Improvement of Thermal Stability via Outer-Loop Ion Pair Interaction of Mutated T1 Lipase from Geobacillus zalihae Strain T1

Abstract
:1. Introduction
2. Results and Discussion
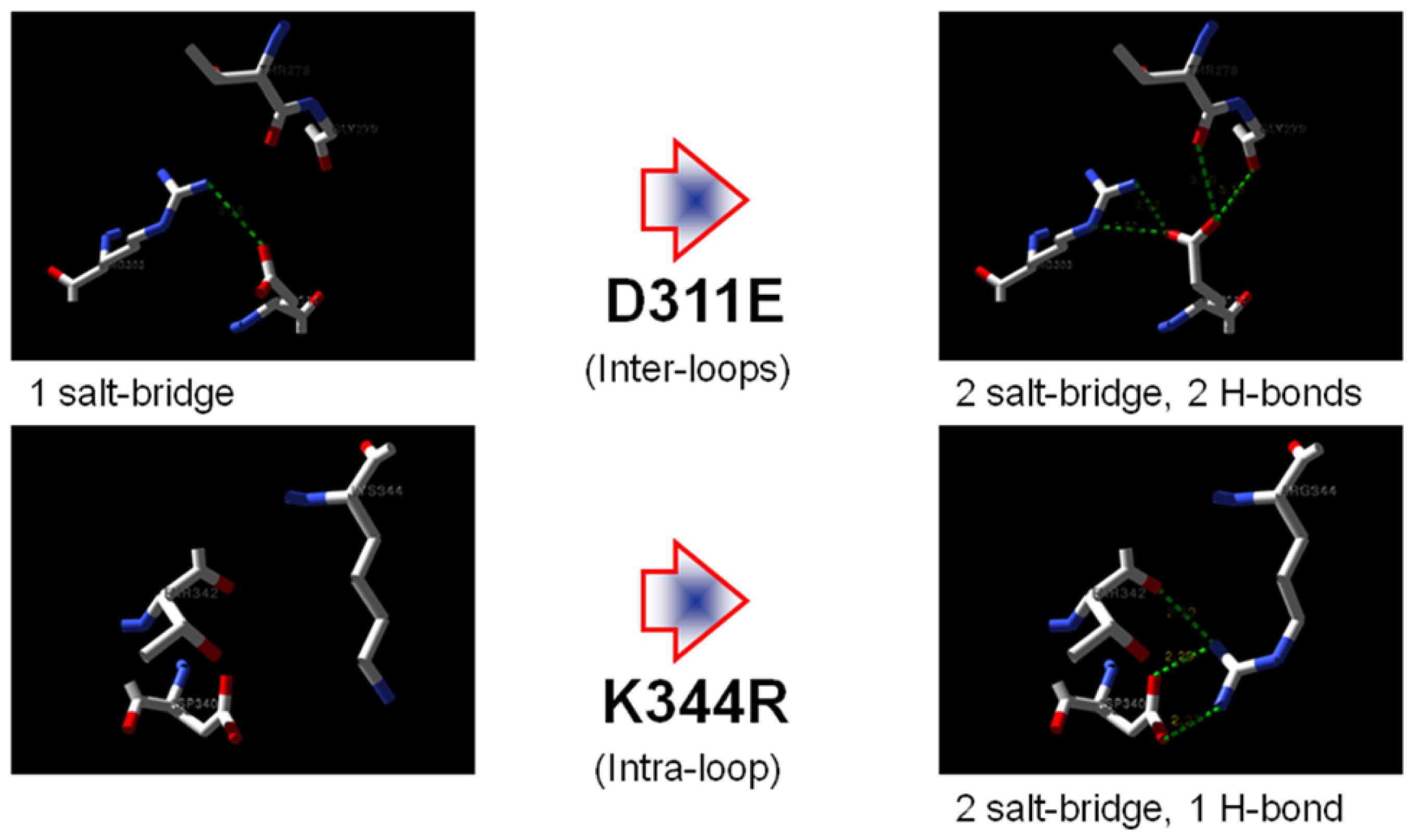
2.1. Rational Design of Mutant Lipases
2.2. Prediction of Protein Stability Changes upon Point Mutation
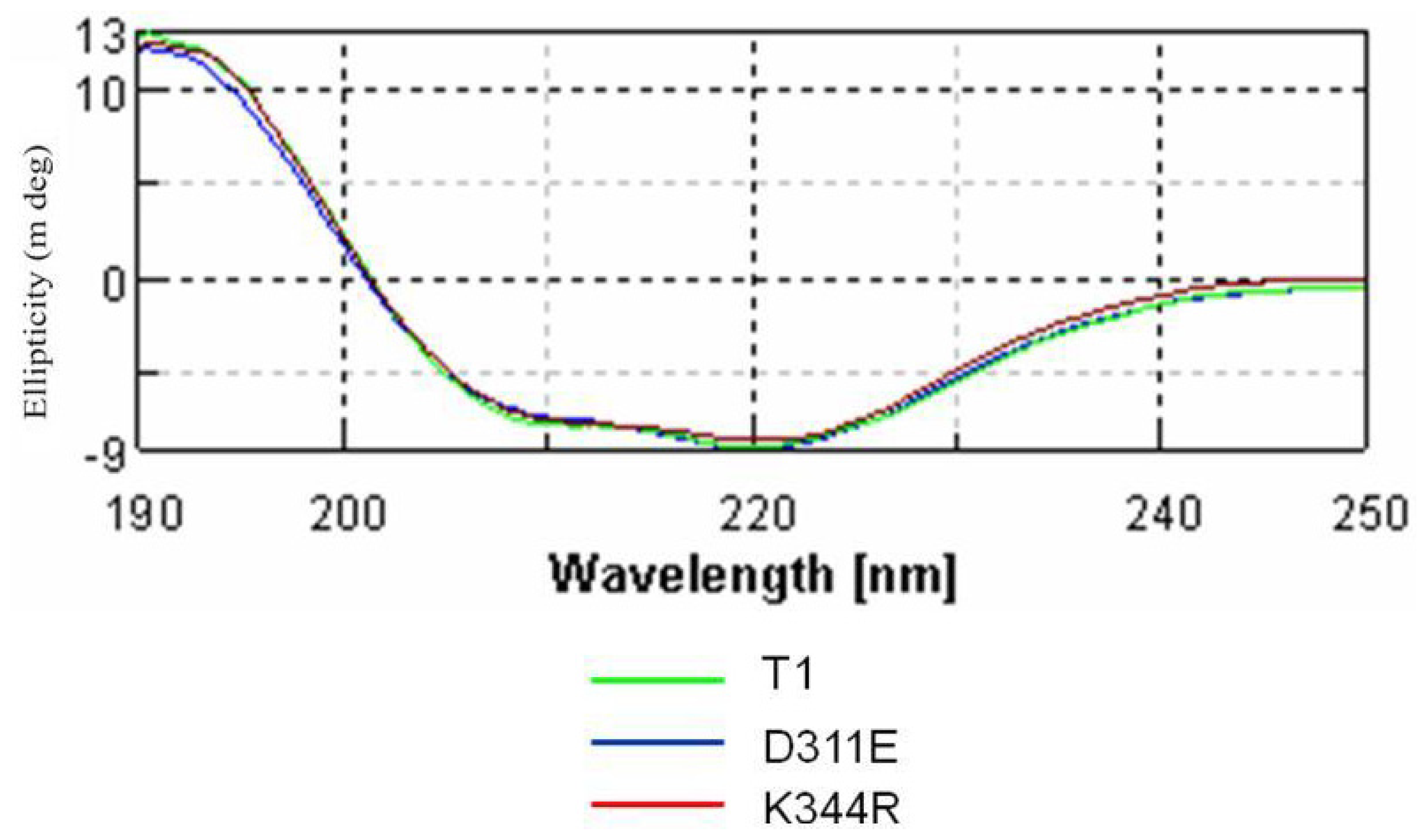
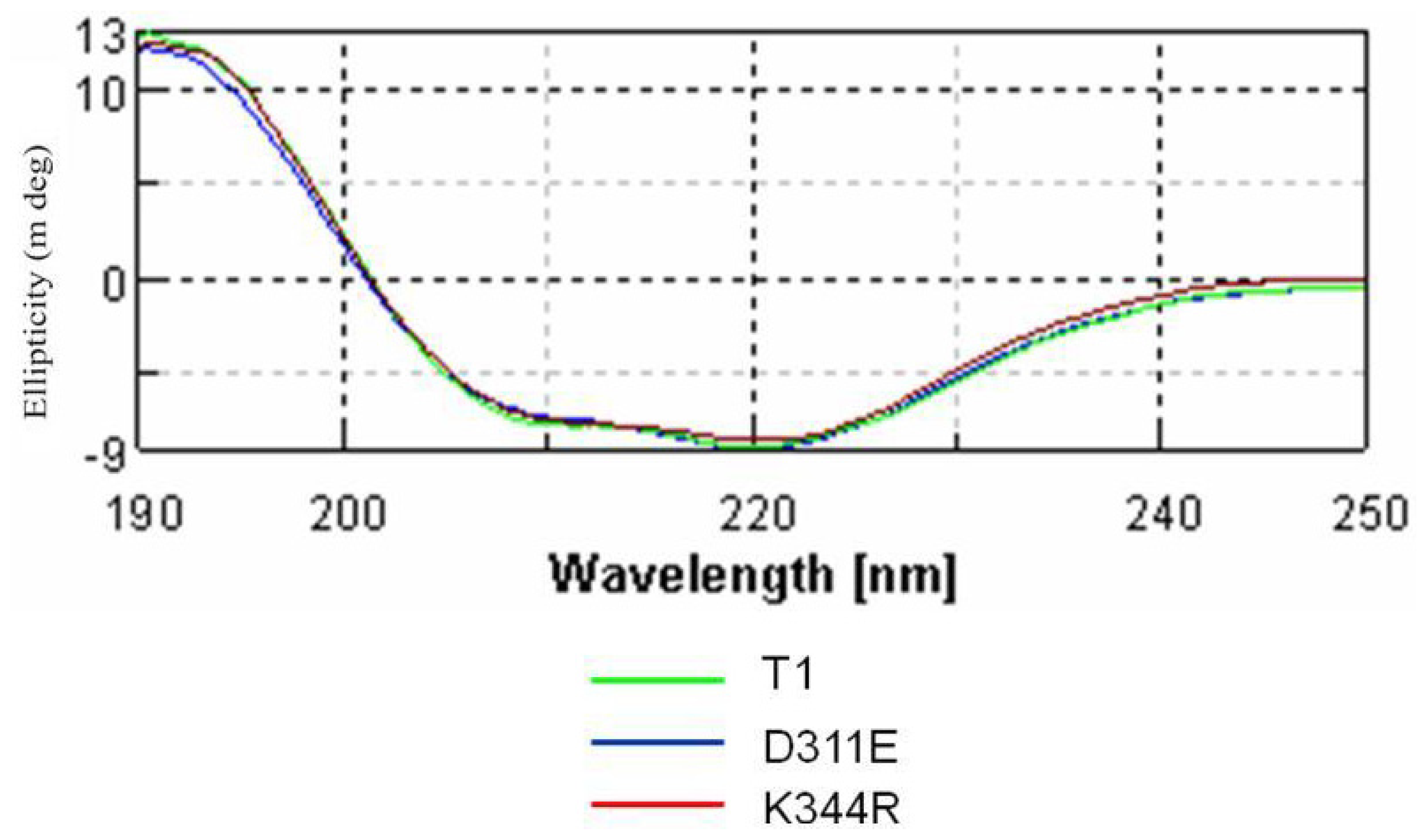
2.3. Circular Dichroism Analysis
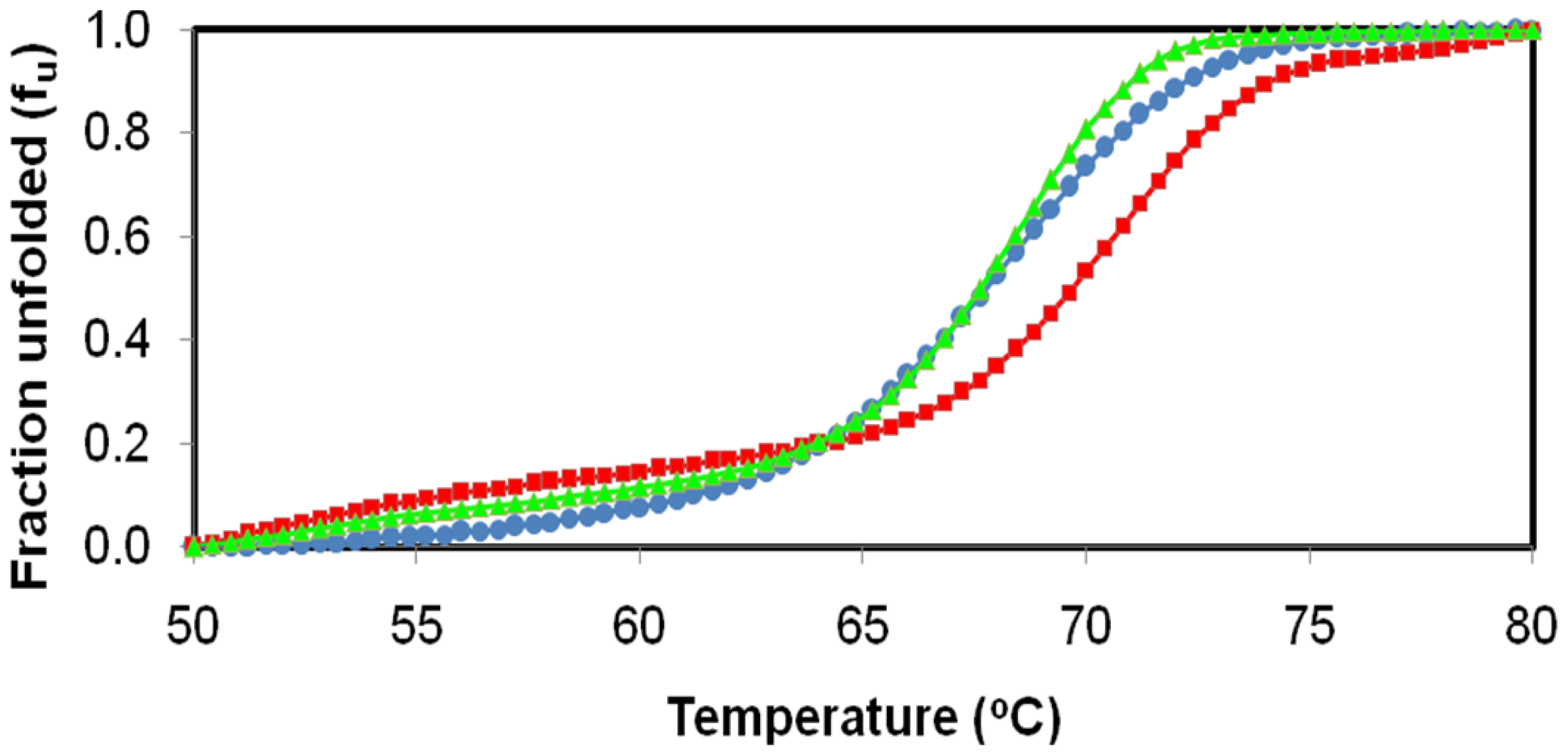
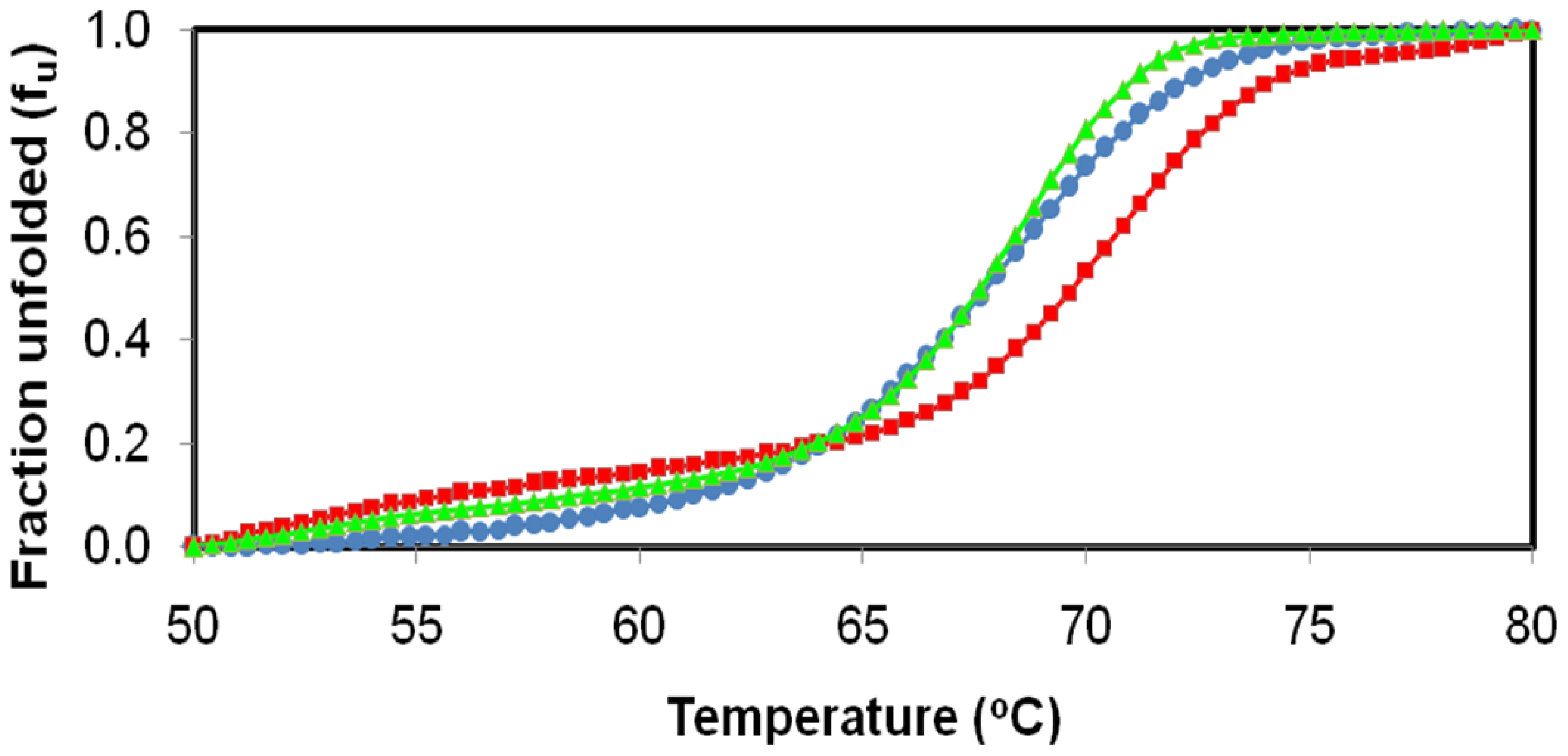
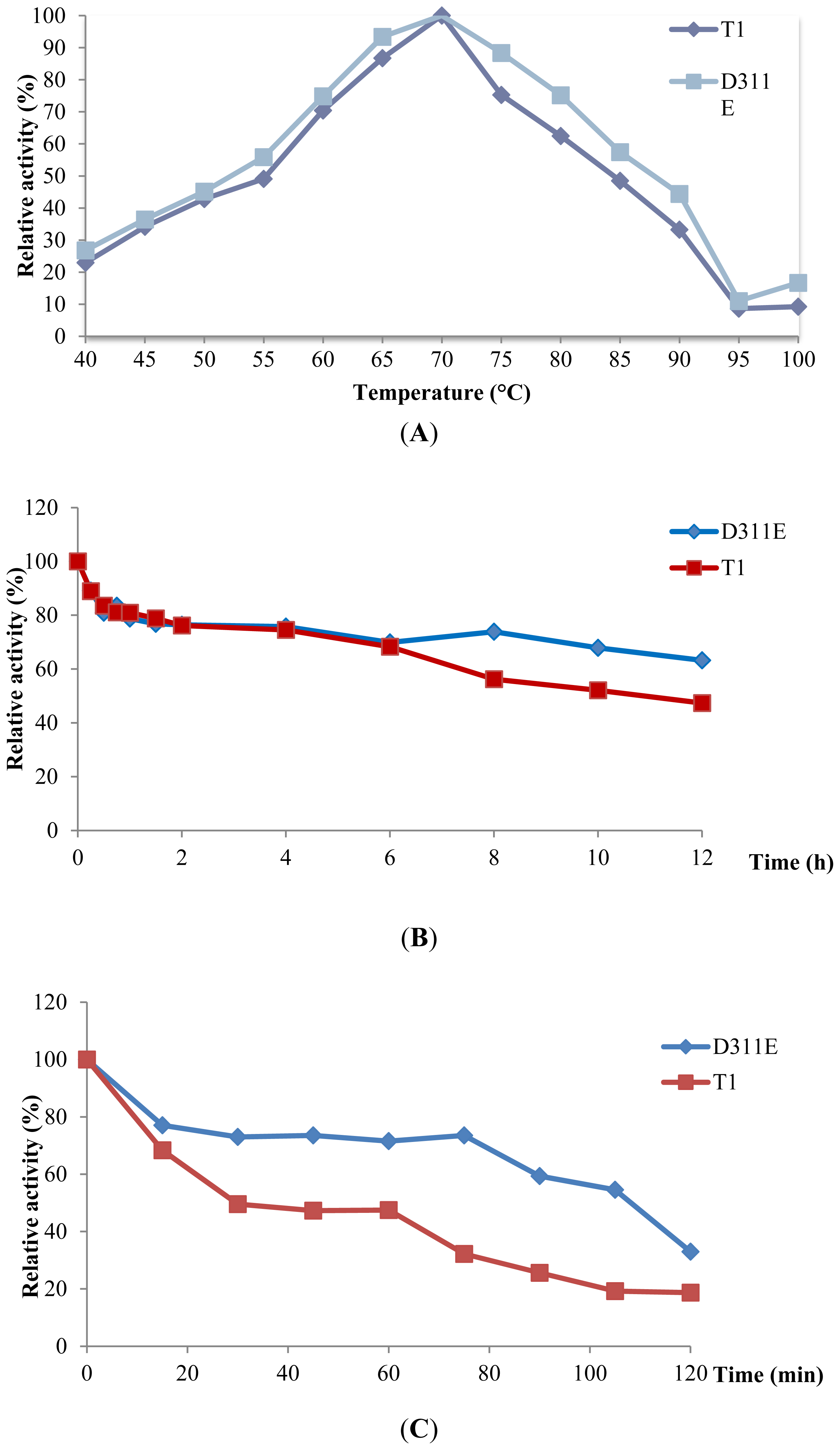
2.4. Effect of Temperature on D311E and T1 Lipase Activity and Stability
2.5. Crystallization of D311E lipase
2.6. X-Ray Data Collection
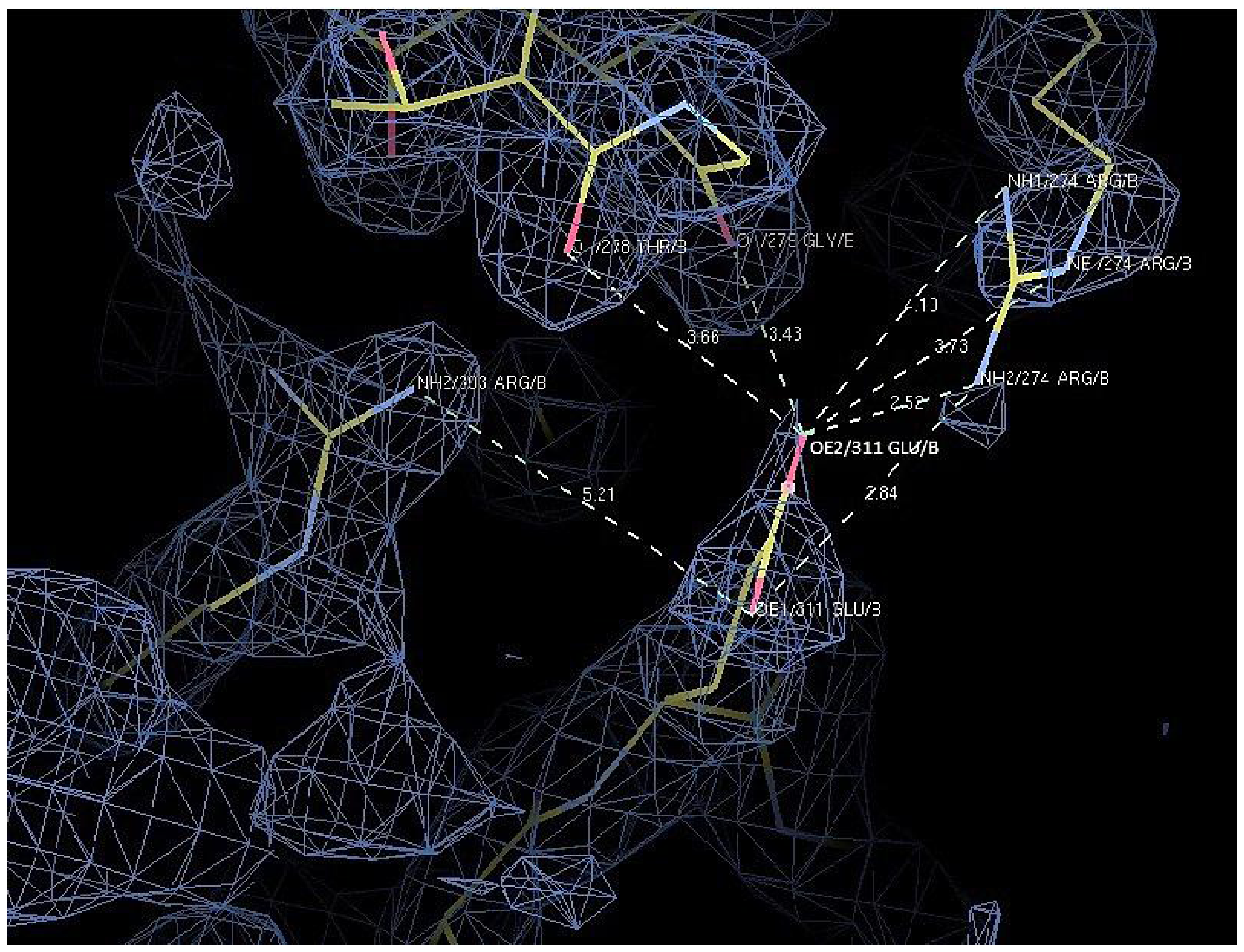
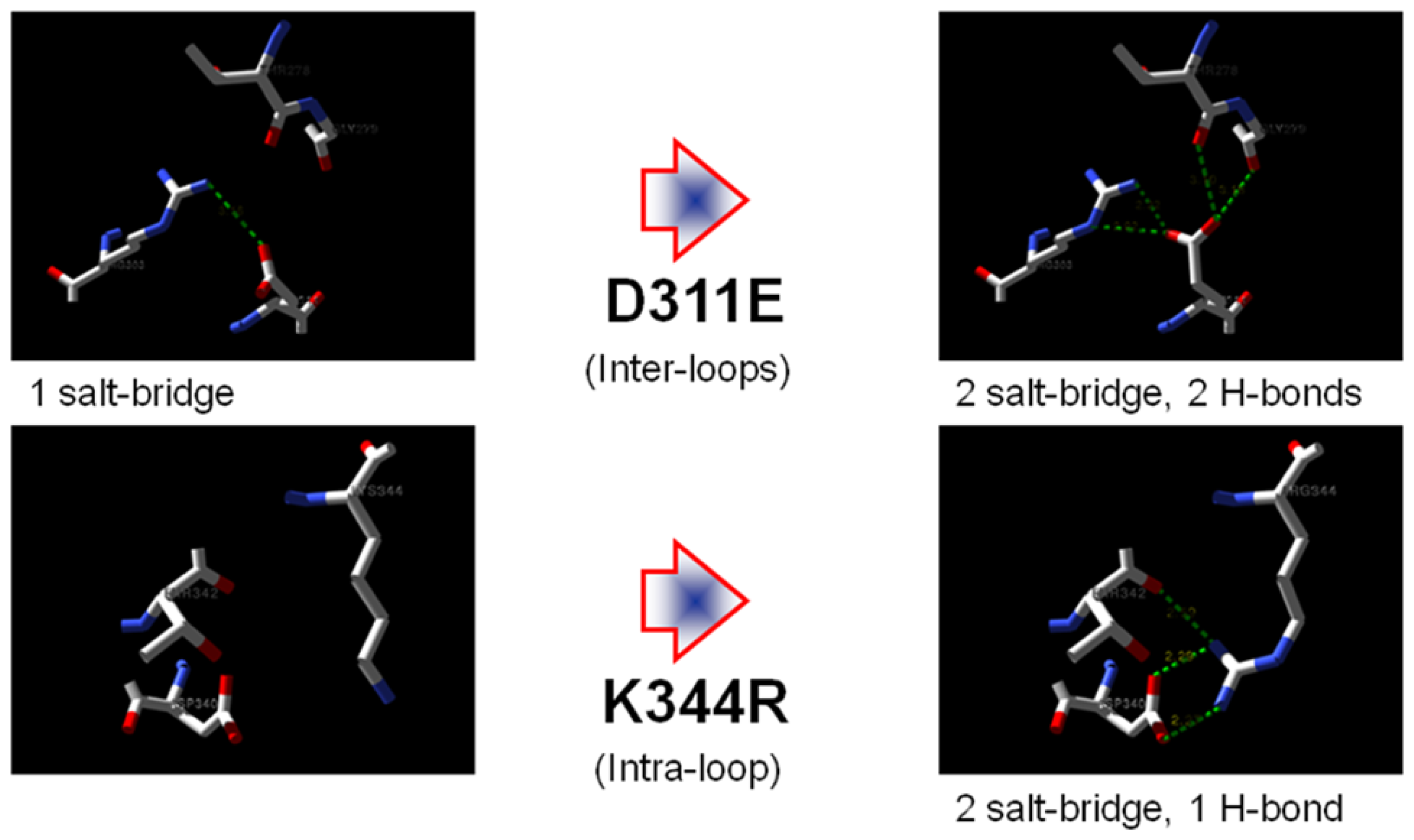
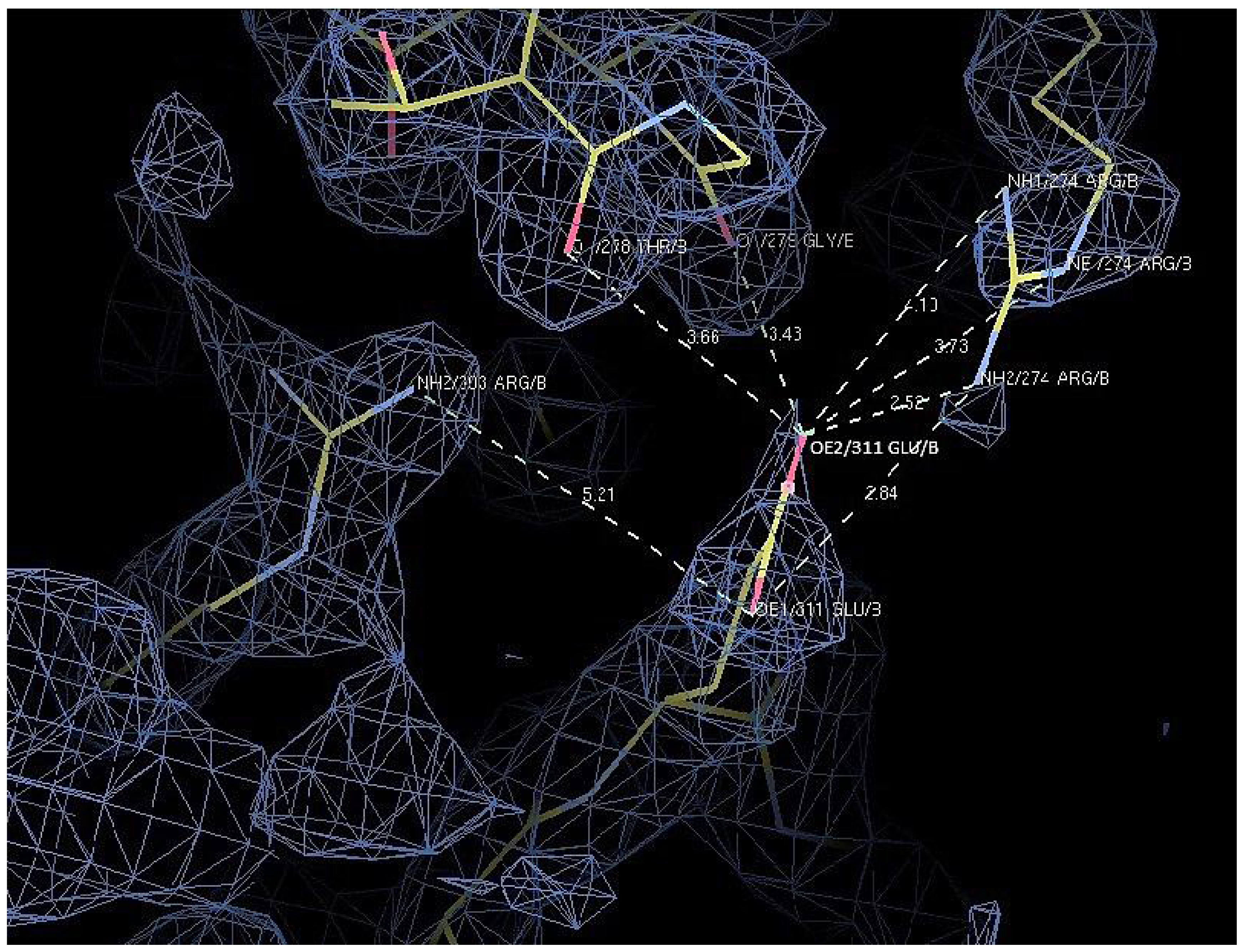
2.7. Structural Analysis
3. Experimental Section
3.1. Site Directed Mutagenesis
3.2. Prediction of Mutants Stability
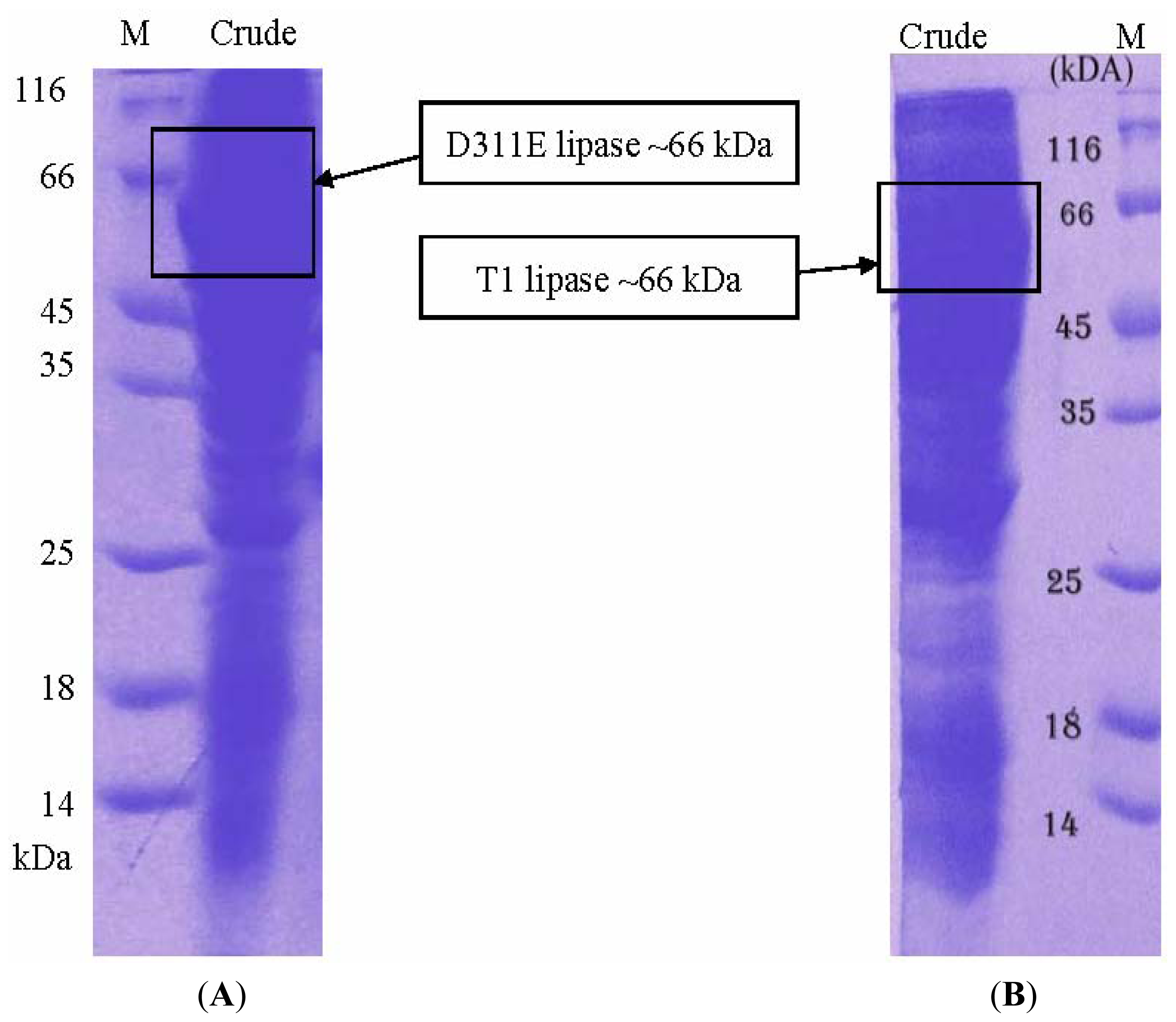
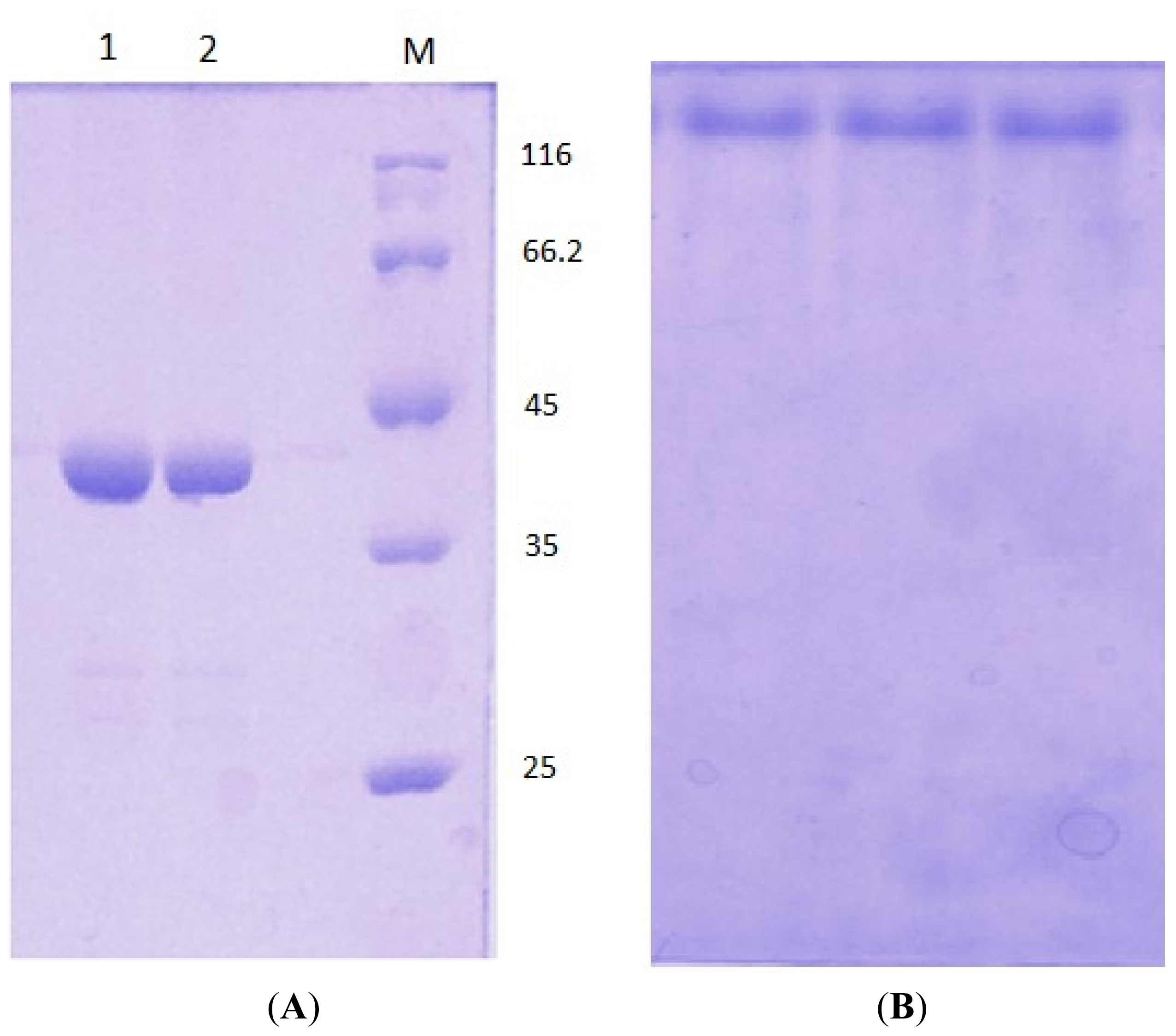
3.3. Protein Expression and Purification of T1 Lipase, D311E Lipase and K344R Lipase
3.4. Circular Dichroism Spectral Analysis
3.5. Electrophoresis
3.6. Effect of Temperature on D311E and T1 Lipase Activity and Stability
3.7. Crystallization of D311E Lipase
3.8. X-Ray Data Collection
4. Conclusions
Acknowledgments
References
- Togan, V.; Daloglu, A.T. Optimization of 3D trusses with adaptive approach in genetic algorithms. Eng. Struct 2006, 28, 1019–1027. [Google Scholar]
- Gandhi, N.N. Applications of lipase. J. Am. Chem. Soc 1997, 74, 621–634. [Google Scholar]
- Guncheva, M.; Zhiryakova, D. Catalytic properties and potential applications of Bacillus lipases. J. Mol. Catal. B Enzym 2011, 68, 1–21. [Google Scholar]
- Maciver, B.; McHale, R.H.; Saul, D.J.; Bergquist, P.L. Cloning and sequencing of a serine proteinase gene from a thermophilic Bacillus species and its expression in Escherichia coli. Appl. Environ. Microbiol 1994, 60, 3981–3988. [Google Scholar]
- Petrounia, I.P.; Arnold, F.H. Designed evolution of enzymatic properties. Curr. Opin. Biotechnol 2000, 11, 325–330. [Google Scholar]
- Rodrigues, R.C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R. Coupling chemical modification and immobilization to improve the catalytic performance of enzymes. Adv. Synth. Catal 2011, 353, 2216–2238. [Google Scholar]
- Ó’Fágáin, C. Enzyme stabilization—Recent experimental progress. Enzym. Microb. Technol 2003, 33, 137–149. [Google Scholar]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol 2007, 40, 1451–1463. [Google Scholar]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol 2002, 320, 369–387. [Google Scholar]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure. Protein Sci 2002, 11, 2714–2726. [Google Scholar]
- Kearns-Jonker, Mary; Barteneva, N.; Mencel, R.; Hussain, N.; Shulkin, I.; Xu, A.; Yew, M.; Cramer, D. Use of molecular modeling and site-directed mutagenesis to define the structural basis for the immune response to carbohydrate xenoantigens. BMC Immunol 2007, 8. [Google Scholar] [CrossRef]
- Rahman, R.N.Z.R.A.; Fujiwara, S.; Nakamura, H.; Takagi, M.; Imanaka, T. Ion pairs involved in maintaining a thermostable structure of glutamate dehydrogenase from a hyperthermophilic archaeon. Biochem. Biophys. Res. Commun 1998, 248, 920–926. [Google Scholar]
- Derewenda, Z.S.; Sharp, A.M. News from the interface: The molecular structures of triacylglyceride lipases. Trends Biochem. Sci 1993, 18, 20–25. [Google Scholar]
- Sarda, L.; Desnuelle, P. Action de la lipase pancreatique sur les esters en emulsion. Biochim. Biophys. Acta 1958, 30, 513–521. [Google Scholar]
- Carrasco-López, C.; Godoy, C.; de las Rivas, B.; Fernández-Lorente, G.; Palomo, J.M.; Guisán, J.M.; Fernández-Lafuente, R.; Martínez-Ripoll, M.; Hermoso, J.A. Activation of bacterial thermoalkalophilic lipases is spurred by dramatic structural rearrangements. J. Biol. Chem 2009, 284, 4365–4372. [Google Scholar]
- Matsumura, H.; Yamamoto, T.; Leow, T.C.; Mori, T.; Salleh, A.B.; Basri, M.; Inoue, T.; Kai, Y.; Rahman, R.N.Z.R.A. Novel cation-pi interaction revealed by crystal structure of thermoalkalophilic lipase. Proteins 2008, 70, 592–598. [Google Scholar]
- Tanaka, S.; Igarashi, S.; Ferri, S.; Sode, K. Increasing stability of water-soluble PQQ glucose dehydrogenase by increasing hydrophobic interaction at dimeric interface. BMC Biochem 2005, 6. [Google Scholar] [CrossRef]
- Yang, S.; Zhou, L.; Tang, H.; Pan, J.; Wu, X.; Huang, H.; Yuan, Z. Rational design of a more stable penicillin G acylase against organic cosolvent. J. Mol. Catal. B Enzym 2002, 18, 285–290. [Google Scholar]
- Akanuma, S.; Yamagishi, A.; Tanaka, N.; Oshima, T. Serial increase in the thermal stability of 3-isopropylmalate dehydrogenase from Bacillus subtilis by experimental evolution. Protein Sci 1998, 7, 698–705. [Google Scholar]
- Bauer, D.C.; Bodén, M.; Their, R.; Gillam, E.M. STAR: Predicting recombination sites from amino acid sequence. BMC Bioinform 2006, 7. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant 2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 2005, 33, 306–310. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Capriotti, E.; Altman, R.B. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinform 2011, 12. [Google Scholar] [CrossRef]
- Kawata, T.; Ogino, H. Amino acid residues involved in organic solvent-stability of the LST-03 lipase. Biochem. Biophys. Res. Commun 2010, 400, 384–388. [Google Scholar]
- Vetriani, C.; Maeder, D.L.; Tolliday, N.; Yip, K.S.-P.; Stillman, T.J.; Britton, K.L.; Rice, D.W.; Klump, H.H.; Robb, F.T. Protein thermostability above 100Â °C: A key role for ionic interactions. Proc. Natl. Acad. Sci. USA 1998, 95, 12300–12305. [Google Scholar]
- Kwon, D.; Rhee, J. A simple and rapid colorimetric method for determination of free fatty acids for lipase assay. J. Am. Chem. Soc 1986, 63, 89–92. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]
- Matthews, B.W. Solvent content of protein crystals. J. Mol. Biol 1968, 33, 491–497. [Google Scholar]
- Crick, F.; Kendrew, J. X-ray analysis and protein structure. Adv. Protein Chem 1957, 12, 133–214. [Google Scholar]
- Voorhost, W.G.B.; Warner, A.; de Vos, W.M.; Siezen, R.J. Homology modeling of two subtilisin-like serine protease from the hyperthermophilic archaea Pyrococcus furiosus and Thermococcus stetteri. Protein Eng 1997, 10, 905–914. [Google Scholar]
- Yip, K.S.P.; Stillman, T.J.; Britton, K.L.; Artymiuk, P.J.; Baker, P.J.; Sedelnikova, S.E.; Engel, P.C.; Pasquo, A.; Chiaraluce, R.; Consalvi, V.; Scandurra, R.; Rice, D.W. The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure 1995, 3, 1147–1158. [Google Scholar]
- Leow, T.C.; Salleh, A.B.; Basri, M.; Rahman, R.N.Z.R.A. A thermoalkaliphilic lipase of Geobacillus sp. T1. Extremophiles 2007, 11, 527–535. [Google Scholar]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 1997, 53, 240–255. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | Stability | Reliability Index (kcal/mole) | Relative Solvent Accessibility Area (%) |
|---|---|---|---|
| D311E | Increase | 7.0 | 67.3 |
| K344R | Decrease | 4.0 | 39.9 |
| Strain | ΔS (kJ/mol/K) | ΔH (kJ/mol) |
|---|---|---|
| T1 | −1.22 | −417.34 |
| D311E | −1.88 | −645.98 |
| K344R | −1.83 | −625.30 |
| Purification Steps | Total Activity (U) | Total Protein (mg/mL) | Specific Activity (U/mg) | Recovery (%) | Purification Fold |
|---|---|---|---|---|---|
| Crude | 41,609.00 | 44.88 | 18.54 | 100.00 | 1.00 |
| Affinity 1 | 18,366.50 | 21.67 | 19.78 | 44.14 | 1.83 |
| Affinity 2 | 12,904.74 | 19.56 | 36.65 | 31.01 | 1.98 |
| IEX | 6,538.10 | 3.34 | 195.75 | 15.71 | 10.56 |
| D311E | |
|---|---|
| Unit cell parameters | a = 117.32 Å, b = 81.16 Å, c = 100.14 Å á = 90.00 °C, â = 96.49 °C, ã = 90.00 °C |
| Space group | C2 |
| Wavelength (Å) | 1.54 |
| Resolution range (Å) | 37.57–2.1 (2.2–2.1) |
| No. of observed reflections | 249,261 |
| No. of unique reflections | 88,705 |
| Redundancy (%) | 2.93 (2.15) |
| Completeness (%) | 96.9 (91.4) |
| Mean I/ó (I) | 10.02 (4.01) |
| Molecules per asymmetric units | 2 |
| VM (Å3 Da−1) | 2.75 |
| Solvent content (%) | 55.33 |
| Rmerge (%) | 8.33 (19.46) |
| Refinement statistics | |
| Structure solution method | Molecular replacement |
| Resolution (High) | 2.1 |
| Resolution (Low) | 33.2 |
| Cut-off sigma (F) | 0.0 |
| Number of reflections (Observed) | 50139 |
| Number of reflections (R-free) | 2677 |
| Percent reflections (Observed) | 96.8 |
| R-factor (Observed) | 0.15 |
| R-work | 0.155 |
| R-free | 0.212 |
| Residue | Position | Residue | Position | Distance (Å) | |
|---|---|---|---|---|---|
| T1 | Asp 311 | OD2 | Arg 274 | NH2 | 7.0 |
| Arg 274 | NE | 7.0 | |||
| Arg 274 | NH1 | 8.8 | |||
| Thr 278 | O | 4.8 | |||
| Gly 279 | O | 4.9 | |||
| D311E | Glu 311 | OE1 | Arg 303 | NH2 | 5.2 |
| Arg 274 | NH2 | 3.8 | |||
| OE2 | Arg 274 | NH2 | 2.5 | ||
| Arg 274 | NE | 3.7 | |||
| Arg 274 | NH1 | 4.1 | |||
| Thr 278 | O | 3.7 | |||
| Gly 279 | O | 3.4 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ruslan, R.; Rahman, R.N.Z.R.A.; Leow, T.C.; Ali, M.S.M.; Basri, M.; Salleh, A.B. Improvement of Thermal Stability via Outer-Loop Ion Pair Interaction of Mutated T1 Lipase from Geobacillus zalihae Strain T1. Int. J. Mol. Sci. 2012, 13, 943-960. https://doi.org/10.3390/ijms13010943
Ruslan R, Rahman RNZRA, Leow TC, Ali MSM, Basri M, Salleh AB. Improvement of Thermal Stability via Outer-Loop Ion Pair Interaction of Mutated T1 Lipase from Geobacillus zalihae Strain T1. International Journal of Molecular Sciences. 2012; 13(1):943-960. https://doi.org/10.3390/ijms13010943
Chicago/Turabian StyleRuslan, Rudzanna, Raja Noor Zaliha Raja Abd. Rahman, Thean Chor Leow, Mohd Shukuri Mohamad Ali, Mahiran Basri, and Abu Bakar Salleh. 2012. "Improvement of Thermal Stability via Outer-Loop Ion Pair Interaction of Mutated T1 Lipase from Geobacillus zalihae Strain T1" International Journal of Molecular Sciences 13, no. 1: 943-960. https://doi.org/10.3390/ijms13010943