Structural Elucidation and Bioactivity of Biflavonoids from the Stems of Wikstroemia taiwanensis

Abstract
:1. Introduction
2. Results and Discussion
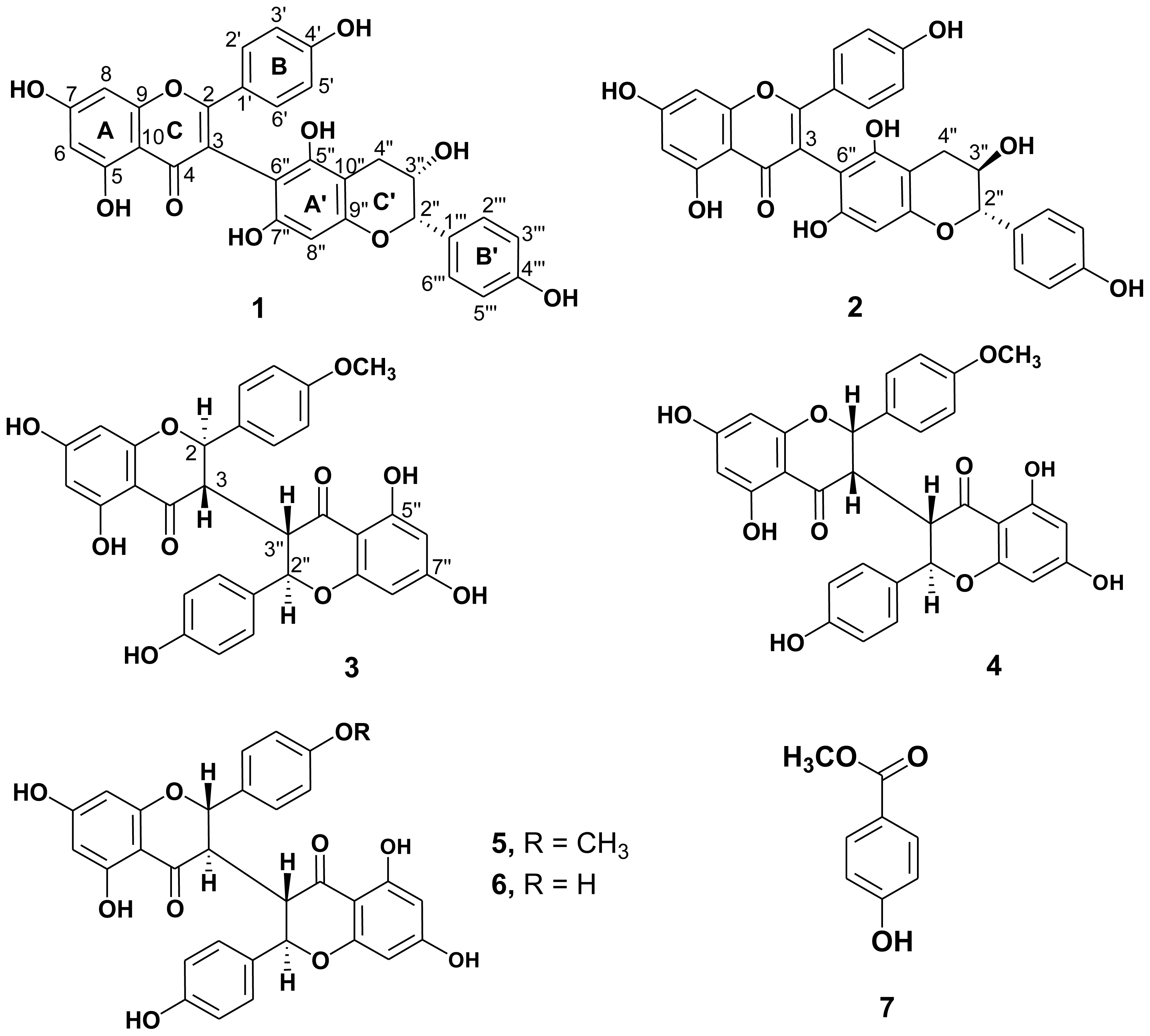
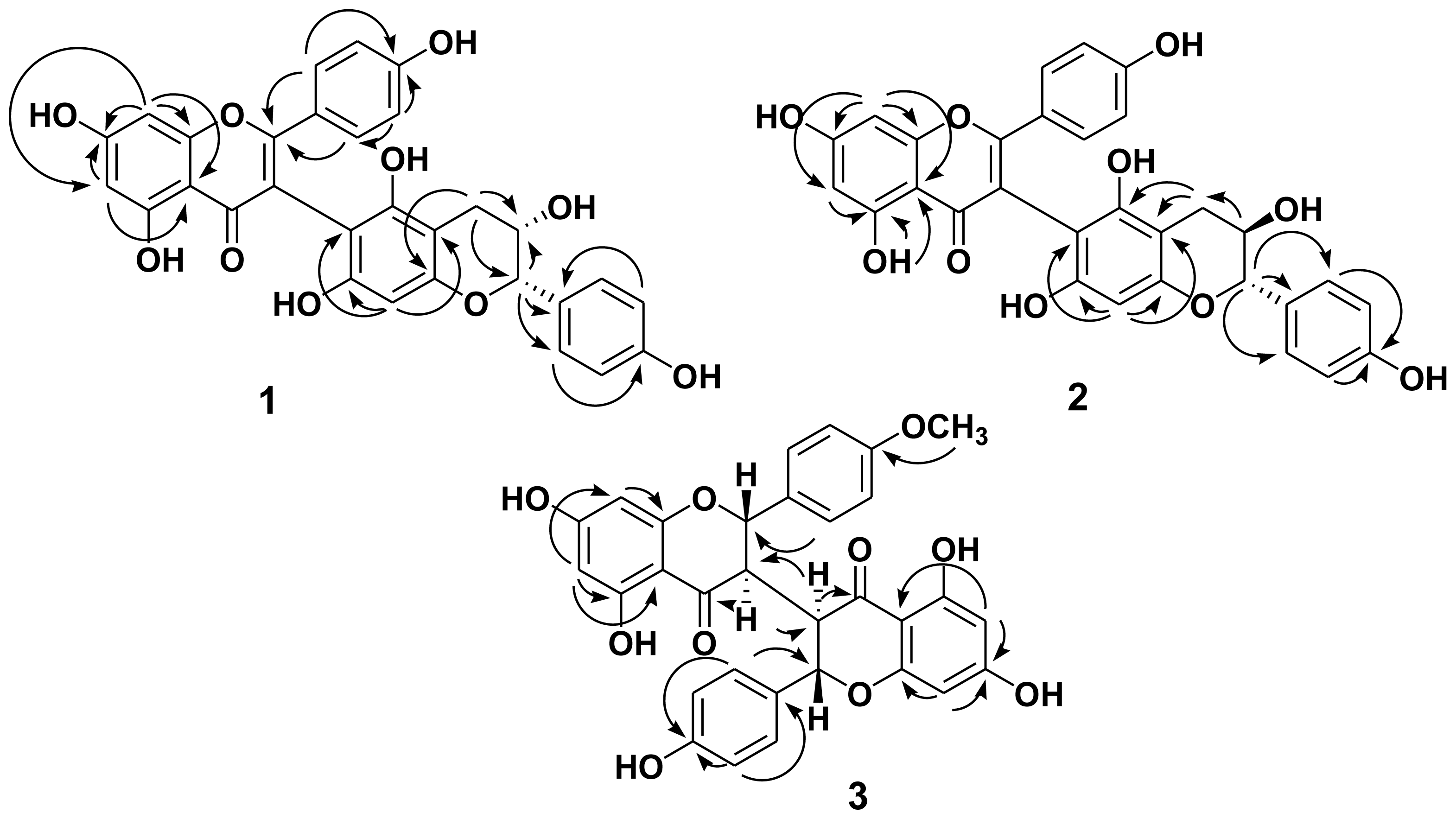
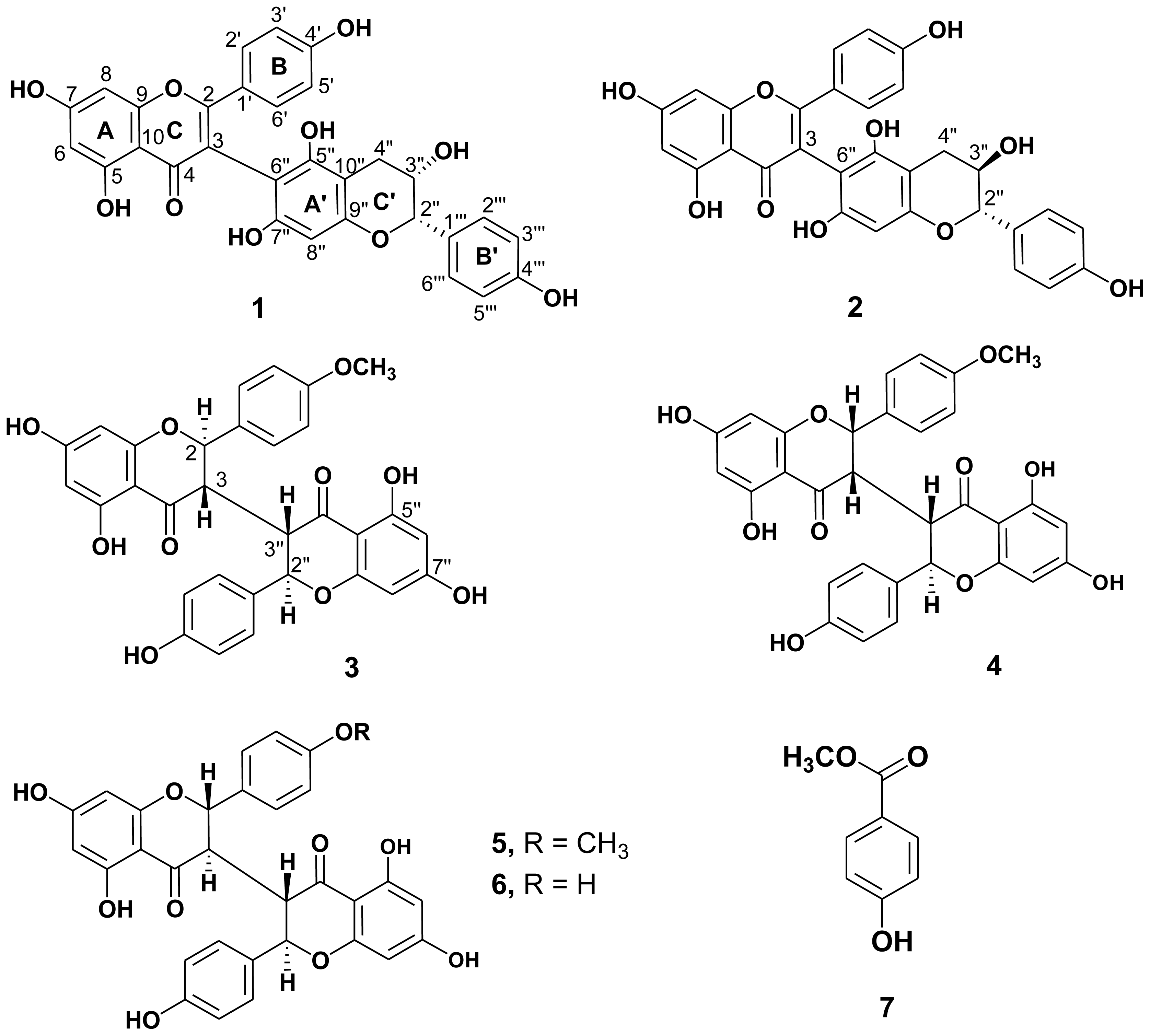
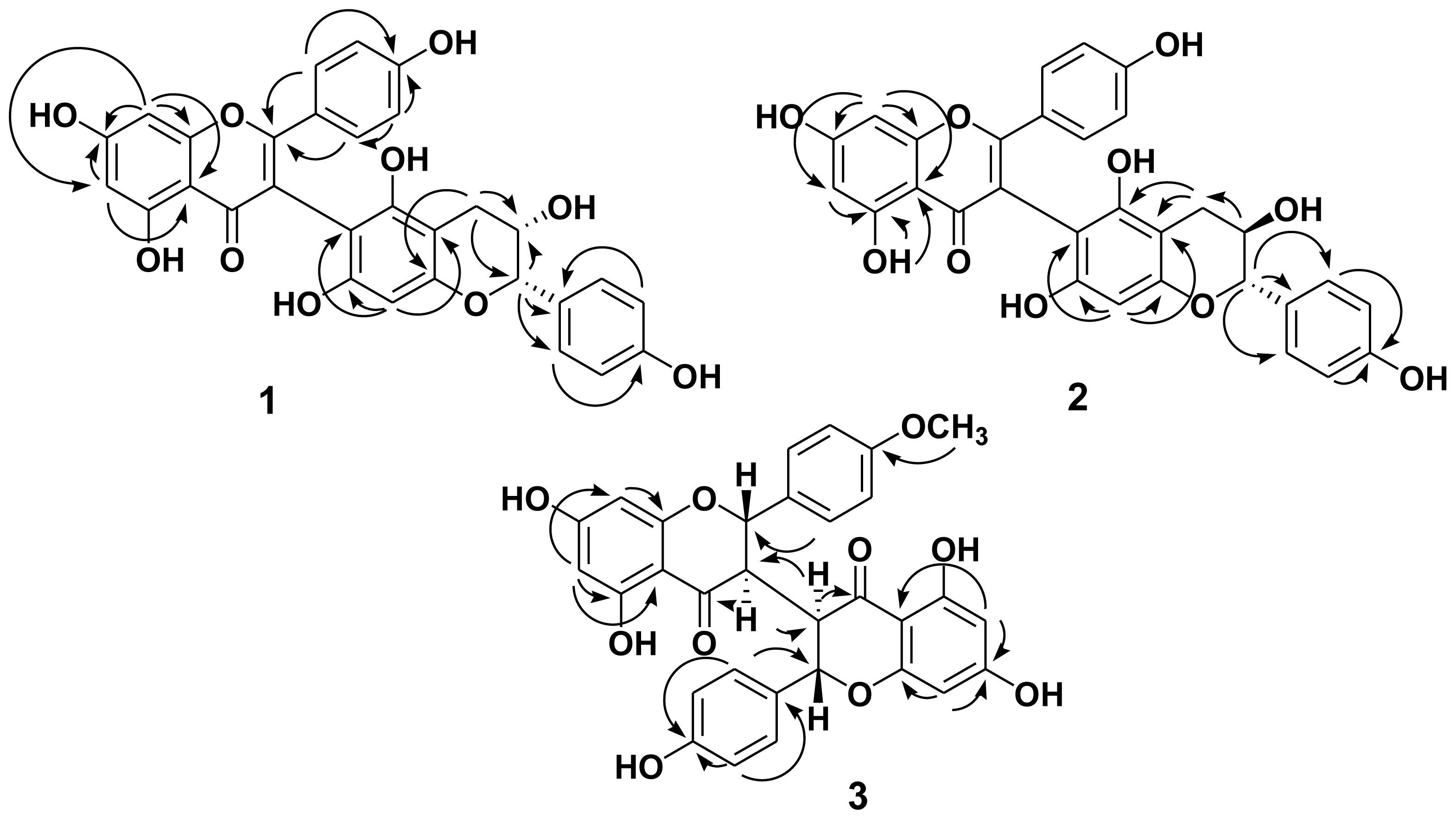
2.1. Structure Elucidation of the New Isolates
2.2. Antitubercular Activity
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Antitubercular Activity Assay
4. Conclusions
References
- The Burden of Disease Caused by TB. In Global Tuberculosis Control 2011; WHO: Geneva, Switzerland, 2011; p. 9.
- Chang, F.Y. Taiwan Tuberculosis Control Report 2010. Centers for Disease Control: Taiwan, China, 2010; p. 4. [Google Scholar]
- Niwa, M.; Jiang, P.F.; Hirata, Y. Two new C-3/C-3″-biflavanones from Wikstroemia sikokiana. Chem. Pharm. Bull 1986, 34, 3631–3634. [Google Scholar]
- Baba, K.; Taniguchi, M.; Kozawa, M. Three biflavonoids from Wikstroemia sikokiana. Phytochemistry 1994, 37, 879–883. [Google Scholar]
- Niwa, M.; Otsuji, S.; Tatematsu, H.; Liu, G.Q.; Chen, X.F.; Hirata, Y. Stereostructures of two biflavanones from Stellera chamaejasme L. Chem. Pharm. Bull 1986, 34, 3249–3251. [Google Scholar]
- Parmar, V.S.; Kumar, A.; Bosht, K.S.; Mukherjee, S.; Prasad, A.K.; Sharma, S.K.; Wengel, J.; Olsen, C.E. Novel chemoselective de-esterification of esters of polyacetoxy aromatic acids by lipases. Tetrahedron 1997, 53, 2163–2176. [Google Scholar]
- Takahashi, H.; Kubota, Y.; Miyazaki, H.; Onda, M. Heterocycles, XV. Enantioselective synthesis of chiral flavanonols and flavan-3,4-diols. Chem. Pharm. Bull 1984, 32, 4852–4857. [Google Scholar]
- Sethi, V.K.; Taneja, S.C.; Dhar, K.L.; Atal, C.K. (−)-Epiafzelechin 5-O-β-d-glucoside from Crataeva religiosa. Phytochemistry 1984, 23, 2402–2403. [Google Scholar]
- Seto, R.; Nakamura, H.; Nanjo, F.; Hara, Y. Preparation of epimers of tea catechins by heat treatment. Biosci. Biotechnol. Biochem 1997, 61, 1434–1439. [Google Scholar]
- Ohmori, K.; Yano, T.; Suzuki, K. General synthesis of epi-series catechins and their 3-gallates: Reverse polarity strategy. Org. Biomol. Chem 2010, 8, 2693–2696. [Google Scholar]
- Yang, G.; Chen, D. Biflavanones, flavonoids, and coumarins from the roots of Stellera chamaejasme and their antiviral effect on hepatitis B virus. Chem. Biodivers 2008, 5, 1419–1424. [Google Scholar]
- Niwa, M.; Tatematsu, H.; Liu, G.Q.; Hirata, Y. Isolation and structures of two new C-3/C-3″-biflavanones, neochamaejasmin A and neochamaejasmin B. Chem. Lett 1984, 539–542. [Google Scholar]
- Liu, G.Q.; Tatematsu, H.; Kurokawa, M.; Niwa, M.; Hirata, Y. Novel C-3/C-3″-biflavanones from Stellera chamaejasme L. Chem. Pharm. Bull 1984, 32, 362–365. [Google Scholar]
- Yang, G.; Liao, Z.; Xu, Z.; Zhang, H.; Chen, D. Antimitotic and antifungal C-3/C-3″-biflavanones from Stellera chamaejasme. Chem. Pharm. Bull 2005, 53, 776–779. [Google Scholar]
- Feng, B.M.; Pei, Y.H.; Hua, H.M. A new biflavonoid from Stellera chmaejasme L. Chin. Chem. Lett 2004, 15, 61–62. [Google Scholar]
- Baba, K.; Taniguchi, M.; Kozawa, M. A spirobiflavonoid genkwanol B from Daphne genkwa. Phytochemistry 1992, 3, 975–980. [Google Scholar]
- Baba, K.; Taniguchi, M.; Kozawa, M. A third spirobiflavonoid genkwanol C from Daphne genkwa. Phytochemistry 1993, 4, 863–867. [Google Scholar]
- Taniguchi, M.; Fujiwara, A.; Baba, K.; Wang, N.H. Two spirobiflavonoid from Daphne acutiloba. Phytochemistry 1998, 49, 913–916. [Google Scholar]
- Nunome, S.; Ishiyama, A.; Kobayashi, M.; Otoguro, K.; Kiyohara, H.; Yamada, H.; Omura, S. In vitro antimalarial activity of biflavonoids from Wikstroemia indica. Planta Med 2004, 70, 76–78. [Google Scholar]
- Hu, K.; Kobayashi, H.; Dong, A.; Iwasaki, S.; Yao, X. Antifungal, antimitotic and anti HIV-1 agents from the roots of Wikstroemia indica. Planta Med 2000, 66, 564–567. [Google Scholar]
- Huang, W.; Zhang, X.; Wang, Y.; Ye, W.; Ooi, V.E.C.; Chung, H.Y.; Li, Y. Antiviral biflavonoids from Radix wikstroemiae (Liaogewanggen). Chin. Med 2010, 5, 1–6. [Google Scholar]
- Inderlied, C.B.; Nash, K.A. Antibiotics in Laboratory Medicine, 4th ed; Williams & Wilkins: Baltimore, MD, USA, 1996; pp. 127–175. [Google Scholar]

{kind=link}
{kind=link}
| 1 a | 2 b | |||
|---|---|---|---|---|
| Position | δH (J in Hz) | δC | δH (J in Hz) | δC |
| 2 | 165.0 | 164.5 | ||
| 3 | 114.2 | 114.4 | ||
| 4 | 183.6 | 183.4 | ||
| 5 | 163.3 | 164.2 | ||
| 6 | 6.18 (d, J = 2.4 Hz) | 99.9 | 6.18 (d, J = 1.2 Hz) | 99.9 |
| 7 | 166.0 | 165.3 | ||
| 8 | 6.32 (d, J = 2.4 Hz) | 94.6 | 6.38 (d, J = 1.2 Hz) | 94.7 |
| 9 | 159.5 | 159.3 | ||
| 10 | 105.0 | 105.7 | ||
| 1′ | 126.0 | 126.4 | ||
| 2′ | 7.26 (d, J = 9.0 Hz) | 131.4 | 7.57 (d, J = 8.8 Hz) | 131.9 |
| 3′ | 6.67 (d, J = 9.0 Hz) | 115.6 | 6.87 (d, J = 8.8 Hz) | 116.4 |
| 4′ | 160.7 | 161.1 | ||
| 5′ | 6.67 (d, J = 9.0 Hz) | 115.6 | 6.87 (d, J = 8.8 Hz) | 116.4 |
| 6′ | 7.26 (d, J = 9.0 Hz) | 131.4 | 7.57 (d, J = 8.8 Hz) | 131.9 |
| 2″ | 4.13 (d, J = 7.8 Hz) | 82.8 | 4.53 (d, J = 8.2 Hz) | 83.5 |
| 3″ | 3.90 (ddd, J = 7.8, 8.4, 6.0 Hz) | 68.9 | 3.68 (br d, J = 8.2 Hz) | 69.2 |
| 4″a | 2.86 (dd, J = 6.0, 16.2 Hz) | 29.1 | 2.91 (dd, J = 16.0, 5.6 Hz) | 30.3 |
| 4″b | 2.46 (dd, J = 8.4, 16.2 Hz) | 2.56 (dd, J = 16.0, 6.8 Hz) | ||
| 5″ | 154.8 | 155.0 | ||
| 6″ | 100.7 | 101.0 | ||
| 7″ | 156.0 | 156.7 | ||
| 8″ | 6.04 (s) | 96.5 | 6.15 (s) | 97.0 |
| 9″ | 157.8 | 157.7 | ||
| 10″ | 101.5 | 101.6 | ||
| 1‴ | 131.6 | 131.9 | ||
| 2‴ | 7.10 (d, J = 9.0 Hz) | 129.5 | 6.87 (d, J = 8.8 Hz) | 130.1 |
| 3‴ | 6.70 (d, J = 9.0 Hz) | 115.8 | 6.65 (d, J = 8.8 Hz) | 116.2 |
| 4‴ | 158.1 | 158.5 | ||
| 5‴ | 6.70 (d, J = 9.0 Hz) | 115.8 | 6.65 (d, J = 8.8 Hz) | 116.2 |
| 6‴ | 7.10 (d, J = 9.0 Hz) | 129.5 | 6.87 (d, J = 8.8 Hz) | 130.1 |
| 3 a | |||
|---|---|---|---|
| Position | δH (J in Hz) | δC | HMBC |
| 2 | 5.70 (d, J = 12.0 Hz) | 82.3 | H-3, H-3″, H-2′, H-6′ |
| 3 | 2.60 (dd, J = 12.0, 18.0 Hz) | 48.5 | H-3″ |
| 4 | 193.0 | H-3 | |
| 5 | 161.8 | H-6 | |
| 6 | 5.46 (br d) | 96.7 | H-8 |
| 7 | 161.9 | H-8 | |
| 8 | 5.54 (s) | 97.5 | H-6 |
| 9 | 163.5 | H-8 | |
| 10 | 99.0 | H-6, H-8 | |
| 1′ | 128.9 | H-2, H-2′, H-3′, H-5′, H-6′ | |
| 2′ | 6.95 (d, J = 8.0 Hz) | 129.1 | H-2, H-3′, H-6′ |
| 3′ | 6.87 (d, J = 8.0 Hz) | 113.8 | H-2′, H-5′, H-6′ |
| 4′ | 159.6 | H-2, H-3′, H-5′, H-6′, Ome | |
| 5′ | 6.87 (d, J = 8.0 Hz) | 113.8 | H-2′, H-3, H-6′ |
| 6′ | 6.95 (d, J = 8.0 Hz) | 129.1 | H-2, H-2′, H-5′ |
| 2′ | 5.64 (d, J = 12.0 Hz) | 82.5 | H-3, H-3″, H-2‴, H-6‴ |
| 3′ | 2.60 (dd, J = 12.0, 18.0 Hz) | 48.6 | H-3 |
| 4″ | 193.0 | H-3″ | |
| 5″ | 161.8 | H-6″ | |
| 6″ | 5.46 (br d) | 96.6 | H-8″ |
| 7″ | 161.9 | H-8″ | |
| 8″ | 5.54 (s) | 97.6 | H-6″ |
| 9″ | 163.5 | H-8″ | |
| 10″ | 99.1 | H-6″, H-8″ | |
| 1‴ | 127.1 | H-2″, H-3‴, H-5‴ | |
| 2‴ | 6.78 (d, J = 8.0 Hz) | 128.9 | H-2″, H-3‴, H-6‴ |
| 3‴ | 6.70 (d, J = 8.0 Hz) | 115.3 | H-2‴, H-5‴ |
| 4‴ | 158.0 | H-2‴, H-3‴, H-5‴, H-6‴ | |
| 5‴ | 6.70 (d, J = 8.0 Hz) | 115.3 | H-3‴, H-6‴ |
| 6‴ | 6.78 (d, J = 8.0 Hz) | 128.9 | H-2″, H-2‴, H-5‴ |
| OMe | 3.79 (s) | 55.1 | |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, L.-Y.; Chen, I.-S.; Peng, C.-F. Structural Elucidation and Bioactivity of Biflavonoids from the Stems of Wikstroemia taiwanensis. Int. J. Mol. Sci. 2012, 13, 1029-1038. https://doi.org/10.3390/ijms13011029
Chen L-Y, Chen I-S, Peng C-F. Structural Elucidation and Bioactivity of Biflavonoids from the Stems of Wikstroemia taiwanensis. International Journal of Molecular Sciences. 2012; 13(1):1029-1038. https://doi.org/10.3390/ijms13011029
Chicago/Turabian StyleChen, Li-Yin, Ih-Sheng Chen, and Chien-Fang Peng. 2012. "Structural Elucidation and Bioactivity of Biflavonoids from the Stems of Wikstroemia taiwanensis" International Journal of Molecular Sciences 13, no. 1: 1029-1038. https://doi.org/10.3390/ijms13011029
APA StyleChen, L.-Y., Chen, I.-S., & Peng, C.-F. (2012). Structural Elucidation and Bioactivity of Biflavonoids from the Stems of Wikstroemia taiwanensis. International Journal of Molecular Sciences, 13(1), 1029-1038. https://doi.org/10.3390/ijms13011029