Genetic Variability and Phylogeny of High Risk HPV Type 16, 18, 31, 33 and 45 L1 Gene in Greek Women


 ,
, 
Abstract
:1. Introduction
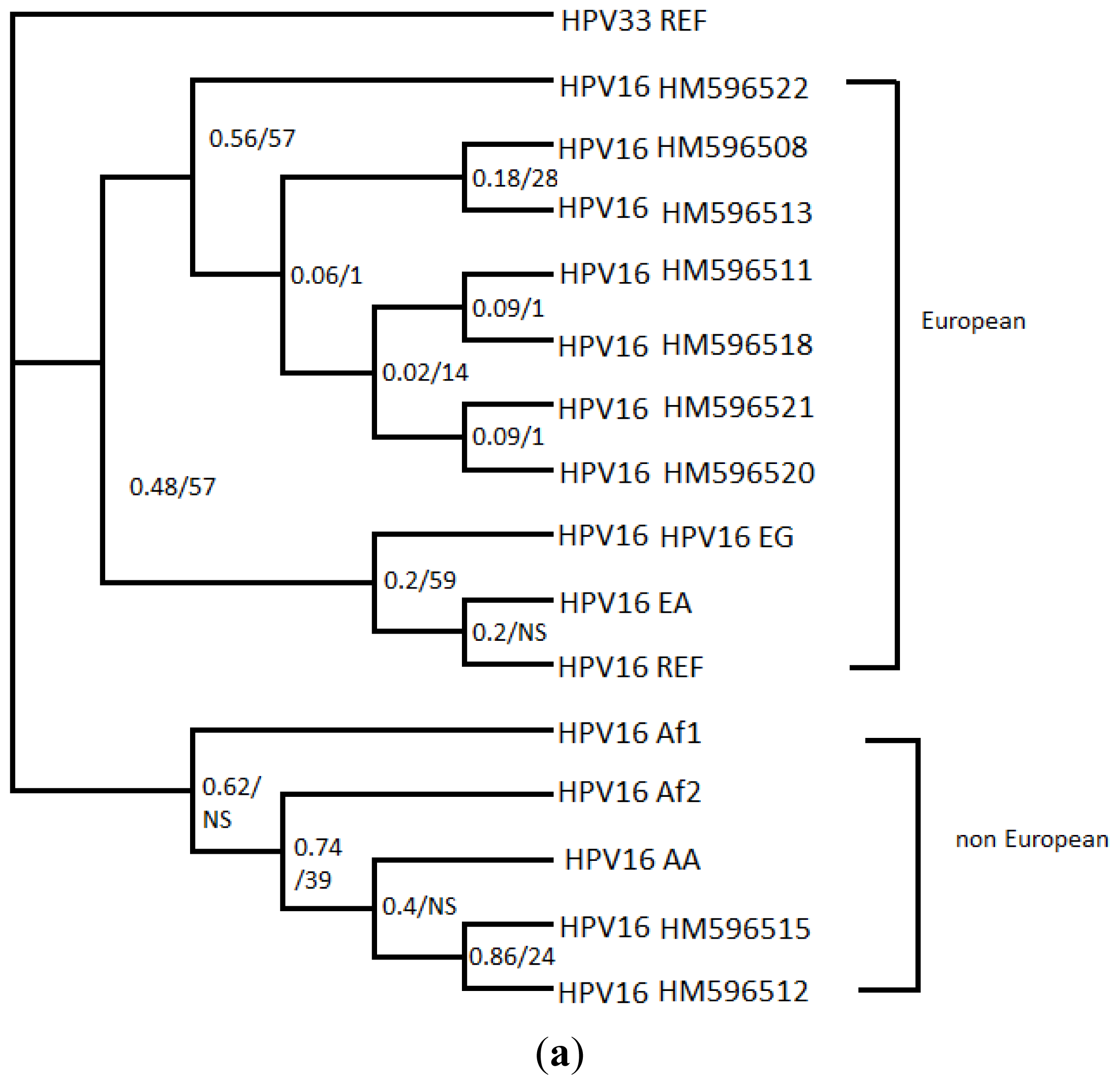
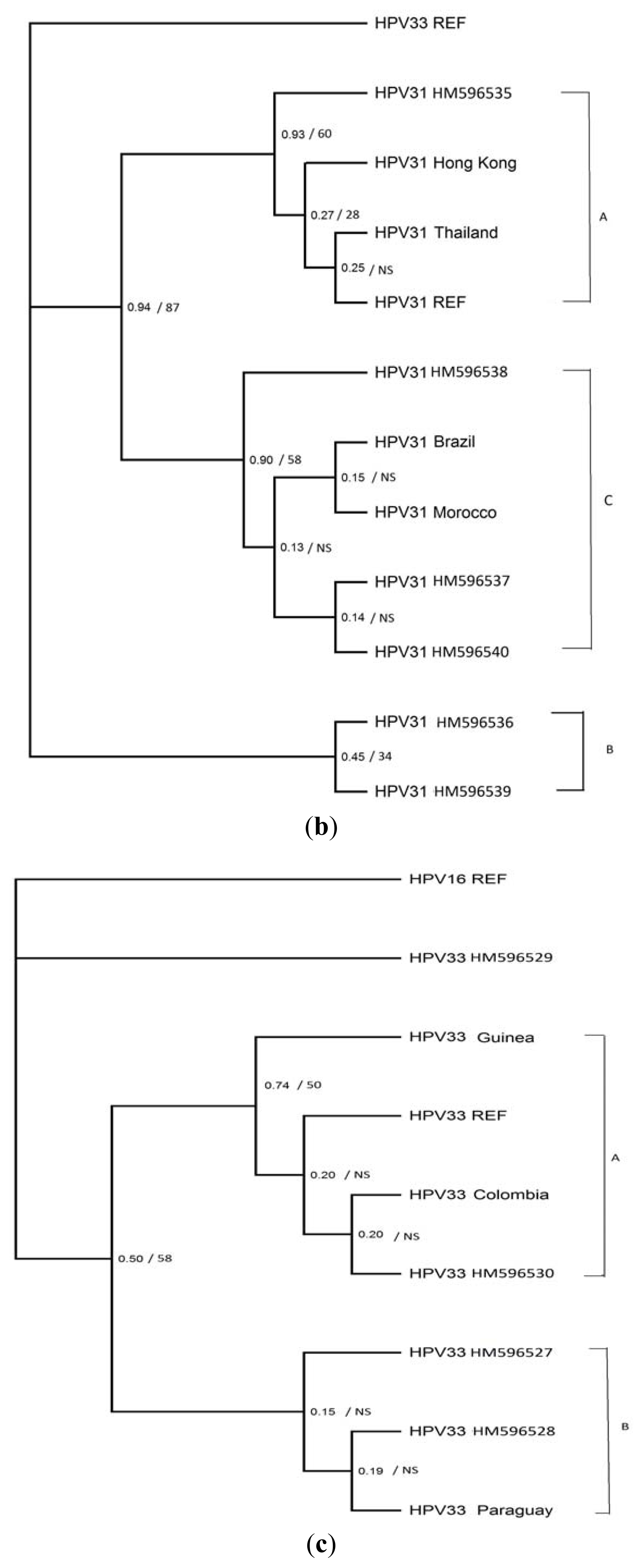
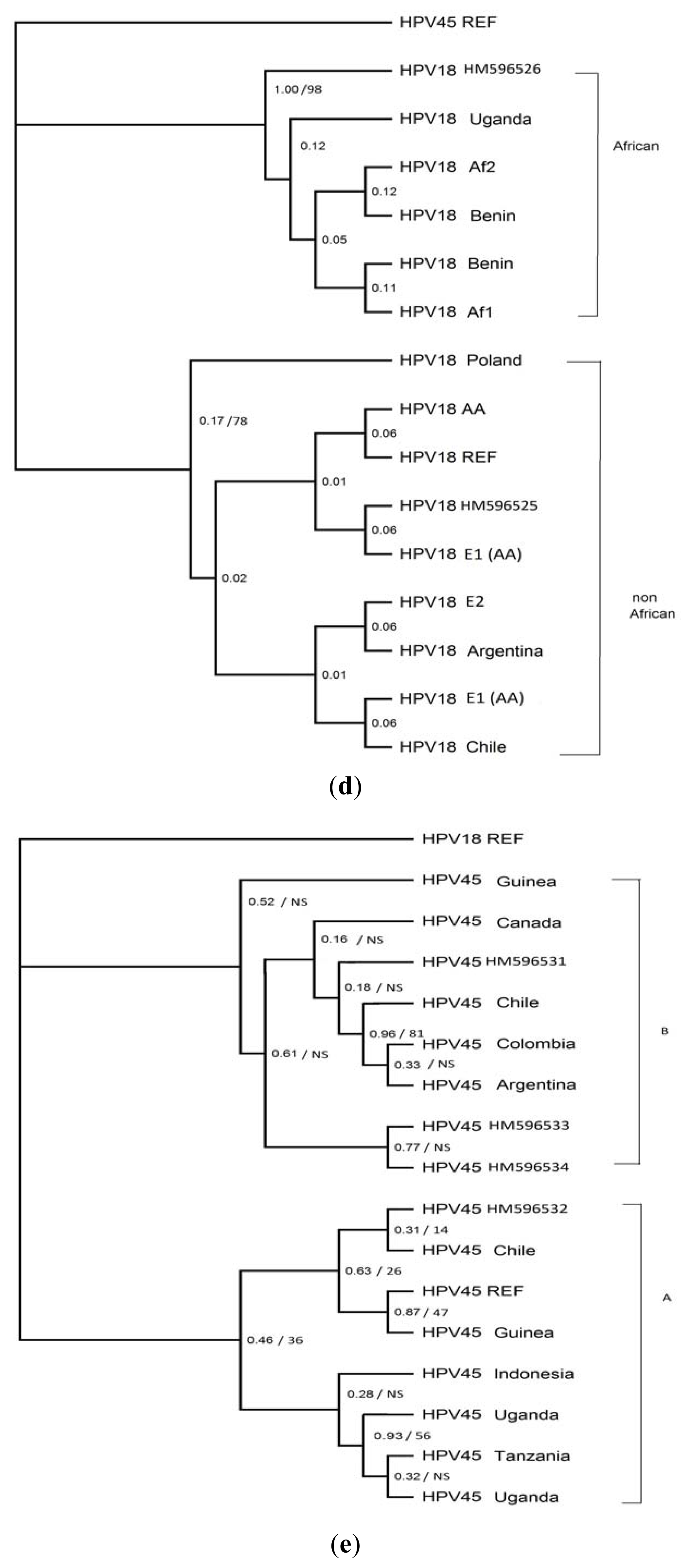
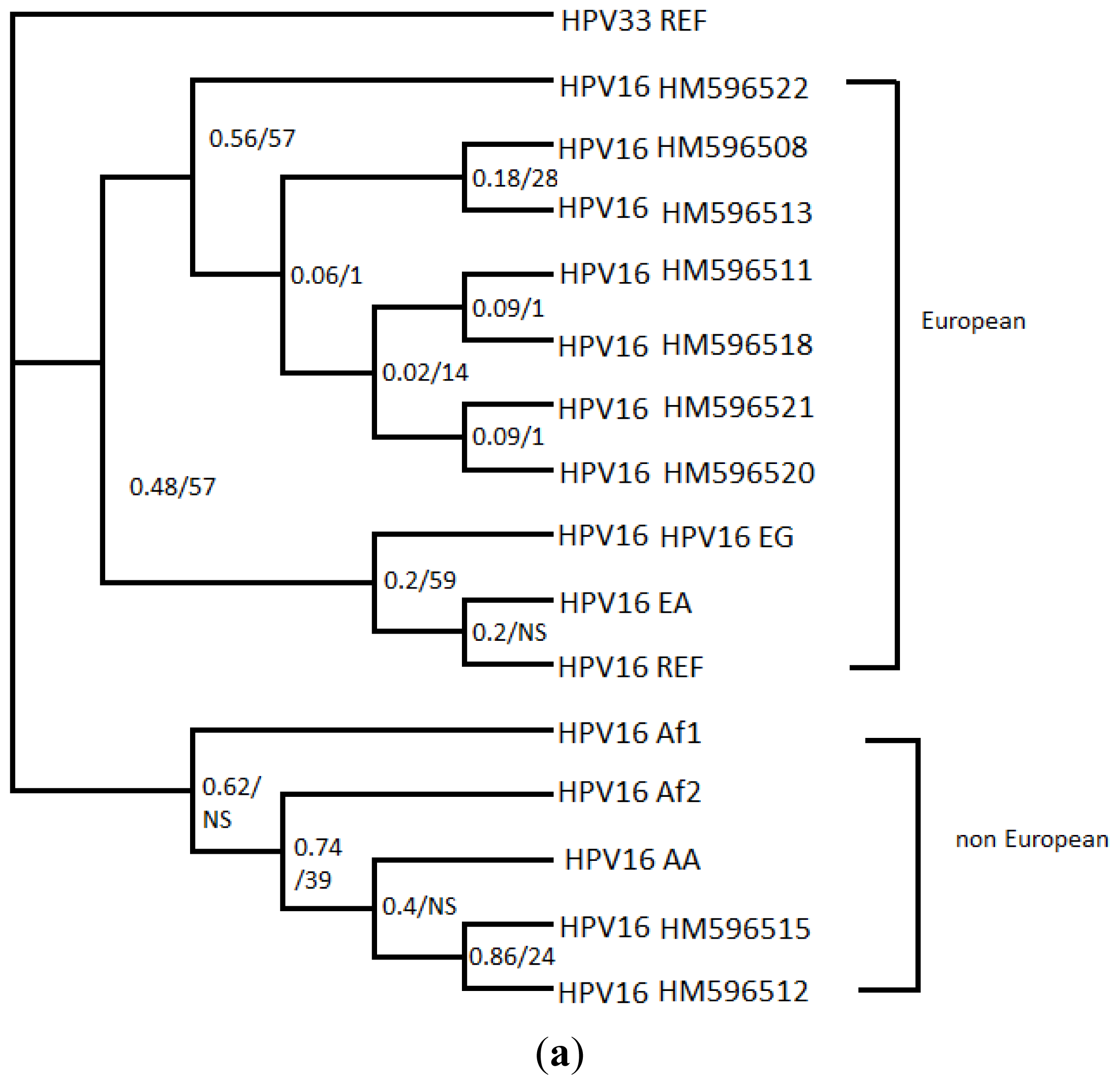
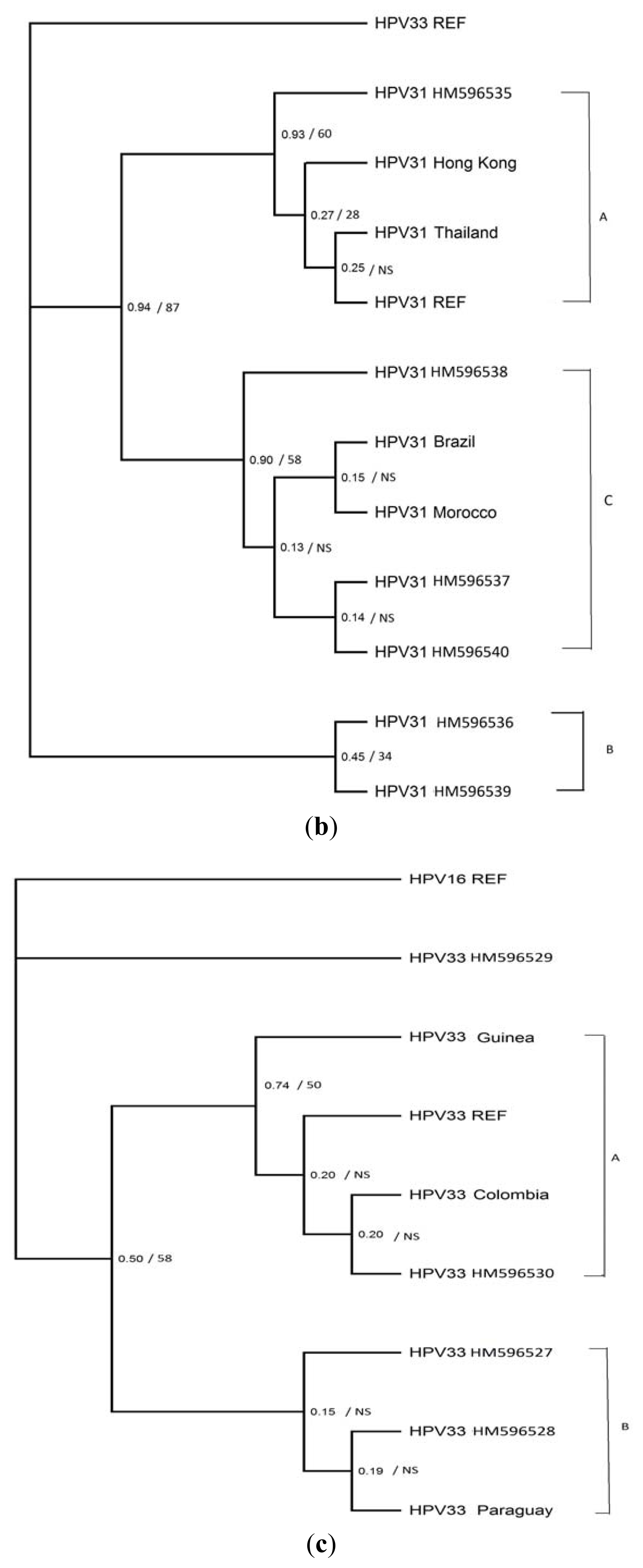
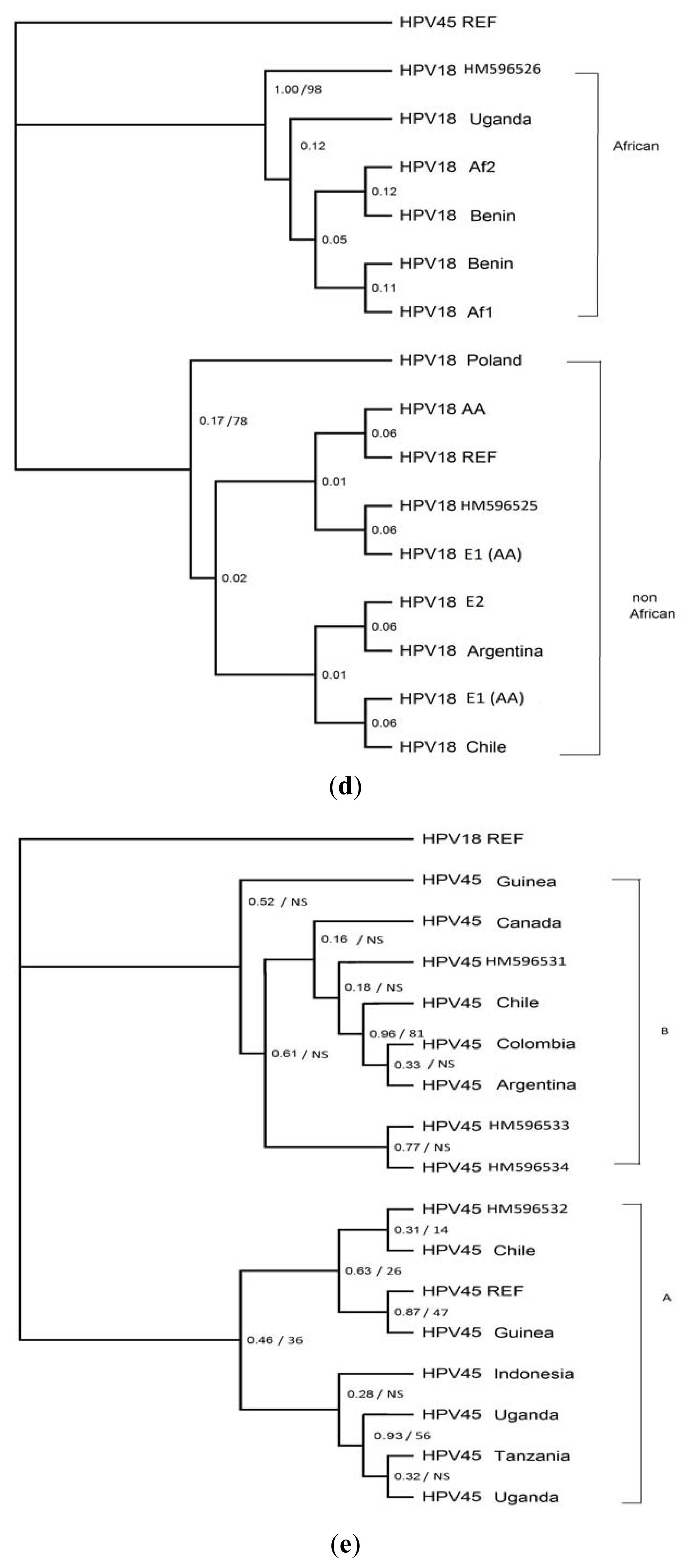
2. Results
3. Experimental Section
3.1. Sample Collection
3.2. DNA Extraction and HPV Typing
3.3. PCR and Sequencing
3.4. Phylogenetic Analysis
4. Discussion
5. Conclusions
Acknowledgments
References
- Li, N.; Franceschi, S.; Howell-Jones, R.; Snijders, P.J.; Clifford, G.M. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: Variation by geographical region, histological type and year of publication. Int. J. Cancer 2011, 128, 927–935. [Google Scholar]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med 2003, 348, 518–527. [Google Scholar]
- Schiffman, M.; Herrero, R.; Desalle, R.; Hildesheim, A.; Wacholder, S.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Morales, J.; Guillen, D.; et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology 2005, 337, 76–84. [Google Scholar]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar]
- Chen, Z.; DeSalle, R.; Schiffman, M.; Herrero, R.; Burk, R.D. Evolutionary dynamics of variant genomes of human papillomavirus types 18, 45, and 97. J. Virol 2009, 83, 1443–1455. [Google Scholar]
- Chen, Z.; Schiffman, M.; Herrero, R.; Desalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Burk, R.D. Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Nylander, J.A.A. Mrmodeltest 2.3. Program Distributed by the Author; Uppsala University: Uppsala, Switzerland, 2004. [Google Scholar]
- Yamada, T.; Wheeler, C.M.; Halpern, A.L.; Stewart, A.C.; Hildesheim, A.; Jenison, S.A. Human papillomavirus type 16 variant lineages in united states populations characterized by nucleotide sequence analysis of the E6, L2, and L1 coding segments. J. Virol 1995, 69, 7743–7753. [Google Scholar]
- Calleja-Macias, I.E.; Villa, L.L.; Prado, J.C.; Kalantari, M.; Allan, B.; Williamson, A.L.; Chung, L.P.; Collins, R.J.; Zuna, R.E.; Dunn, S.T.; et al. Worldwide genomic diversity of the high-risk human papillomavirus types 31, 35, 52, and 58, four close relatives of human papillomavirus type 16. J. Virol 2005, 79, 13630–13640. [Google Scholar]
- Stewart, A.C.; Eriksson, A.M.; Manos, M.M.; Munoz, N.; Bosch, F.X.; Peto, J.; Wheeler, C.M. Intratype variation in 12 human papillomavirus types: A worldwide perspective. J. Virol 1996, 70, 3127–3136. [Google Scholar]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. Mega4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol 2007, 24, 1596–1599. [Google Scholar]
- Swofford, D.L. Paup*: Phylogenetic Analysis Using Parsimony (and Other Methods), Version 4.0b10; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar]
- Xia, X.; Xie, Z. Dambe: Software package for data analysis in molecular biology and evolution. J. Hered 2001, 92, 371–373. [Google Scholar]
- Page, R.D. Treeview: An application to display phylogenetic trees on personal computers. Comput. Appl. Biosci 1996, 12, 357–358. [Google Scholar]
- HPV Sequence Database. Available online: http://hpv-web.lanl.gov/ (accessed on 3 July 2011).
- Bernard, H.U.; Chan, S.Y.; Delius, H. Evolution of papillomaviruses. Curr. Top. Microbiol. Immunol 1994, 186, 33–54. [Google Scholar]
- Zehbe, I.; Wilander, E.; Delius, H.; Tommasino, M. Human papillomavirus 16 E6 variants are more prevalent in invasive cervical carcinoma than the prototype. Cancer Res 1998, 58, 829–833. [Google Scholar]
- Hildesheim, A.; Schiffman, M.; Bromley, C.; Wacholder, S.; Herrero, R.; Rodriguez, A.; Bratti, M.C.; Sherman, M.E.; Scarpidis, U.; Lin, Q.Q.; et al. Human papillomavirus type 16 variants and risk of cervical cancer. J. Natl. Cancer Inst 2001, 93, 315–318. [Google Scholar]
- Pista, A.; Oliveira, A.; Barateiro, A.; Costa, H.; Verdasca, N.; Paixao, M.T. Molecular variants of human papillomavirus type 16 and 18 and risk for cervical neoplasia in portugal. J. Med. Virol 2007, 79, 1889–1897. [Google Scholar]
- Ellis, J.R.; Keating, P.J.; Baird, J.; Hounsell, E.F.; Renouf, D.V.; Rowe, M.; Hopkins, D.; Duggan-Keen, M.F.; Bartholomew, J.S.; Young, L.S.; et al. The association of an HPV16 oncogene variant with HLA-B7 has implications for vaccine design in cervical cancer. Nat. Med 1995, 1, 464–470. [Google Scholar]
- Cheng, G.; Icenogle, J.P.; Kirnbauer, R.; Hubbert, N.L.; St. Louis, M.E.; Han, C.; Svare, E.I.; Kjaer, S.K.; Lowy, D.R.; Schiller, J.T. Divergent human papillomavirus type 16 variants are serologically cross-reactive. J. Infect. Dis 1995, 172, 1584–1587. [Google Scholar]
- Hildesheim, A. Human papillomavirus variants: Implications for natural history studies and vaccine development efforts. J. Natl. Cancer Inst 1997, 89, 752–753. [Google Scholar]
- Park, J.S.; Hwang, E.S.; Lee, C.J.; Kim, C.J.; Rha, J.G.; Kim, S.J.; Namkoong, S.E.; Um, S.J. Mutational and functional analysis of HPV-16 URR derived from Korean cervical neoplasia. Gynecol. Oncol 1999, 74, 23–29. [Google Scholar]
- Kammer, C.; Warthorst, U.; Torrez-Martinez, N.; Wheeler, C.M.; Pfister, H. Sequence analysis of the long control region of human papillomavirus type 16 variants and functional consequences for p97 promoter activity. J. Gen. Virol 2000, 81, 1975–1981. [Google Scholar]
- Ho, L.; Chan, S.Y.; Burk, R.D.; Das, B.C.; Fujinaga, K.; Icenogle, J.P.; Kahn, T.; Kiviat, N.; Lancaster, W.; Mavromara-Nazos, P.; et al. The genetic drift of human papillomavirus type 16 is a means of reconstructing prehistoric viral spread and the movement of ancient human populations. J. Virol 1993, 67, 6413–6423. [Google Scholar]
- Wheeler, C.M.; Yamada, T.; Hildesheim, A.; Jenison, S.A. Human papillomavirus type 16 sequence variants: Identification by E6 and L1 lineage-specific hybridization. J. Clin. Microbiol 1997, 35, 11–19. [Google Scholar]
- Chen, Z.; Terai, M.; Fu, L.; Herrero, R.; DeSalle, R.; Burk, R.D. Diversifying selection in human papillomavirus type 16 lineages based on complete genome analyses. J. Virol 2005, 79, 7014–7023. [Google Scholar]
- Bosch, F.X.; Manos, M.M.; Munoz, N.; Sherman, M.; Jansen, A.M.; Peto, J.; Schiffman, M.H.; Moreno, V.; Kurman, R.; Shah, K.V. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. International biological study on cervical cancer (IBSCC) study group. J. Natl. Cancer Inst 1995, 87, 796–802. [Google Scholar]
- Wu, Y.; Chen, Y.; Li, L.; Yu, G.; Zhang, Y.; He, Y. Associations of high-risk hpv types and viral load with cervical cancer in china. J. Clin. Virol 2006, 35, 264–269. [Google Scholar]
- Das, B.C.; Sharma, J.K.; Gopalkrishna, V.; Das, D.K.; Singh, V.; Gissmann, L.; zur Hausen, H.; Luthra, U.K. A high frequency of human papillomavirus DNA sequences in cervical carcinomas of Indian women as revealed by southern blot hybridization and polymerase chain reaction. J. Med. Virol 1992, 36, 239–245. [Google Scholar]
- Tornesello, M.L.; Duraturo, M.L.; Buonaguro, L.; Vallefuoco, G.; Piccoli, R.; Palmieri, S.; Buonaguro, F.M. Prevalence of human papillomavirus genotypes and their variants in high risk West Africa women immigrants in South Italy. Infect. Agent Cancer 2007, 2. [Google Scholar] [CrossRef]
- Pillai, M.R.; Hariharan, R.; Babu, J.M.; Lakshmi, S.; Chiplunkar, S.V.; Patkar, M.; Tongaonkar, H.; Dinshaw, K.; Jayshree, R.S.; Reddy, B.K.; et al. Molecular variants of HPV-16 associated with cervical cancer in indian population. Int. J. Cancer 2009, 125, 91–103. [Google Scholar]
- Daponte, A.; Pournaras, S.; Mademtzis, I.; Hadjichristodoulou, C.; Kostopoulou, E.; Maniatis, A.N.; Messinis, I.E. Evaluation of high-risk human papillomavirus types PCR detection in paired urine and cervical samples of women with abnormal cytology. J. Clin. Virol 2006, 36, 189–193. [Google Scholar]
- Konidaris, S.; Kouskouni, E.E.; Panoskaltsis, T.; Kreatsas, G.; Patsouris, E.S.; Sarivalassis, A.; Nonni, A.; Lazaris, A.C. Human papillomavirus infection in malignant and benign gynaecological conditions: A study in Greek women. Health Care Women Int 2007, 28, 182–191. [Google Scholar]
- Kroupis, C.; Thomopoulou, G.; Papathomas, T.G.; Vourlidis, N.; Lazaris, A.C. Population-based study of human papillomavirus infection and cervical neoplasia in Athens, Greece. Epidemiol. Infect 2007, 135, 943–950. [Google Scholar]
- Yapijakis, C.; Adamopoulou, M.; Antonopoulos, G.; Koufaliotis, N.; Vairaktaris, E. Prevalence of HPV types in a cohort of Greeks with clinical indication of infection. Anticancer Res 2008, 28, 2233–2237. [Google Scholar]
- Villa, L.L.; Sichero, L.; Rahal, P.; Caballero, O.; Ferenczy, A.; Rohan, T.; Franco, E.L. Molecular variants of human papillomavirus types 16 and 18 preferentially associated with cervical neoplasia. J. Gen. Virol 2000, 81, 2959–2968. [Google Scholar]
- Tornesello, M.L.; Duraturo, M.L.; Salatiello, I.; Buonaguro, L.; Losito, S.; Botti, G.; Stellato, G.; Greggi, S.; Piccoli, R.; Pilotti, S.; et al. Analysis of human papillomavirus type-16 variants in Italian women with cervical intraepithelial neoplasia and cervical cancer. J. Med. Virol 2004, 74, 117–126. [Google Scholar]
- Ong, C.K.; Chan, S.Y.; Campo, M.S.; Fujinaga, K.; Mavromara-Nazos, P.; Labropoulou, V.; Pfister, H.; Tay, S.K.; ter Meulen, J.; Villa, L.L.; et al. Evolution of human papillomavirus type 18: An ancient phylogenetic root in Africa and intratype diversity reflect coevolution with human ethnic groups. J. Virol 1993, 67, 6424–6431. [Google Scholar]
- Altekruse, S.F.; Lacey, J.V., Jr; Brinton, L.A.; Gravitt, P.E.; Silverberg, S.G.; Barnes, W.A., Jr; Greenberg, M.D.; Hadjimichael, O.C.; McGowan, L.; Mortel, R.; et al. Comparison of human papillomavirus genotypes, sexual, and reproductive risk factors of cervical adenocarcinoma and squamous cell carcinoma: Northeastern United States. Am. J. Obstet. Gynecol 2003, 188, 657–663. [Google Scholar]
- Burk, R.D.; Terai, M.; Gravitt, P.E.; Brinton, L.A.; Kurman, R.J.; Barnes, W.A.; Greenberg, M.D.; Hadjimichael, O.C.; Fu, L.; McGowan, L.; et al. Distribution of human papillomavirus types 16 and 18 variants in squamous cell carcinomas and adenocarcinomas of the cervix. Cancer Res 2003, 63, 7215–7220. [Google Scholar]
- Schlecht, N.F.; Burk, R.D.; Palefsky, J.M.; Minkoff, H.; Xue, X.; Massad, L.S.; Bacon, M.; Levine, A.M.; Anastos, K.; Gange, S.J.; et al. Variants of human papillomaviruses 16 and 18 and their natural history in human immunodeficiency virus-positive women. J. Gen. Virol 2005, 86, 2709–2720. [Google Scholar]
- Xi, L.F.; Koutsky, L.A.; Hildesheim, A.; Galloway, D.A.; Wheeler, C.M.; Winer, R.L.; Ho, J.; Kiviat, N.B. Risk for high-grade cervical intraepithelial neoplasia associated with variants of human papillomavirus types 16 and 18. Cancer Epidemiol. Biomark. Prev 2007, 16, 4–10. [Google Scholar]
- Cerqueira, D.M.; Raiol, T.; Veras, N.M.; von Gal Milanezi, N.; Amaral, F.A.; de Macedo Brigido, M.; Martins, C.R. New variants of human papillomavirus type 18 identified in central Brazil. Virus Genes 2008, 37, 282–287. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| WNL | ASCUS | LGSIL | HGSIL | SCC | Total | |
|---|---|---|---|---|---|---|
| HPV 16 variants (sample) | n = 11(10.6) | n = 5 (4.8) | n = 33 (31.7) | n = 49 (47.1) | n = 6 (5.8) | n = 104 |
| E (HPV16 HM596508) | 1 (2.0) | 1 (0.96) | ||||
| E (HPV16 HM596511) | 1 (20.0) | 1 (0.96) | ||||
| E (HPV16 HM596513) | 1 (20.0) | 1 (0.96) | ||||
| E (HPV16 HM596521) | 3 (9.1) | 3 (2.88) | ||||
| E (HPV16 HM596522) | 1 (3.0) | 1 (2.0) | 2 (1.92) | |||
| E (HPV16 HM596518) | 1 (3.0) | 1 (0.96) | ||||
| E (HPV16 HM596520) | 10 (90.1) | 3 (60.0) | 27 (81.7) | 42 (85.7) | 6 (100) | 88 (84.6) |
| Non E (HPV16 HM596515) | 1 (9.1) | 1 (2.0) | 2 (1.92) | |||
| Non E (HPV16 HM596512) | 1 (3.0) | 4 (8.1) | 5(4.82) | |||
| HPV 31 variants (sample) | n = 5 (12.5) | n = 6 (15) | n = 23 (57.5) | n = 5 (12.5) | 40 | |
| A (HPV31 HM596535) | 1 (4.3) | 1 (20.0) | 2(5) | |||
| B (HPV31 HM596536) | 1 (20) | 1 (2.5) | ||||
| B (HPV31 HM596539) | 2 (40.0) | 1 (16.7) | 2 (8.6) | 1 (20.0) | 6 (15) | |
| C (HPV31 HM596537) | 1 (4,3) | 1 (2.5) | ||||
| C (HPV31 HM596538) | 4 (17.4) | 4 (10) | ||||
| C (HPV31 HM596540) | 2 (40.0) | 5 (83.3) | 15 (65.2) | 3 (60.0) | 26 (65) | |
| HPV 33 variants (sample) | n = 1 (14.3) | n = 1 (14.3) | n = 2 (40.0) | n = 3 (42.9) | 7 | |
| A (HPV33 HM596530) | 1 (50.0) | 3 (100.0) | 4 (57.1) | |||
| B (HPV33 HM596527) | 1 (100.0) | 1 (14.3) | ||||
| B (HPV33 HM596528) | 1 (100.0) | 1 (14.3) | ||||
| C (HPV33 HM596529) | 1 (50.0) | 1 (14.3) | ||||
| HPV 18 variants (sample) | n = 3 (60.0) | n = 2 (40.0) | 5 | |||
| Non Af (HPV18 HM596525) | 2 (66.7) | 2 (100.0) | 4 (80.0) | |||
| Af (HPV18 HM596526) | 1 (33.3) | 1 (20.0) | ||||
| HPV 45 variants (sample) | n = 1 (25.0) | n = 2 (50.0) | n = 1 (25.0) | 4 | ||
| A (HPV45 HM596532) | 1 (100.0) | 1 (25.0) | ||||
| B (HPV45 HM596531) | 1 (50.0) | 1 (25.0) | ||||
| B (HPV45 HM596533) | 1 (100.0) | 1 (25.0) | ||||
| B (HPV45 HM596534) | 1 (50.0) | 1 (25.0) | ||||
| Isolates with identicqal sequences (a) | HPV16 (104 samples) | 6592 | 6595 | 6694 | 6695 | 6721 | 6803 | 6824 | 6854 | 6865 | 6888 | 6914 | 6973 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HPV16 Ref (K02718) | C | C | A | A | G | A | T | C | C | C | A | C | |
| Non-synonymous mutations | E352D | T353P | T389S | S396P S396A | T417I | ||||||||
| 1 | HPV16 HM596508 | T | T | . | . | . | . | C | . | . | . | . | . |
| 1 | HPV16 HM596511 | T | T | . | . | . | . | . | . | . | . | G | . |
| 5 | HPV16 HM596512 | T | T | . | C | A | . | . | T | T | . | . | T |
| 1 | HPV16 HM596513 | T | T | . | . | . | . | G | . | . | . | . | . |
| 3 | HPV16 HM596521 | T | T | . | . | . | . | . | . | . | T | . | NI |
| 2 | HPV16 HM596522 | T | T | . | . | . | . | . | T | . | . | . | . |
| 2 | HPV16 HM596515 | T | T | . | C | A | T | . | T | T | . | . | T |
| 1 | HPV16 HM596518 | T | T | C | . | . | . | . | . | . | . | . | . |
| 88 | HPV16 HM596520 | T | T | . | . | . | . | . | . | . | . | . | . |
| Isolates with identicqal sequences (b) | HPV 31(40 samples) | 6511 | 6568 | 6586 | 6647 | 6664 | 6703 | 6772 | 6796 | 6817 | 6846 | 6862 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HPV31 Ref (J04353) | C | T | T | A | T | A | G | G | C | C | C | |
| Non-synonymous mutations | T432S | |||||||||||
| 2 | HPV31 HM596535 | T | . | . | . | . | . | . | . | A | G | . |
| 1 | HPV31 HM596536 | T | C | . | C | . | . | A | A | A | . | T |
| 1 | HPV31 HM596537 | T | . | G | . | . | G | . | A | A | . | . |
| 4 | HPV31 HM596538 | T | . | G | . | C | . | . | A | A | . | . |
| 6 | HPV31 HM596539 | T | C | . | . | . | . | A | A | A | . | T |
| 26 | HPV31 HM596540 | T | . | G | . | . | . | . | A | A | . | . |
| Isolates with identicqal sequences (c) | HPV33 (7 samples) | 6664 | 6673 | 6748 | 6841 |
|---|---|---|---|---|---|
| HPV33 Ref (M12732) | A | A | A | C | |
| Non-synonymous mutations | E385D | ||||
| 1 | HPV33 HM596527 | G | . | C | . |
| 1 | HPV33 HM596528 | G | . | . | . |
| 1 | HPV33 HM596529 | G | G | . | T |
| 4 | HPV33 HM596530 | . | . | . | . |
| Isolates with identicqal sequences (d) | HPV18 (5 samples) | 6579 | 6581 | 6626 | 6719 | 6749 | 6842 | 6917 |
|---|---|---|---|---|---|---|---|---|
| HPV18 Ref (X05015) | G | T | C | G | G | C | G | |
| Non-synonymous mutations | V384I | |||||||
| 4 | HPV18 HM596525 | . | . | . | . | . | G | . |
| 1 | HPV18 HM596526 | A | C | T | A | A | G | A |
| Isolates with identicqal sequences (e) | HPV45 (4 samples) | 6615 | 6665 | 6676 | 6687 | 6705 | 6816 | 6837 | 6861 | 6867 |
|---|---|---|---|---|---|---|---|---|---|---|
| HPV45 Ref (X74479) | A | A | A | C | G | A | C | G | T | |
| Non-synonymous mutations | N383T | S389G | Q392H | |||||||
| 1 | HPV45 HM596531 | . | . | G | T | C | G | A | A | C |
| 1 | HPV45 HM596532 | . | . | G | . | . | G | . | A | . |
| 1 | HPV45 HM596533 | G | . | G | T | C | G | A | A | . |
| 1 | HPV45 HM596534 | G | C | G | T | C | G | A | A | . |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ntova, C.K.; Kottaridi, C.; Chranioti, A.; Spathis, A.; Kassanos, D.; Paraskevaidis, E.; Karakitsos, P. Genetic Variability and Phylogeny of High Risk HPV Type 16, 18, 31, 33 and 45 L1 Gene in Greek Women. Int. J. Mol. Sci. 2012, 13, 1-17. https://doi.org/10.3390/ijms13010001
Ntova CK, Kottaridi C, Chranioti A, Spathis A, Kassanos D, Paraskevaidis E, Karakitsos P. Genetic Variability and Phylogeny of High Risk HPV Type 16, 18, 31, 33 and 45 L1 Gene in Greek Women. International Journal of Molecular Sciences. 2012; 13(1):1-17. https://doi.org/10.3390/ijms13010001
Chicago/Turabian StyleNtova, Chara Kleio, Christine Kottaridi, Aikaterini Chranioti, Aris Spathis, Dimitrios Kassanos, Evangelos Paraskevaidis, and Petros Karakitsos. 2012. "Genetic Variability and Phylogeny of High Risk HPV Type 16, 18, 31, 33 and 45 L1 Gene in Greek Women" International Journal of Molecular Sciences 13, no. 1: 1-17. https://doi.org/10.3390/ijms13010001