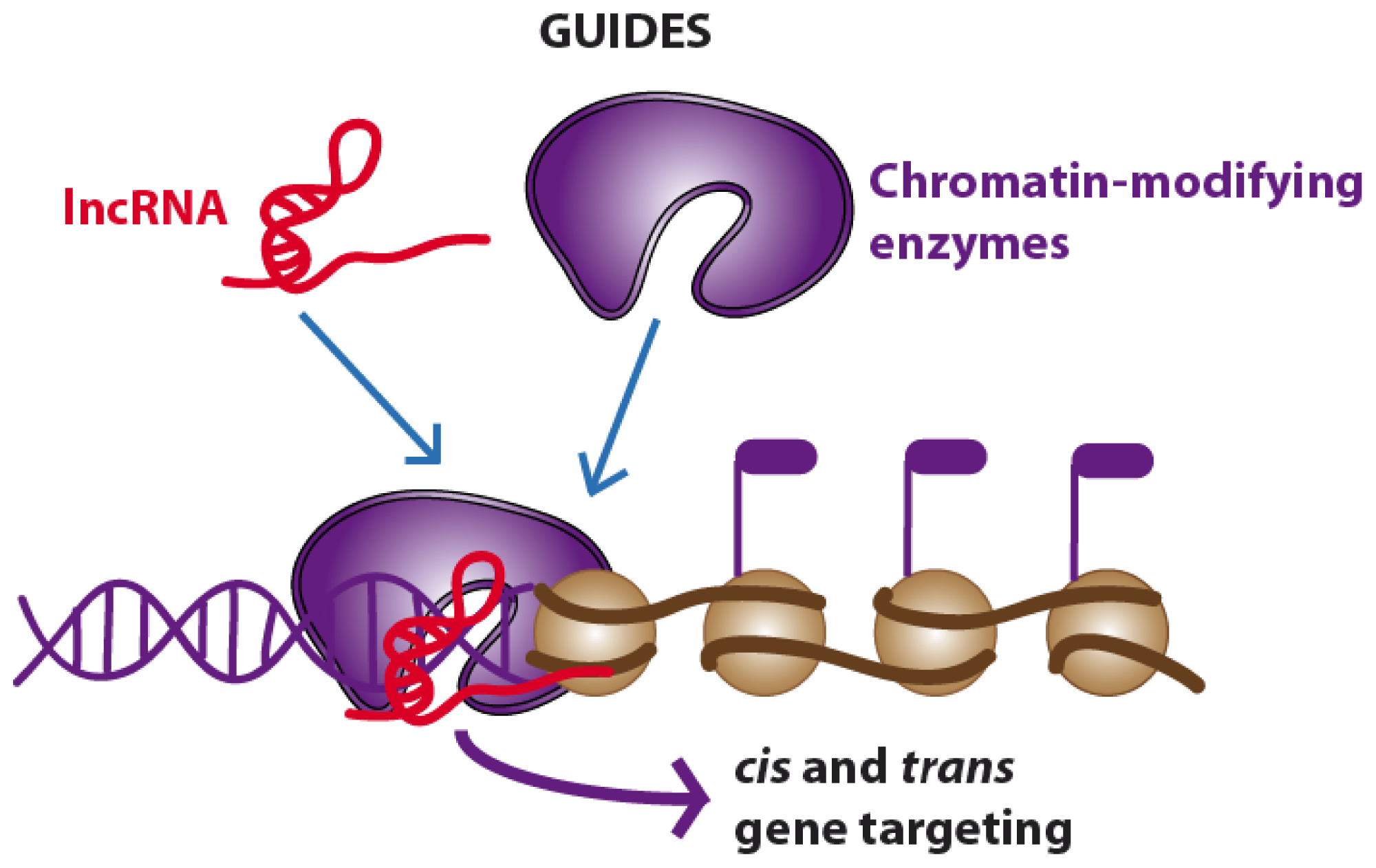
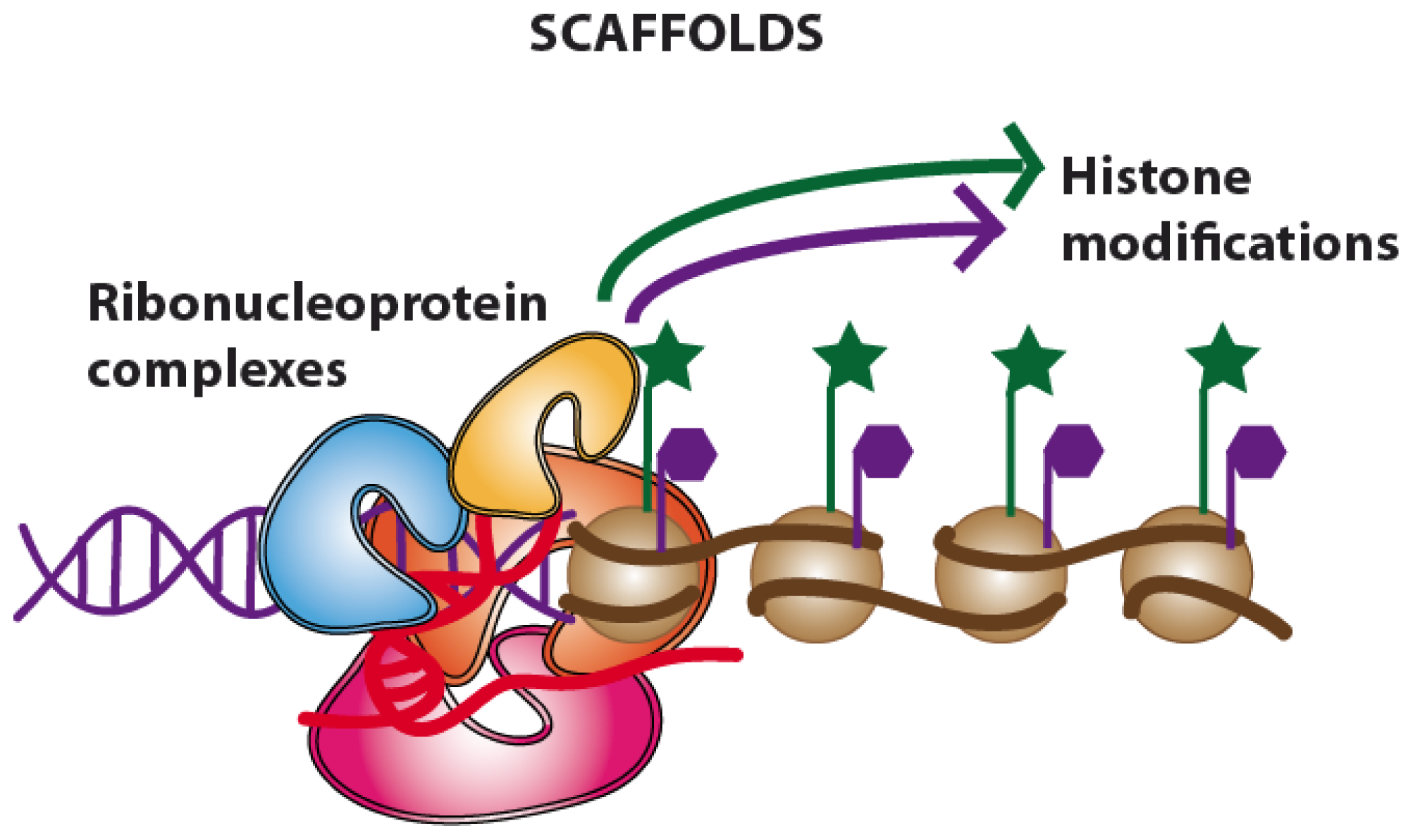
2.3. Guides and Scaffolds. Long Non-Coding RNAs Acting as “Molecular Assemblers”
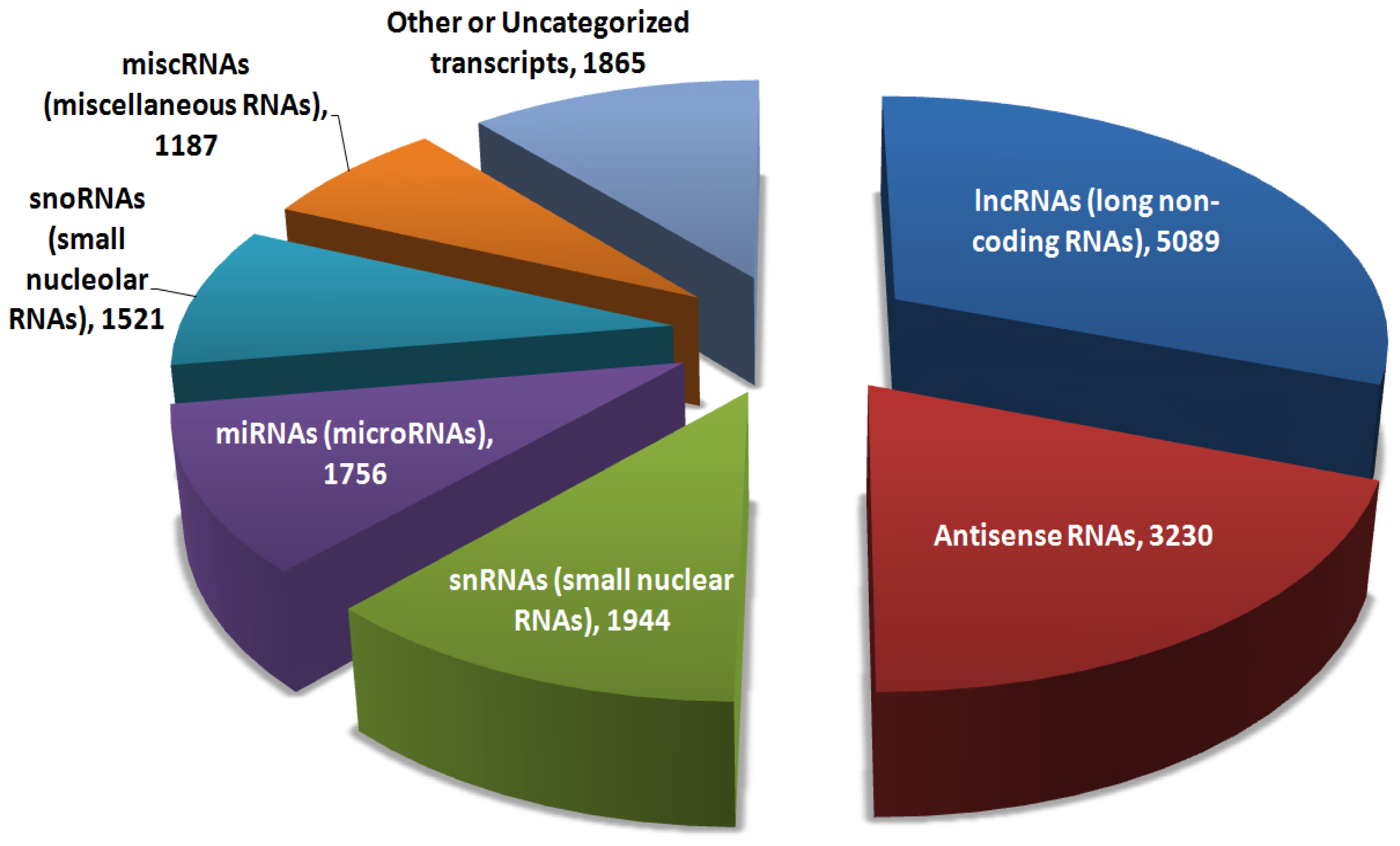
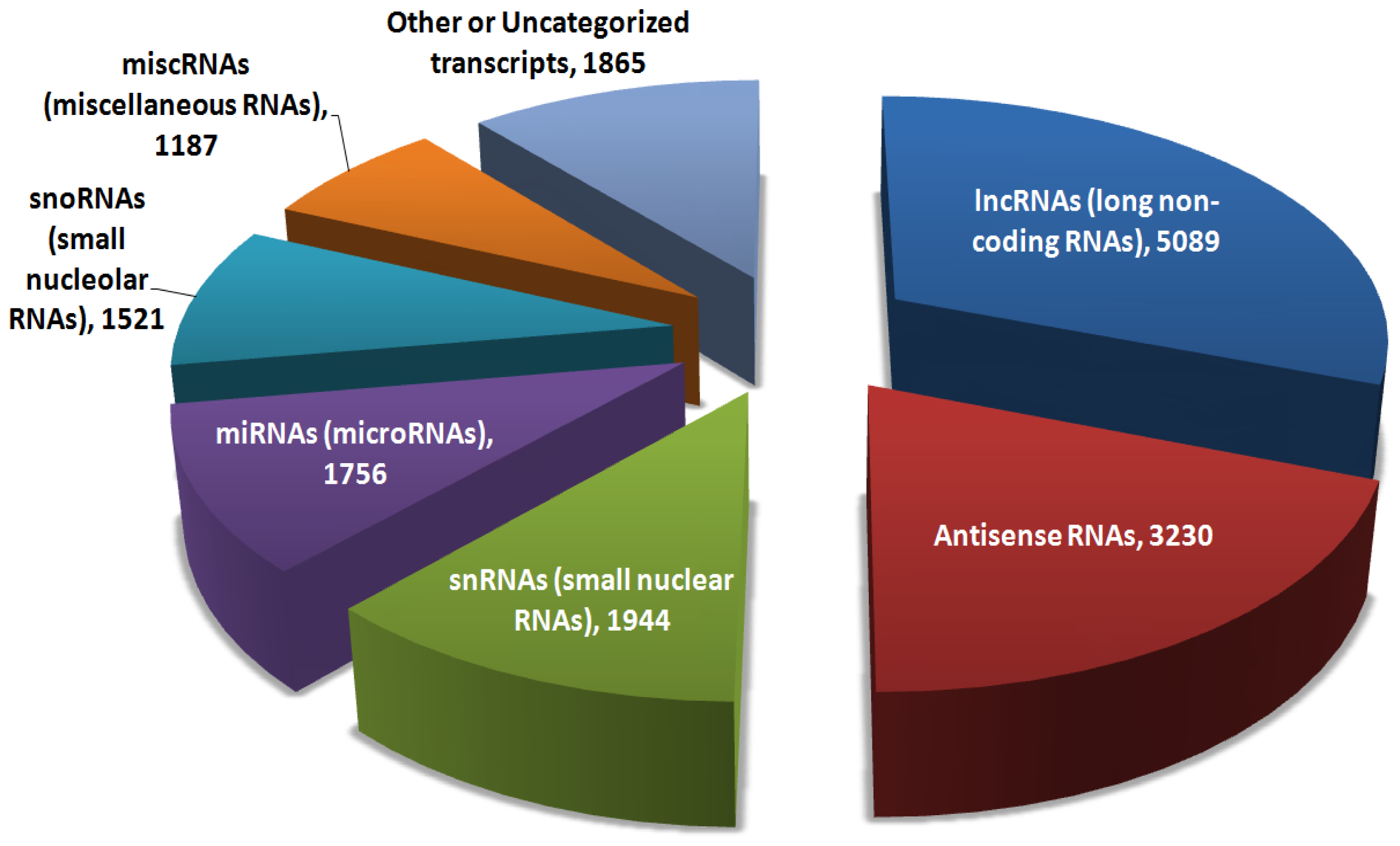
3. Functional Analysis of Long Non-Coding RNAs (lncRNAs)
3.1. Databases and Public Repositories
3.2. Annotation Tools and Other Bioinformatics Tools
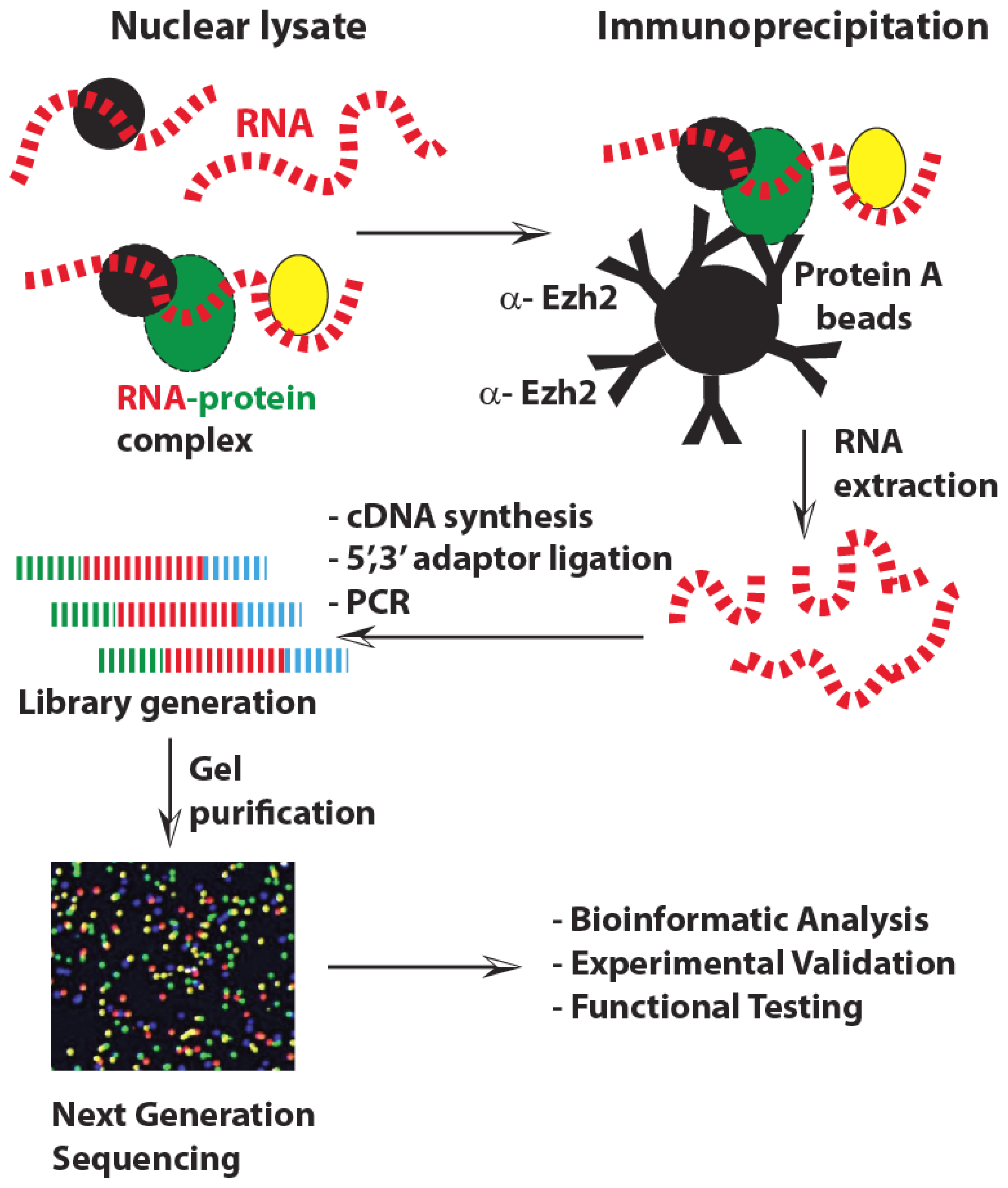
3.3. Biological and Bioinformatics Approaches for lncRNA Discovery
5. Conclusions
Acknowledgments
References
- ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640.
- ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816.
- Mattick, J.S. The genetic signatures of noncoding RNAs. PLoS Genet 2009, 5, e1000459. [Google Scholar]
- Mattick, J.S. The functional genomics of noncoding RNA. Science 2005, 309, 1527–1528. [Google Scholar]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet 2006, 15, R17–R29. [Google Scholar]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet 2009, 10, 155–159. [Google Scholar]
- Nagano, T.; Fraser, P. No-nonsense functions for long noncoding RNAs. Cell 2011, 145, 178–181. [Google Scholar]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev 2009, 23, 1494–1504. [Google Scholar]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar]
- De Santa, F.; Barozzi, I.; Mietton, F.; Ghisletti, S.; Polletti, S.; Tusi, B.K.; Muller, H.; Ragoussis, J.; Wei, C.L.; Natoli, G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 2010, 8, e1000384. [Google Scholar]
- Orom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar]
- Amaral, P.P.; Clark, M.B.; Gascoigne, D.K.; Dinger, M.E.; Mattick, J.S. lncRNAdb: A reference database for long noncoding RNAs. Nucleic Acids Res 2011, 39, D146–D151. [Google Scholar]
- Baker, M. Long noncoding RNAs: the search for function. Nat. Meth 2011, 8, 379–383. [Google Scholar]
- He, S.; Su, H.; Liu, C.; Skogerbo, G.; He, H.; He, D.; Zhu, X.; Liu, T.; Zhao, Y.; Chen, R. MicroRNA-encoding long non-coding RNAs. BMC Genomics 2008, 9, 236. [Google Scholar]
- Kanduri, C.; Whitehead, J.; Mohammad, F. The long and the short of it: RNA-directed chromatin asymmetry in mammalian X-chromosome inactivation. FEBS Lett 2009, 583, 857–864. [Google Scholar]
- Lee, J.T. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev 2009, 23, 1831–1842. [Google Scholar]
- Mohammad, F.; Mondal, T.; Kanduri, C. Epigenetics of imprinted long noncoding RNAs. Epigenetics 2009, 4, 277–286. [Google Scholar]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar]
- Sasaki, Y.T.; Ideue, T.; Sano, M.; Mituyama, T.; Hirose, T. MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc. Natl. Acad. Sci. USA 2009, 106, 2525–2530. [Google Scholar]
- Schoeftner, S.; Blasco, M.A. A “higher order” of telomere regulation: Telomere heterochromatin and telomeric RNAs. EMBO J 2009, 28, 2323–2336. [Google Scholar]
- Wong, L.H.; Brettingham-Moore, K.H.; Chan, L.; Quach, J.M.; Anderson, M.A.; Northrop, E.L.; Hannan, R.; Saffery, R.; Shaw, M.L.; Williams, E.; et al. Centromere RNA is a key component for the assembly of nucleoproteins at the nucleolus and centromere. Genome Res 2007, 17, 1146–1160. [Google Scholar]
- Ferri, F.; Bouzinba-Segard, H.; Velasco, G.; Hube, F.; Francastel, C. Non-coding murine centromeric transcripts associate with and potentiate Aurora B kinase. Nucleic Acids Res 2009, 37, 5071–5080. [Google Scholar]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar]
- Huarte, M.; Rinn, J.L. Large non-coding RNAs: Missing links in cancer? Hum. Mol. Genet 2010, 19, R152–R161. [Google Scholar]
- Zong, X.; Tripathi, V.; Prasanth, K.V. RNA splicing control: Yet another gene regulatory role for long nuclear noncoding RNAs. RNA Biol 2011, 8, 968–977. [Google Scholar]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar]
- Wu, Q.; Kim, Y.C.; Lu, J.; Xuan, Z.; Chen, J.; Zheng, Y.; Zhou, T.; Zhang, M.Q.; Wu, C.I.; Wang, S.M. Poly A-transcripts expressed in HeLa cells. PLoS One 2008, 3, e2803. [Google Scholar]
- Martone, R.; Euskirchen, G.; Bertone, P.; Hartman, S.; Royce, T.E.; Luscombe, N.M.; Rinn, J.L.; Nelson, F.K.; Miller, P.; Gerstein, M.; et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. USA 2003, 100, 12247–12252. [Google Scholar]
- Prasanth, K.V.; Spector, D.L. Eukaryotic regulatory RNAs: An answer to the “genome complexity” conundrum. Genes Dev 2007, 21, 11–42. [Google Scholar]
- Dieci, G.; Fiorino, G.; Castelnuovo, M.; Teichmann, M.; Pagano, A. The expanding RNA polymerase III transcriptome. Trends Genet 2007, 23, 614–622. [Google Scholar]
- Amaral, P.P.; Mattick, J.S. Noncoding RNA in development. Mamm. Genome 2008, 19, 454–492. [Google Scholar]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Fraser, P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008, 322, 1717–1720. [Google Scholar]
- Pontier, D.B.; Gribnau, J. Xist regulation and function explored. Hum. Genet 2011, 130, 223–236. [Google Scholar]
- Gontan, C.; Jonkers, I.; Gribnau, J. Long noncoding RNAs and X chromosome inactivation. Prog. Mol. Subcell. Biol 2011, 51, 43–64. [Google Scholar]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet 2011, 43, 621–629. [Google Scholar]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 2007, 318, 798–801. [Google Scholar]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res 2010, 38, 5797–5806. [Google Scholar]
- Spitale, R.C.; Tsai, M.C.; Chang, H.Y. RNA templating the epigenome: Long noncoding RNAs as molecular scaffolds. Epigenetics 2011, 6, 539–543. [Google Scholar]
- Collins, K. Physiological assembly and activity of human telomerase complexes. Mech. Ageing Dev 2008, 129, 91–98. [Google Scholar]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar]
- Szymanski, M.; Erdmann, V.A.; Barciszewski, J. Noncoding regulatory RNAs database. Nucleic Acids Res 2003, 31, 429–431. [Google Scholar]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res 2003, 31, 439–441. [Google Scholar]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res 2009, 37, D136–D140. [Google Scholar]
- Pang, K.C.; Stephen, S.; Engstrom, P.G.; Tajul-Arifin, K.; Chen, W.; Wahlestedt, C.; Lenhard, B.; Hayashizaki, Y.; Mattick, J.S. RNAdb—A comprehensive mammalian noncoding RNA database. Nucleic Acids Res 2005, 33, D125–D130. [Google Scholar]
- Pang, K.C.; Stephen, S.; Dinger, M.E.; Engstrom, P.G.; Lenhard, B.; Mattick, J.S. RNAdb 2.0—An expanded database of mammalian non-coding RNAs. Nucleic Acids Res 2007, 35, D178–D182. [Google Scholar]
- Imanishi, T.; Itoh, T.; Suzuki, Y.; O’Donovan, C.; Fukuchi, S.; Koyanagi, K.O.; Barrero, R.A.; Tamura, T.; Yamaguchi-Kabata, Y.; Tanino, M.; et al. Integrative annotation of 21,037 human genes validated by full-length cDNA clones. PLoS Biol 2004, 2, e162. [Google Scholar]
- Yamasaki, C.; Koyanagi, K.O.; Fujii, Y.; Itoh, T.; Barrero, R.; Tamura, T.; Yamaguchi-Kabata, Y.; Tanino, M.; Takeda, J.; Fukuchi, S.; et al. Investigation of protein functions through data-mining on integrated human transcriptome database, H-Invitational database (H-InvDB). Gene 2005, 364, 99–107. [Google Scholar]
- Liu, C.; Bai, B.; Skogerbo, G.; Cai, L.; Deng, W.; Zhang, Y.; Bu, D.; Zhao, Y.; Chen, R. NONCODE: An integrated knowledge database of non-coding RNAs. Nucleic Acids Res 2005, 33, D112–D115. [Google Scholar]
- He, S.; Liu, C.; Skogerbo, G.; Zhao, H.; Wang, J.; Liu, T.; Bai, B.; Zhao, Y.; Chen, R. NONCODE v2.0: Decoding the non-coding. Nucleic Acids Res 2008, 36, D170–D172. [Google Scholar]
- Bu, D.; Yu, K.; Sun, S.; Xie, C.; Skogerbo, G.; Miao, R.; Xiao, H.; Liao, Q.; Luo, H.; Zhao, G.; et al. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res 2011. [Google Scholar] [CrossRef]
- Kin, T.; Yamada, K.; Terai, G.; Okida, H.; Yoshinari, Y.; Ono, Y.; Kojima, A.; Kimura, Y.; Komori, T.; Asai, K. fRNAdb: A platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res 2007, 35, D145–D148. [Google Scholar]
- Mituyama, T.; Yamada, K.; Hattori, E.; Okida, H.; Ono, Y.; Terai, G.; Yoshizawa, A.; Komori, T.; Asai, K. The Functional RNA Database 3.0: databases to support mining and annotation of functional RNAs. Nucleic Acids Res 2009, 37, D89–D92. [Google Scholar]
- Zhang, Y.; Guan, D.G.; Yang, J.H.; Shao, P.; Zhou, H.; Qu, L.H. ncRNAimprint: A comprehensive database of mammalian imprinted noncoding RNAs. RNA 2010, 16, 1889–1901. [Google Scholar]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Crowe, M.L.; Grimmond, S.M.; Mattick, J.S. NRED: A database of long noncoding RNA expression. Nucleic Acids Res 2009, 37, D122–D126. [Google Scholar]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011, 25, 1915–1927. [Google Scholar]
- Risueno, A.; Fontanillo, C.; Dinger, M.E.; De Las Rivas, J. GATExplorer: Genomic and transcriptomic explorer; mapping expression probes to gene loci, transcripts, exons and ncRNAs. BMC Bioinforma 2010, 11, 221. [Google Scholar]
- Liao, Q.; Xiao, H.; Bu, D.; Xie, C.; Miao, R.; Luo, H.; Zhao, G.; Yu, K.; Zhao, H.; Skogerbo, G.; et al. ncFANs: A web server for functional annotation of long non-coding RNAs. Nucleic Acids Res 2011, 39, W118–W124. [Google Scholar]
- Deng, W.; Zhu, X.; Skogerbø, G.; Zhao, Y.; Fu, Z.; Wang, Y.; He, H.; Cai, L.; Sun, H.; Liu, C.; et al. Organization of the Caenorhabditis elegans small non-coding transcriptome: genomic features, biogenesis, and expression. Genome Res 2006, 16, 20–29. [Google Scholar]
- Maeda, N.; Kasukawa, T.; Oyama, R.; Gough, J.; Frith, M.; Engström, P.G.; Lenhard, B.; Aturaliya, R.N.; Batalov, S.; Beisel, K.W.; et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genet 2006, 2, e62. [Google Scholar]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 2006, 34, D140–D144. [Google Scholar]
- Lestrade, L.; Weber, M.J. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res 2006, 34, D158–D162. [Google Scholar]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Rudnev, D.; Evangelista, C.; Kim, I.F.; Soboleva, A.; Tomashevsky, M.; Edgar, R. NCBI GEO: mining tens of millions of expression profiles—database and tools update. Nucleic Acids Res 2007, 35, D760–D765. [Google Scholar]
- Royo, H.; Cavaille, J. Non-coding RNAs in imprinted gene clusters. Biol. Cell 2008, 100, 149–166. [Google Scholar]
- Sahoo, T.; del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet 2008, 40, 719–721. [Google Scholar]
- Bliek, J.; Terhal, P.; van den Bogaard, M.J.; Maas, S.; Hamel, B.; Salieb-Beugelaar, G.; Simon, M.; Letteboer, T.; van der Smagt, J.; Kroes, H.; Mannens, M. Hypomethylation of the H19 gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am. J. Hum. Genet 2006, 78, 604–614. [Google Scholar]
- Zhang, X.; Zhou, Y.; Mehta, K.R.; Danila, D.C.; Scolavino, S.; Johnson, S.R.; Klibanski, A. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab 2003, 88, 5119–5126. [Google Scholar]
- Koerner, M.V.; Pauler, F.M.; Huang, R.; Barlow, D.P. The function of non-coding RNAs in genomic imprinting. Development 2009, 136, 1771–1783. [Google Scholar]
- Liao, Q.; Liu, C.; Yuan, X.; Kang, S.; Miao, R.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res 2011, 39, 3864–3878. [Google Scholar]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar]
- Garmire, L.X.; Garmire, D.G.; Huang, W.; Yao, J.; Glass, C.K.; Subramaniam, S. A global clustering algorithm to identify long intergenic non-coding RNA—with applications in mouse macrophages. PLoS One 2011, 6, e24051. [Google Scholar]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar]
- Bernstein, E.; Allis, C.D. RNA meets chromatin. Genes Dev 2005, 19, 1635–1655. [Google Scholar]
- Kanhere, A.; Viiri, K.; Araujo, C.C.; Rasaiyaah, J.; Bouwman, R.D.; Whyte, W.A.; Pereira, C.F.; Brookes, E.; Walker, K.; Bell, G.W.; et al. Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol. Cell 2010, 38, 675–688. [Google Scholar]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar]
- Cloonan, N.; Forrest, A.R.R.; Kolle, G.; Gardiner, B.B.A.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioinformatics Resources | Year | Web Link | Reference |
|---|---|---|---|
| Databases and public repositories | |||
| NcRNAdb | 2003 | http://biobases.ibch.poznan.pl/ncRNA | [47] |
| Rfam | 2003 | http://www.sanger.ac.uk/Software/Rfam | [48,49] |
| RNAdb | 2005 | http://research.imb.uq.edu.au/RNAdb | [50] |
| RNAdb 2.0 | 2007 | http://research.imb.uq.edu.au/RNAdb | [51] |
| H-InvDB | 2004 | http://www.h-invitational.jp | [52] |
| H-InvDB rel 5.0 | 2005 | http://www.h-invitational.jp | [53] |
| NONCODE | 2005 | http://www.noncode.org | [54] |
| NONCODE v2.0 | 2008 | http://www.noncode.org | [55] |
| NONCODE v3.0 | 2011 | http://www.noncode.org | [56] |
| fRNAdb | 2007 | http://www.ncrna.org/frnadb | [57] |
| fRNAdb 3.0 | 2009 | http://www.ncrna.org/frnadb | [58] |
| ncRNAimprint | 2010 | http://rnaqueen.sysu.edu.cn/ncRNAimprint/ | [59] |
| NRED | 2009 | http://jsm-research.imb.uq.edu.au/NRED | [60] |
| lncRNAdb | 2011 | http://www.lncrnadb.org/ | [13] |
| Human Body Map lincRNAs | 2011 | http://www.broadinstitute.org/genome_bio/human_lincrnas/ | [61] |
| Annotation tools and other bioinformatics tools | |||
| GATExplorer | 2010 | http://bioinfow.dep.usal.es/xgate/ | [62] |
| ncFANs | 2011 | http://www.ebiomed.org/ncFANs/ | [63] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sacco, L.D.; Baldassarre, A.; Masotti, A. Bioinformatics Tools and Novel Challenges in Long Non-Coding RNAs (lncRNAs) Functional Analysis. Int. J. Mol. Sci. 2012, 13, 97-114. https://doi.org/10.3390/ijms13010097
Sacco LD, Baldassarre A, Masotti A. Bioinformatics Tools and Novel Challenges in Long Non-Coding RNAs (lncRNAs) Functional Analysis. International Journal of Molecular Sciences. 2012; 13(1):97-114. https://doi.org/10.3390/ijms13010097
Chicago/Turabian StyleSacco, Letizia Da, Antonella Baldassarre, and Andrea Masotti. 2012. "Bioinformatics Tools and Novel Challenges in Long Non-Coding RNAs (lncRNAs) Functional Analysis" International Journal of Molecular Sciences 13, no. 1: 97-114. https://doi.org/10.3390/ijms13010097