Disruption of Axonal Transport in Motor Neuron Diseases

Abstract
:
1. Introduction
2. Motor Proteins and Motor Neuron Disease
2.1. Dynein/Dynactin Complex
2.2. Kinesin
3. Axonal Transport Defects and Cargo Accumulation
3.1. Neurofilaments
3.2. Mitochondria
3.3. Autophagosomes
4. MND and Axonal Transport Defects
ALS
SBMA
SMA
HSP
5. Conclusions
Acknowledgments
References
- Holzbaur, E.L. Motor neurons rely on motor proteins. Trends Cell Biol 2004, 14, 233–240. [Google Scholar]
- Chao, M.V. Retrograde transport redux. Neuron 2003, 39, 1–2. [Google Scholar]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci 2005, 118, 5411–5419. [Google Scholar]
- Hollenbeck, P.J. The pattern and mechanism of mitochondrial transport in axons. Front Biosci 1996, 1, d91–d102. [Google Scholar]
- El-Kadi, A.M.; Soura, V.; Hafezparast, M. Defective axonal transport in motor neuron disease. J. Neurosci. Res 2007, 85, 2557–2566. [Google Scholar]
- Fichera, M.; Lo Giudice, M.; Falco, M.; Sturnio, M.; Amata, S.; Calabrese, O.; Bigoni, S.; Calzolari, E.; Neri, M. Evidence of kinesin heavy chain (kif5a) involvement in pure hereditary spastic paraplegia. Neurology 2004, 63, 1108–1110. [Google Scholar]
- Hafezparast, M.; Klocke, R.; Ruhrberg, C.; Marquardt, A.; Ahmad-Annuar, A.; Bowen, S.; Lalli, G.; Witherden, A.S.; Hummerich, H.; Nicholson, S.; et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 2003, 300, 808–812. [Google Scholar]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet 2003, 33, 455–456. [Google Scholar]
- Pfister, K.K.; Fisher, E.M.; Gibbons, I.R.; Hays, T.S.; Holzbaur, E.L.; McIntosh, J.R.; Porter, M.E.; Schroer, T.A.; Vaughan, K.T.; Witman, G.B.; et al. Cytoplasmic dynein nomenclature. J. Cell Biol 2005, 171, 411–413. [Google Scholar]
- King, S.M. The dynein microtubule motor. Biochim. Biophys. Acta 2000, 1496, 60–75. [Google Scholar]
- Holzbaur, E.L.; Vallee, R.B. Dyneins: Molecular structure and cellular function. Annu. Rev. Cell Biol 1994, 10, 339–372. [Google Scholar]
- Kieran, D.; Hafezparast, M.; Bohnert, S.; Dick, J.R.; Martin, J.; Schiavo, G.; Fisher, E.M.; Greensmith, L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J. Cell Biol 2005, 169, 561–567. [Google Scholar]
- Ori-McKenney, K.M.; Xu, J.; Gross, S.P.; Vallee, R.B. A cytoplasmic dynein tail mutation impairs motor processivity. Nat. Cell Biol 2010, 12, 1228–1234. [Google Scholar]
- Chen, X.J.; Levedakou, E.N.; Millen, K.J.; Wollmann, R.L.; Soliven, B.; Popko, B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J. Neurosci 2007, 27, 14515–14124. [Google Scholar]
- Gill, S.R.; Schroer, T.A.; Szilak, I.; Steuer, E.R.; Sheetz, M.P.; Cleveland, D.W. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J. Cell Biol 1991, 115, 1639–1650. [Google Scholar]
- Karki, S.; Holzbaur, E.L. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J. Biol. Chem 1995, 270, 28806–28811. [Google Scholar]
- Haghnia, M.; Cavalli, V.; Shah, S.B.; Schimmelpfeng, K.; Brusch, R.; Yang, G.; Herrera, C.; Pilling, A.; Goldstein, L.S. Dynactin is required for coordinated bidirectional motility, but not for dynein membrane attachment. Mol. Biol. Cell 2007, 18, 2081–2089. [Google Scholar]
- Levy, J.R.; Sumner, C.J.; Caviston, J.P.; Tokito, M.K.; Ranganathan, S.; Ligon, L.A.; Wallace, K.E.; LaMonte, B.H.; Harmison, G.G.; Puls, I.; et al. A motor neuron disease-associated mutation in p150glued perturbs dynactin function and induces protein aggregation. J. Cell Biol 2006, 172, 733–745. [Google Scholar]
- Chevalier-Larsen, E.S.; Wallace, K.E.; Pennise, C.R.; Holzbaur, E.L. Lysosomal proliferation and distal degeneration in motor neurons expressing the g59s mutation in the p150glued subunit of dynactin. Hum. Mol. Genet 2008, 17, 1946–1955. [Google Scholar]
- Laird, F.M.; Farah, M.H.; Ackerley, S.; Hoke, A.; Maragakis, N.; Rothstein, J.D.; Griffin, J.; Price, D.L.; Martin, L.J.; Wong, P.C. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J. Neurosci 2008, 28, 1997–2005. [Google Scholar]
- Lai, C.; Lin, X.; Chandran, J.; Shim, H.; Yang, W.J.; Cai, H. The g59s mutation in p150 (glued) causes dysfunction of dynactin in mice. J. Neurosci 2007, 27, 13982–13990. [Google Scholar]
- Miki, H.; Setou, M.; Kaneshiro, K.; Hirokawa, N. All kinesin superfamily protein, kif, genes in mouse and human. Proc. Natl. Acad. Sci. USA 2001, 98, 7004–7011. [Google Scholar]
- Hirokawa, N.; Takemura, R. Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci 2005, 6, 201–214. [Google Scholar]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar]
- Reid, E.; Kloos, M.; Ashley-Koch, A.; Hughes, L.; Bevan, S.; Svenson, I.K.; Graham, F.L.; Gaskell, P.C.; Dearlove, A.; Pericak-Vance, M.A.; et al. A kinesin heavy chain (kif5a) mutation in hereditary spastic paraplegia (spg10). Am. J. Hum. Genet 2002, 71, 1189–1194. [Google Scholar]
- Song, H.; Endow, S.A. Decoupling of nucleotide- and microtubule-binding sites in a kinesin mutant. Nature 1998, 396, 587–590. [Google Scholar]
- Hirokawa, N.; Noda, Y. Intracellular transport and kinesin superfamily proteins, kifs: Structure, function, and dynamics. Physiol. Rev 2008, 88, 1089–1118. [Google Scholar]
- Shi, P.; Strom, A.L.; Gal, J.; Zhu, H. Effects of als-related SOD1 mutants on dynein- and kif5-mediated retrograde and anterograde axonal transport. Biochim. Biophys. Acta 2010, 1802, 707–716. [Google Scholar]
- Morfini, G.; Pigino, G.; Szebenyi, G.; You, Y.; Pollema, S.; Brady, S.T. Jnk mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat. Neurosci 2006, 9, 907–916. [Google Scholar]
- Lee, M.K.; Cleveland, D.W. Neuronal intermediate filaments. Annu. Rev. Neurosci 1996, 19, 187–217. [Google Scholar]
- Garcia, M.L.; Lobsiger, C.S.; Shah, S.B.; Deerinck, T.J.; Crum, J.; Young, D.; Ward, C.M.; Crawford, T.O.; Gotow, T.; Uchiyama, Y.; et al. Nf-m is an essential target for the myelin-directed “Outside-in” Signaling cascade that mediates radial axonal growth. J. Cell Biol 2003, 163, 1011–1020. [Google Scholar]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in als. Annu. Rev. Neurosci 2004, 27, 723–749. [Google Scholar]
- Xia, C.H.; Roberts, E.A.; Her, L.S.; Liu, X.; Williams, D.S.; Cleveland, D.W.; Goldstein, L.S. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain kif5a. J. Cell Biol 2003, 161, 55–66. [Google Scholar]
- Koehnle, T.J.; Brown, A. Slow axonal transport of neurofilament protein in cultured neurons. J. Cell Biol 1999, 144, 447–458. [Google Scholar]
- Mersiyanova, I.V.; Perepelov, A.V.; Polyakov, A.V.; Sitnikov, V.F.; Dadali, E.L.; Oparin, R.B.; Petrin, A.N.; Evgrafov, O.V. A new variant of charcot-marie-tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am. J. Hum. Genet 2000, 67, 37–46. [Google Scholar]
- Perez-Olle, R.; Jones, S.T.; Liem, R.K. Phenotypic analysis of neurofilament light gene mutations linked to charcot-marie-tooth disease in cell culture models. Hum. Mol. Genet 2004, 13, 2207–2220. [Google Scholar]
- Perez-Olle, R.; Lopez-Toledano, M.A.; Goryunov, D.; Cabrera-Poch, N.; Stefanis, L.; Brown, K.; Liem, R.K. Mutations in the neurofilament light gene linked to charcot-marie-tooth disease cause defects in transport. J. Neurochem 2005, 93, 861–874. [Google Scholar]
- Magrane, J.; Manfredi, G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid. Redox. Signal 2009, 11, 1615–1626. [Google Scholar]
- Bilsland, L.G.; Sahai, E.; Kelly, G.; Golding, M.; Greensmith, L.; Schiavo, G. Deficits in axonal transport precede als symptoms in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 20523–20528. [Google Scholar]
- Collard, J.F.; Cote, F.; Julien, J.P. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 1995, 375, 61–64. [Google Scholar]
- Sasaki, S.; Iwata, M. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology 1996, 47, 535–540. [Google Scholar]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial als-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci 1998, 18, 3241–3250. [Google Scholar]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol 2007, 66, 10–16. [Google Scholar]
- Zhu, Y.B.; Sheng, Z.H. Increased axonal mitochondrial mobility does not slow als-like disease in mutant SOD1 mice. J. Biol. Chem 2011, 26, 23432–23440. [Google Scholar]
- d’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Berghe, P.V.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. Hdac6 inhibitors reverse axonal loss in a mouse model of mutant hspb1-induced charcot-marie-tooth disease. Nat. Med 2011, 17, 968–974. [Google Scholar]
- Ravikumar, B.; Acevedo-Arozena, A.; Imarisio, S.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Brown, S.D.; Rubinsztein, D.C. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet 2005, 37, 771–776. [Google Scholar]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—a novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol 2005, 171, 87–98. [Google Scholar]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: a critical update. J. Cell Mol. Med 2010, 14, 457–487. [Google Scholar]
- Sapp, E.; Schwarz, C.; Chase, K.; Bhide, P.G.; Young, A.B.; Penney, J.; Vonsattel, J.P.; Aronin, N.; DiFiglia, M. Huntingtin localization in brains of normal and huntington’s disease patients. Ann. Neurol 1997, 42, 604–612. [Google Scholar]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct 2008, 33, 109–122. [Google Scholar]
- Yang, Y.; Xu, K.; Koike, T.; Zheng, X. Transport of autophagosomes in neurites of pc12 cells during serum deprivation. Autophagy 2008, 4, 243–245. [Google Scholar]
- Yue, Z.; Wang, Q.J.; Komatsu, M. Neuronal autophagy: Going the distance to the axon. Autophagy 2008, 4, 94–96. [Google Scholar]
- Katsumata, K.; Nishiyama, J.; Inoue, T.; Mizushima, N.; Takeda, J.; Yuzaki, M. Dynein- and activity-dependent retrograde transport of autophagosomes in neuronal axons. Autophagy 2010, 6, 378–385. [Google Scholar]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol 2011, 70, 349–359. [Google Scholar]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar]
- Otomo, A.; Hadano, S.; Okada, T.; Mizumura, H.; Kunita, R.; Nishijima, H.; Showguchi-Miyata, J.; Yanagisawa, Y.; Kohiki, E.; Suga, E.; et al. Als2, a novel guanine nucleotide exchange factor for the small gtpase rab5, is implicated in endosomal dynamics. Hum. Mol. Genet 2003, 12, 1671–1687. [Google Scholar]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein vapb causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet 2004, 75, 822–831. [Google Scholar]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. Als phenotypes with mutations in chmp2b (charged multivesicular body protein 2b). Neurology 2006, 67, 1074–1077. [Google Scholar]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in chmp2b in lower motor neuron predominant amyotrophic lateral sclerosis (als). PLoS One 2010, 5. [Google Scholar] [CrossRef]
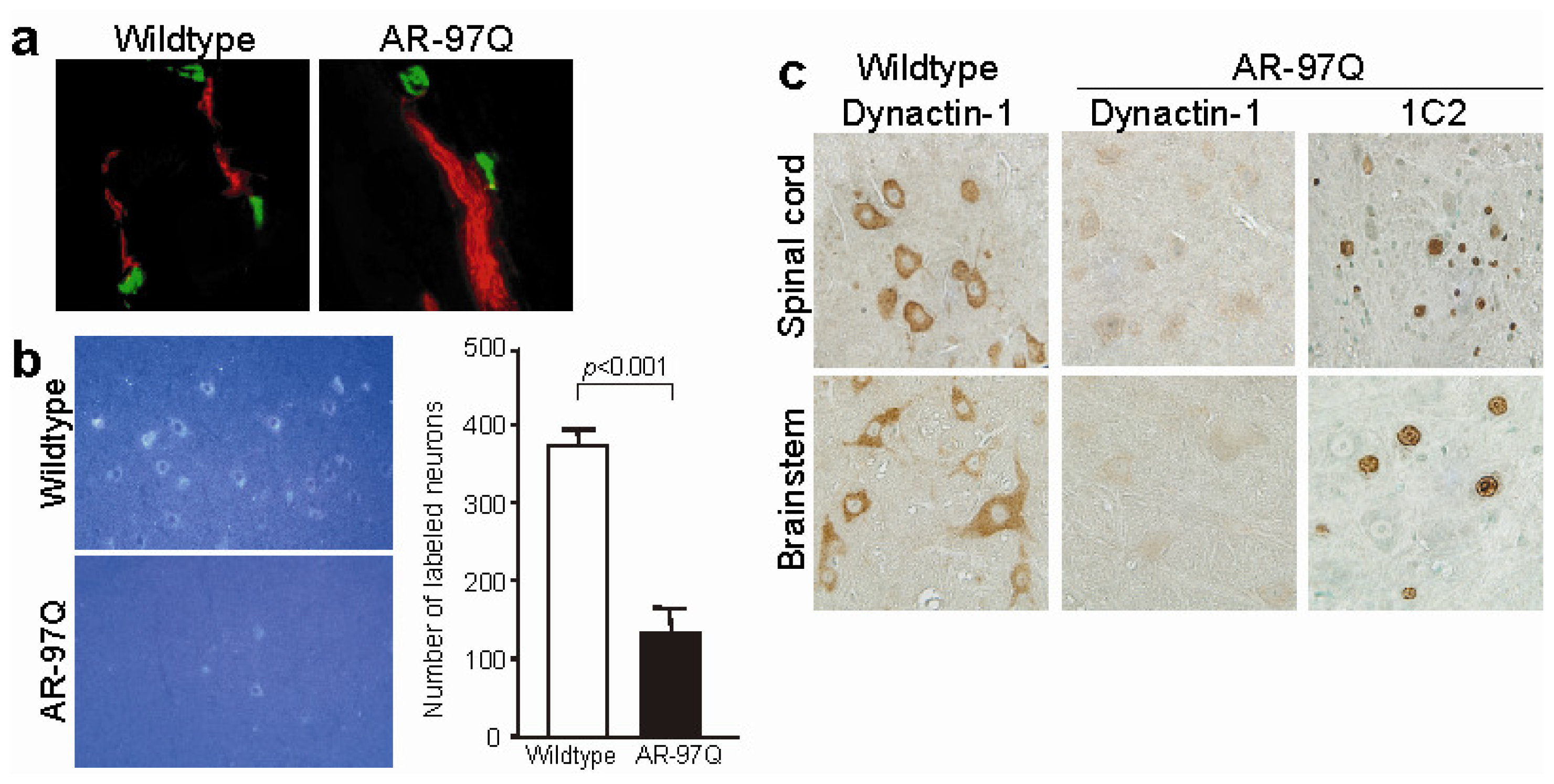
- Katsuno, M.; Adachi, H.; Minamiyama, M.; Waza, M.; Tokui, K.; Banno, H.; Suzuki, K.; Onoda, Y.; Tanaka, F.; Doyu, M.; et al. Reversible disruption of dynactin-1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration. J. Neurosci 2006, 26, 12106–12117. [Google Scholar]
- Sobue, G.; Hashizume, Y.; Mukai, E.; Hirayama, M.; Mitsuma, T.; Takahashi, A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain 1989, 112, 209–232. [Google Scholar]
- Zhao, X.; Alvarado, D.; Rainier, S.; Lemons, R.; Hedera, P.; Weber, C.H.; Tukel, T.; Apak, M.; Heiman-Patterson, T.; Ming, L.; et al. Mutations in a newly identified gtpase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet 2001, 29, 326–331. [Google Scholar]
- Rismanchi, N.; Soderblom, C.; Stadler, J.; Zhu, P.P.; Blackstone, C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet 2008, 17, 1591–1604. [Google Scholar]
- Hazan, J.; Fonknechten, N.; Mavel, D.; Paternotte, C.; Samson, D.; Artiguenave, F.; Davoine, C.S.; Cruaud, C.; Durr, A.; Wincker, P.; et al. Spastin, a new aaa protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet 1999, 23, 296–303. [Google Scholar]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med 2001, 344, 1688–1700. [Google Scholar]
- Ince, P.G.; Highley, J.R.; Kirby, J.; Wharton, S.B.; Takahashi, H.; Strong, M.J.; Shaw, P.J. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol 2011, 122, 657–671. [Google Scholar]
- Wroe, R.; Wai-Ling Butler, A.; Andersen, P.M.; Powell, J.F.; Al-Chalabi, A. ALSOD: the Amyotrophic Lateral Sclerosis Online Database. Amyotroph. Lateral. Scler 2008, 9, 249–250. [Google Scholar]
- Sasaki, S.; Warita, H.; Abe, K.; Iwata, M. Impairment of axonal transport in the axon hillock and the initial segment of anterior horn neurons in transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol 2005, 110, 48–56. [Google Scholar]
- Jiang, Y.M.; Yamamoto, M.; Kobayashi, Y.; Yoshihara, T.; Liang, Y.; Terao, S.; Takeuchi, H.; Ishigaki, S.; Katsuno, M.; Adachi, H.; et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol 2005, 57, 236–251. [Google Scholar]
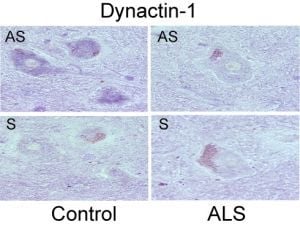
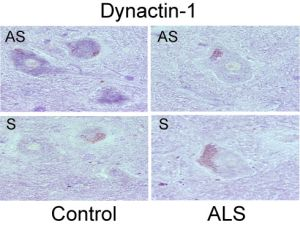
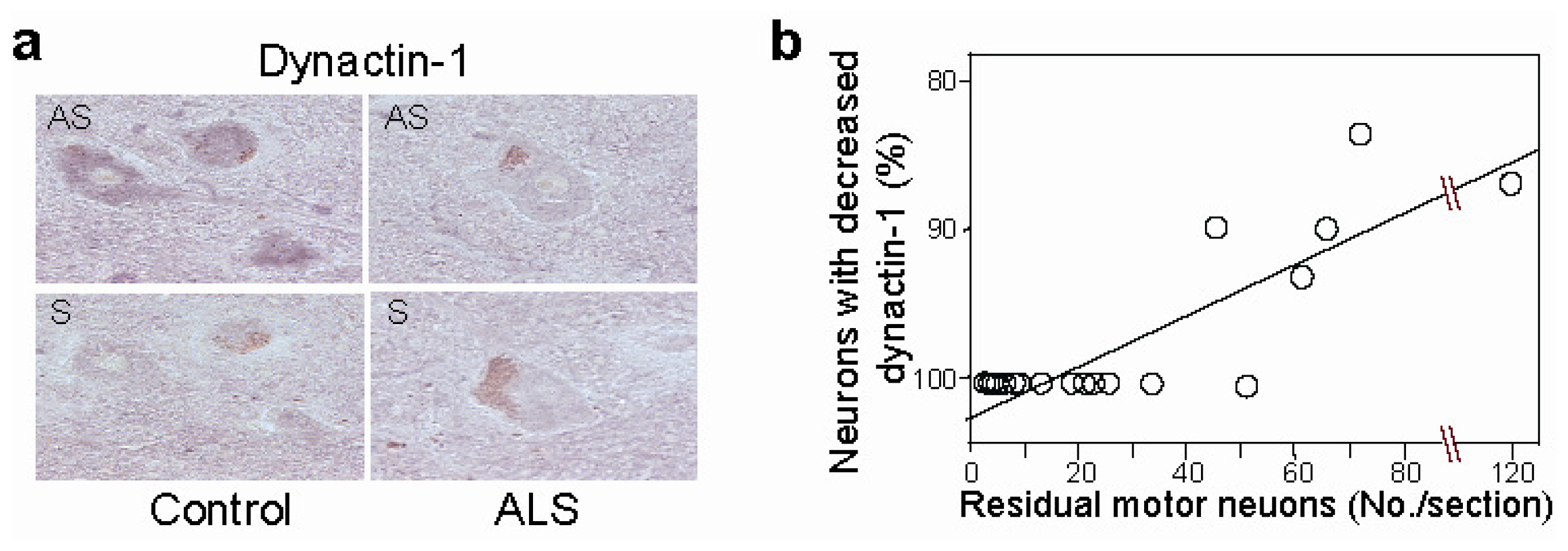
- Jiang, Y.M.; Yamamoto, M.; Tanaka, F.; Ishigaki, S.; Katsuno, M.; Adachi, H.; Niwa, J.; Doyu, M.; Yoshida, M.; Hashizume, Y.; et al. Gene expressions specifically detected in motor neurons (dynactin-1, early growth response 3, acetyl-coa transporter, death receptor 5, and cyclin c) differentially correlate to pathologic markers in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol 2007, 66, 617–627. [Google Scholar]
- Kennedy, W.R.; Alter, M.; Sung, J.H. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology 1968, 18, 671–680. [Google Scholar]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in x-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar]
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet 2005, 6, 743–755. [Google Scholar]
- Cha, J.H. Transcriptional dysregulation in huntington’s disease. Trends Neurosci 2000, 23, 387–392. [Google Scholar]
- Kemp, M.Q.; Poort, J.L.; Baqri, R.M.; Lieberman, A.P.; Breedlove, S.M.; Miller, K.E.; Jordan, C.L. Impaired motoneuronal retrograde transport in two models of SBMA implicates two sites of androgen action. Hum. Mol. Genet 2011, 22, 4475–4490. [Google Scholar]
- Malik, B.; Nirmalananthan, N.; Bilsland, L.G.; La Spada, A.R.; Hanna, M.G.; Schiavo, G.; Gallo, J.M.; Greensmith, L. Absence of disturbed axonal transport in spinal and bulbar muscular atrophy. Hum. Mol. Genet 2011, 20, 1776–1786. [Google Scholar]
- Lorson, C.L.; Rindt, H.; Shababi, M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum. Mol. Genet 2010, 19, R111–R118. [Google Scholar]
- Kong, L.; Wang, X.; Choe, D.W.; Polley, M.; Burnett, B.G.; Bosch-Marcé, M.; Griffin, J.W.; Rich, M.M.; Sumner, C.J. Impaired synaptic release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci 2009, 29, 842–851. [Google Scholar]
- Dale, J.M.; Shen, H.; Barry, D.M.; Garcia, V.B.; Rose, F.F., Jr; Lorsen, C.L.; Garcia, M.L. The spinal muscular atrophy mouse model, SMAΔ7, displays altered axonal transport without global neurofilament alterations. Acta Neuropathol 2011, 122, 331–341. [Google Scholar]
- Blackstone, C.; O’Kane, C.J.; Reid, E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat. Rev. Neurosci 2011, 12, 31–42. [Google Scholar]


{kind=link}
{kind=link}
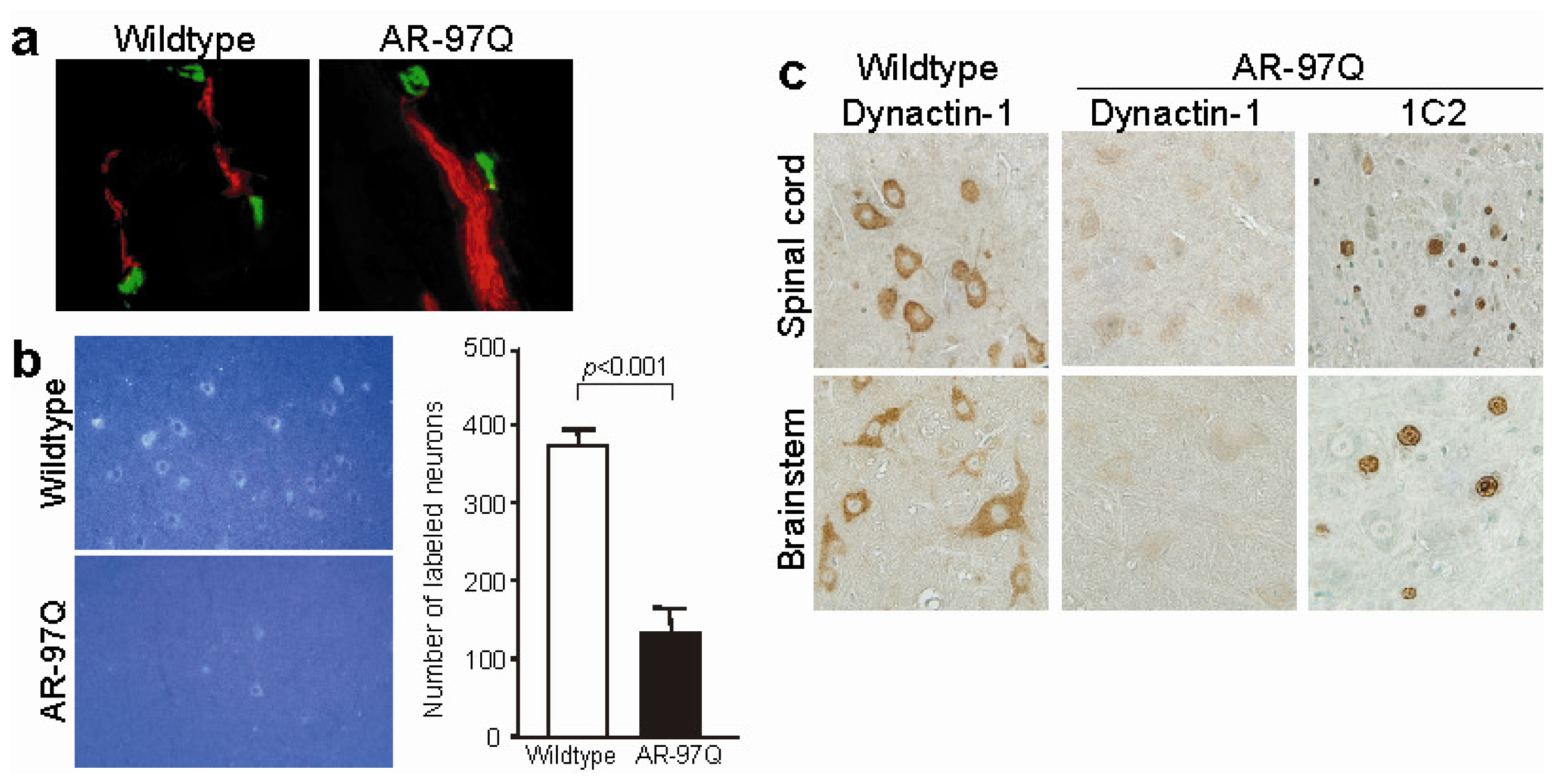{kind=link}
| MND type | Gene symbol | Protein | Protein function | Phenotype | Ref. |
|---|---|---|---|---|---|
| ALS1 | SOD1 | Cu/Zn superoxide dismutase | Detoxification enzyme | Varies among mutations from typical ALS type to atypical ALS | [57] |
| ALS2 | ALS2 | Alsin | Guanine nucleotide exchange factor (GEF) signaling; controlling endosomal dynamics | Juvenile onset, progressive muscle weakness and paralysis | [58] |
| ALS8 and SMA | VAPB | Synaptobrevin-associated membrane protein B (VAPB) | Vesicular trafficking; acts during ER-Golgi transport and secretion | Adult onset, slowly progressive upper and lower motor neuron disease. Phenotype varies from SMA type to ALS type | [59] |
| Lower motor neuron disease | DCTN1 | Dynactin-1 (p150glued) | Retrograde axonal transport | Slowly progressive lower motor neuron disease | [8,18] |
| ALS | CHMP2B | Charged multivesicular body protein 2B (CHMP2B) | Vesicular trafficking; acts as a component of the ESCRTIII (endosomal secretory complex required for transport) complex | Lower dominant motor neuron disease | [60,61] |
| SBMA | AR | Androgen receptor | DNA-binding transcription factor | Slowly progressive lower motor neuron disease | [29,62,63] |
| SPG3 | ATL1 | Atlastin | Vesicular trafficking; a member of GTPase family, essential for axon formation and elongation | Early-onset pure, slow progression HSP | [64,65] |
| SPG4 | SPAST | Spastin | An ATPase belonging to the AAA family acting for microtubule dynamics | Mainly pure HSP with variable onset | [66] |
| SPG10 | Kif5A | Kinesin (K1F5A) | Anterograde axonal transport | Early onset progressive weakness and leg spasticity | [33] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ikenaka, K.; Katsuno, M.; Kawai, K.; Ishigaki, S.; Tanaka, F.; Sobue, G. Disruption of Axonal Transport in Motor Neuron Diseases. Int. J. Mol. Sci. 2012, 13, 1225-1238. https://doi.org/10.3390/ijms13011225
Ikenaka K, Katsuno M, Kawai K, Ishigaki S, Tanaka F, Sobue G. Disruption of Axonal Transport in Motor Neuron Diseases. International Journal of Molecular Sciences. 2012; 13(1):1225-1238. https://doi.org/10.3390/ijms13011225
Chicago/Turabian StyleIkenaka, Kensuke, Masahisa Katsuno, Kaori Kawai, Shinsuke Ishigaki, Fumiaki Tanaka, and Gen Sobue. 2012. "Disruption of Axonal Transport in Motor Neuron Diseases" International Journal of Molecular Sciences 13, no. 1: 1225-1238. https://doi.org/10.3390/ijms13011225