FK506-Binding Protein 22 from a Psychrophilic Bacterium, a Cold Shock-Inducible Peptidyl Prolyl Isomerase with the Ability to Assist in Protein Folding


Abstract
:1. Introduction
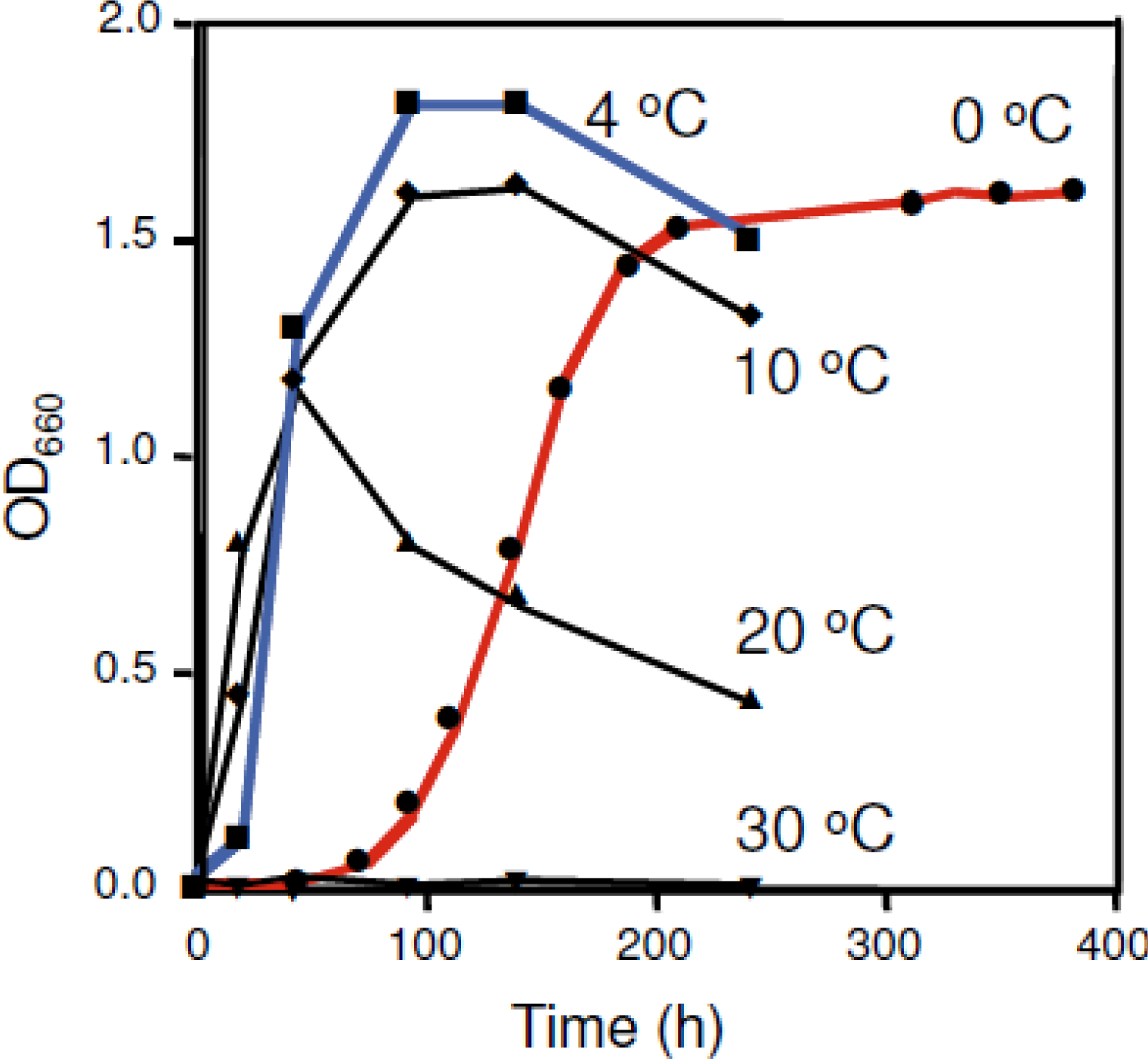
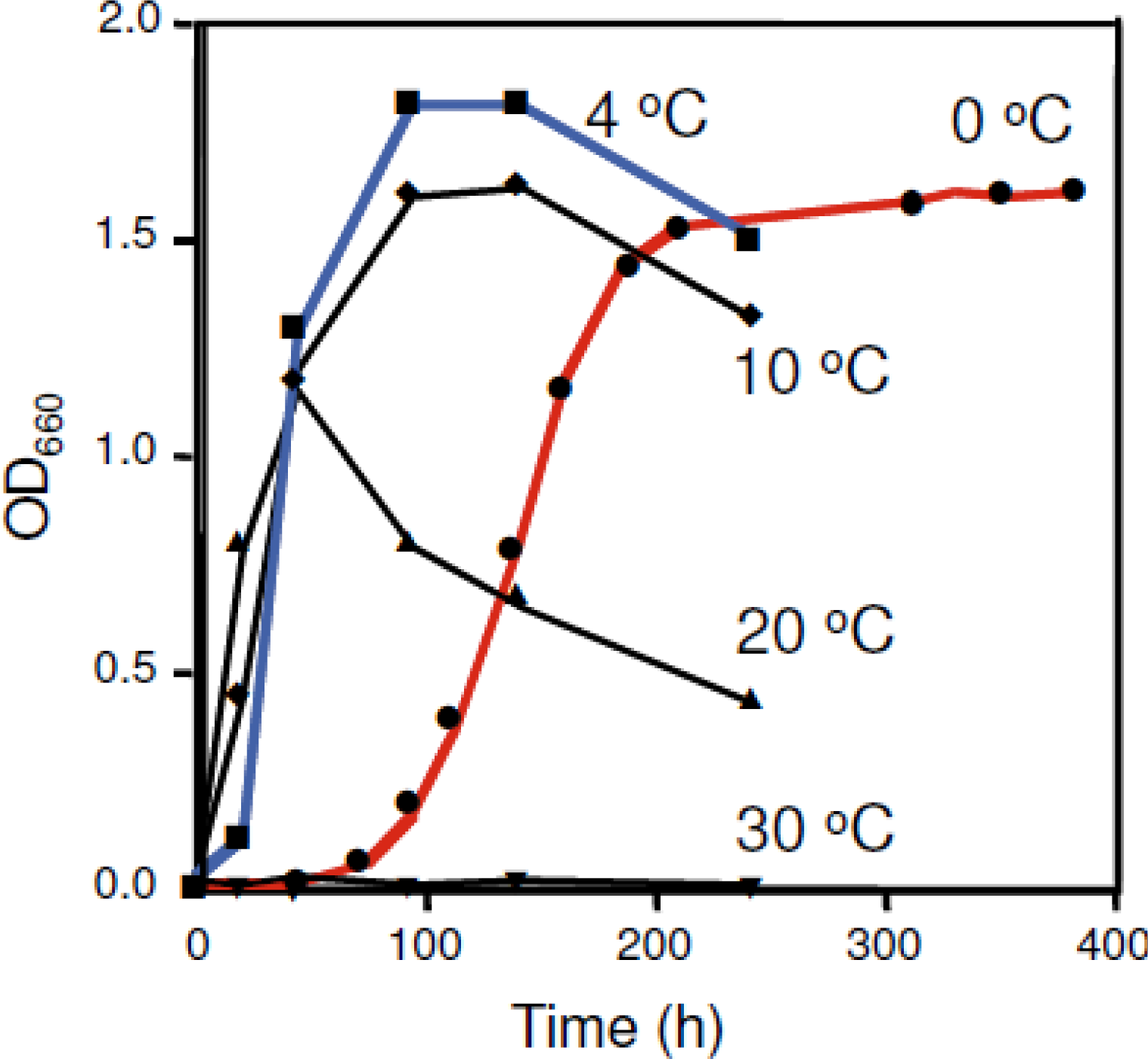
2. Shewanella sp. SIB1 as a Psychrophilic Bacterium
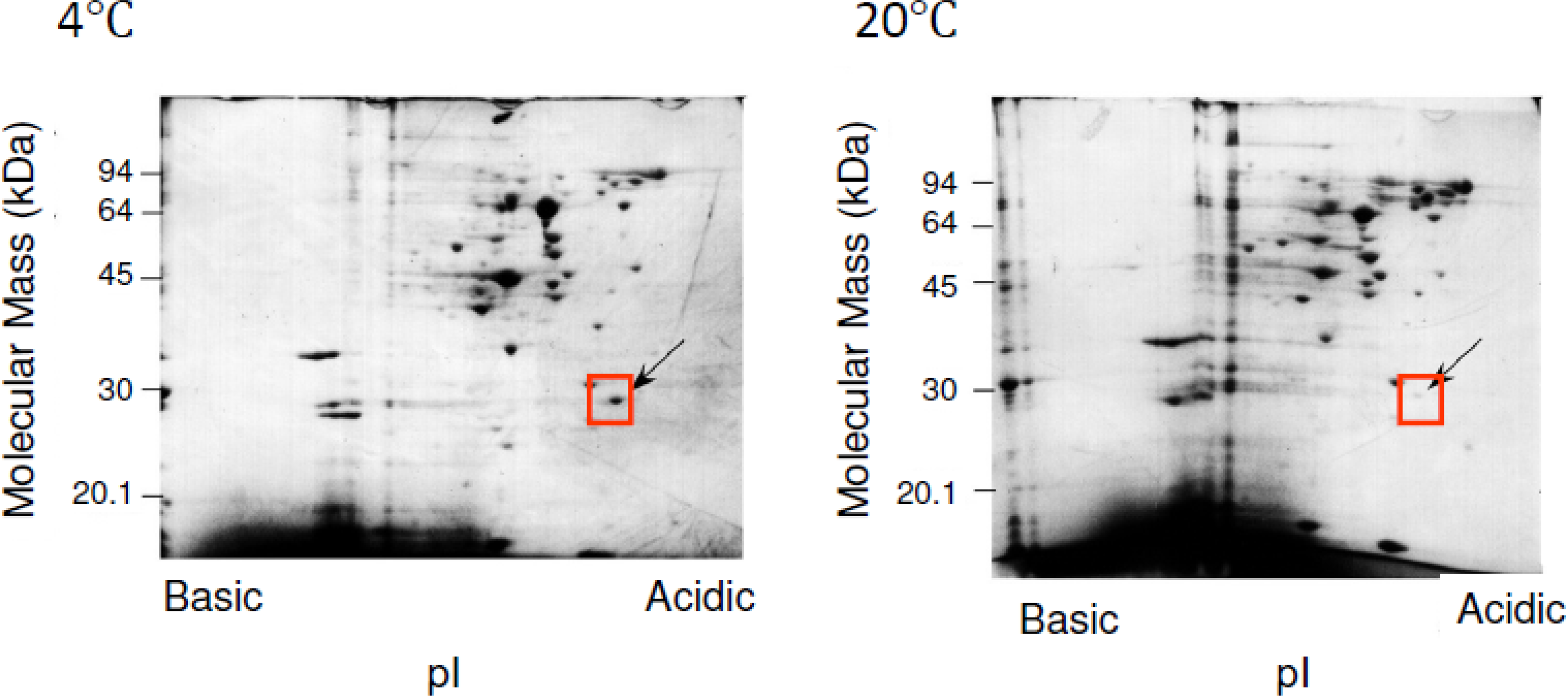
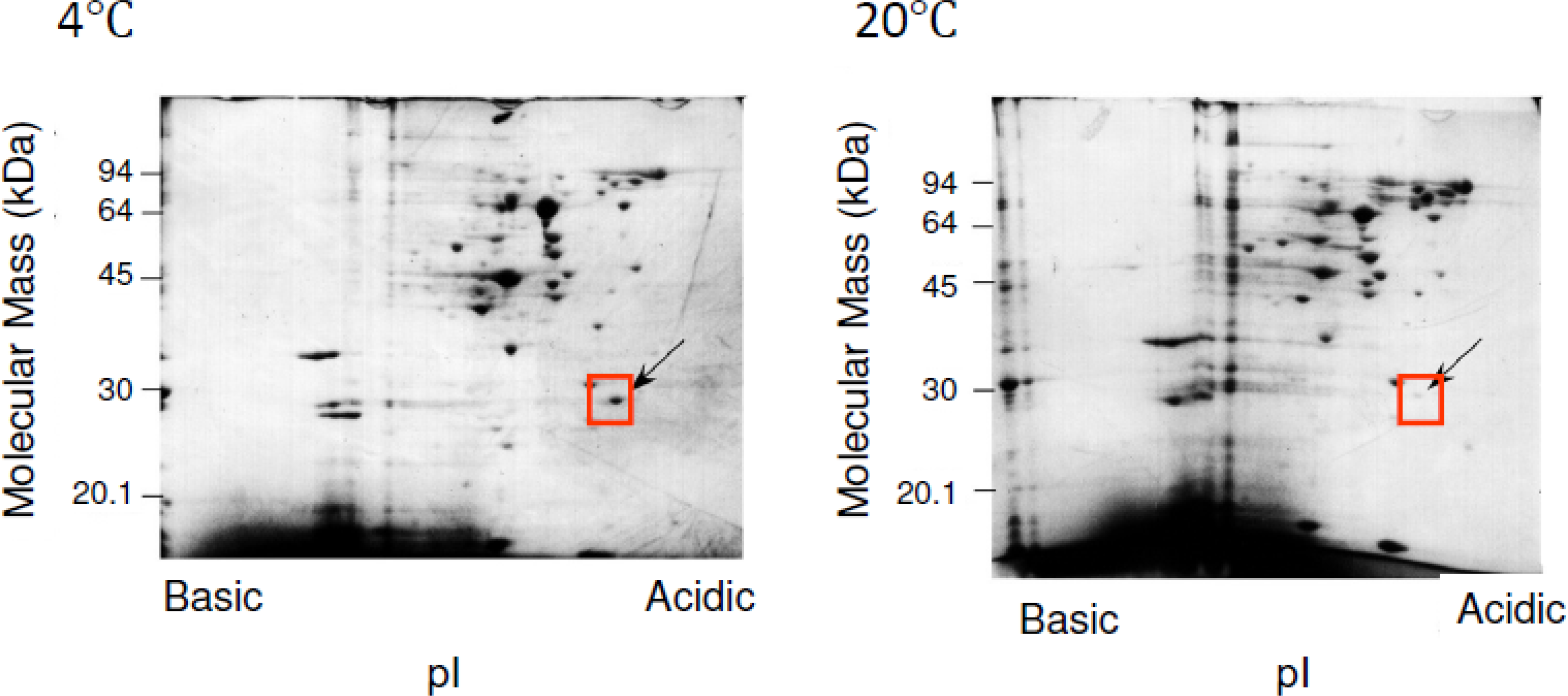
3. SIB1 FKBP22 as a Cold Shock Protein
4. Characterization of the Activity and Stability of SIB1 FKBP22 as a Cold-Adapted Enzyme
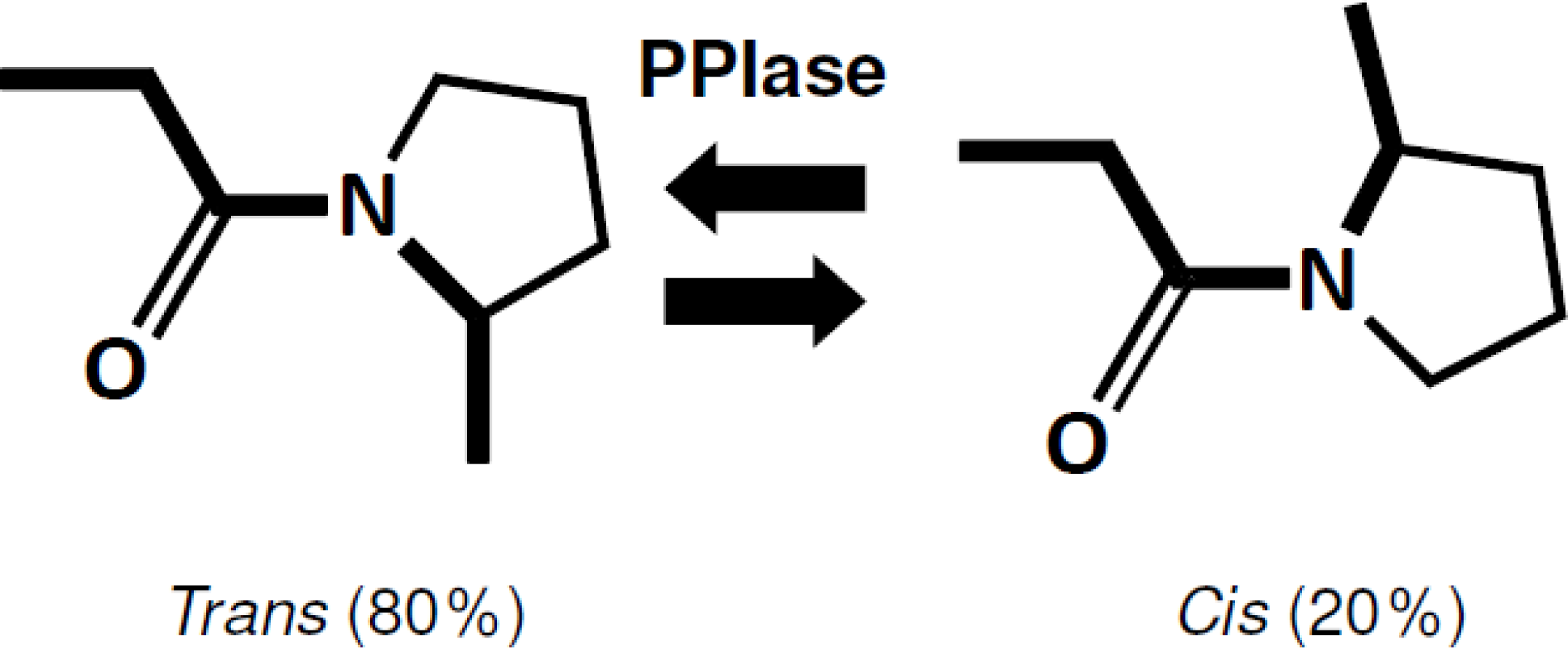
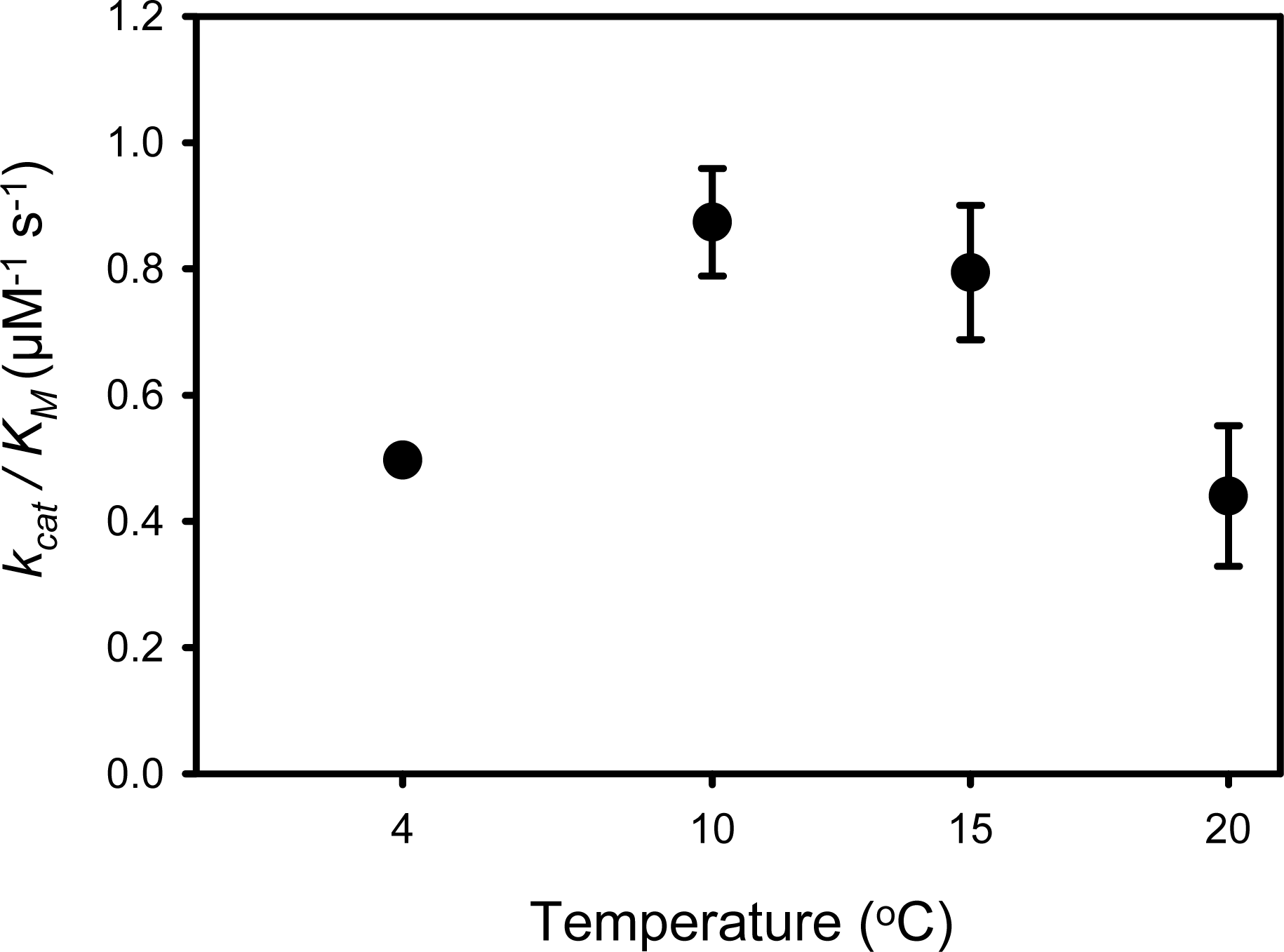
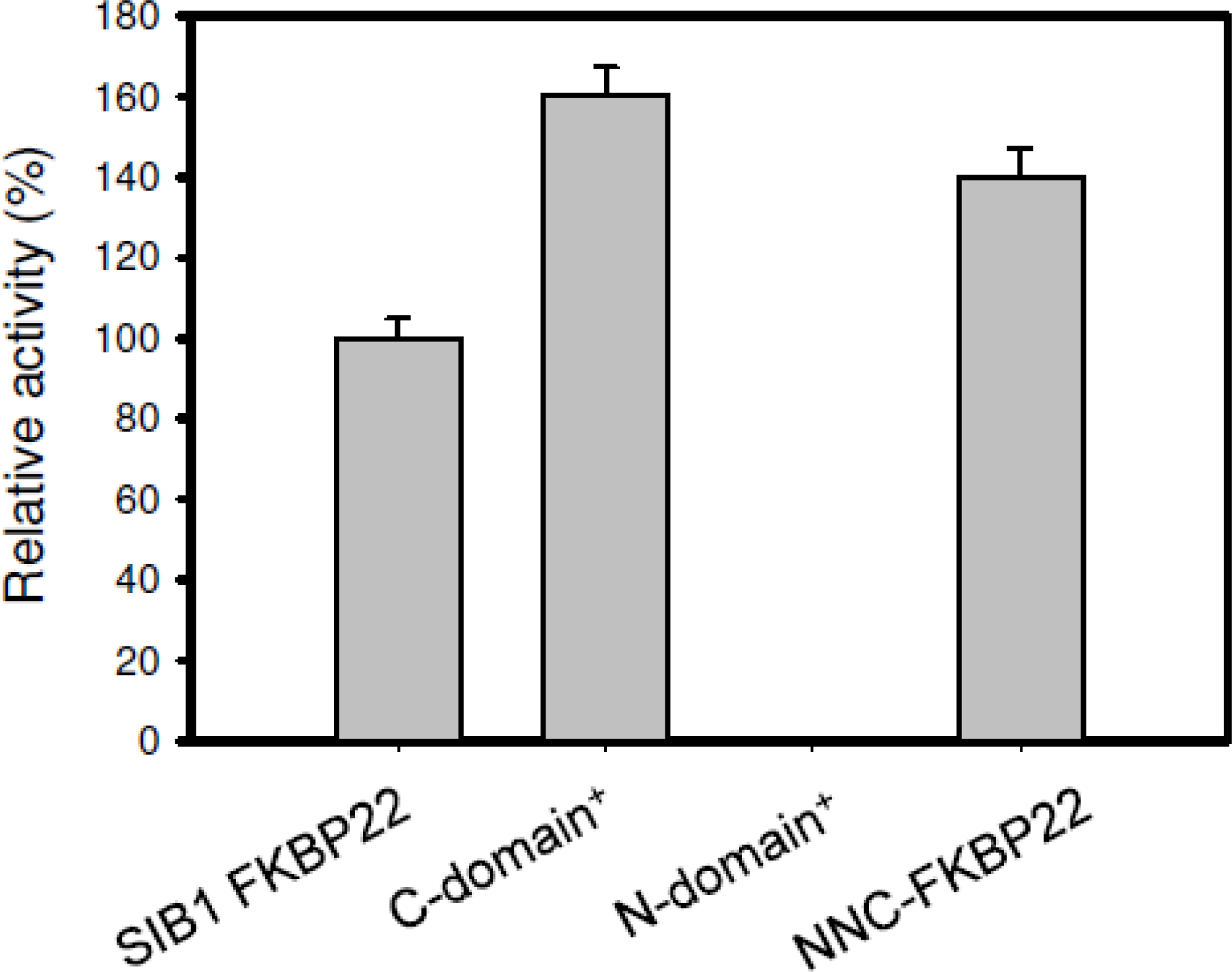
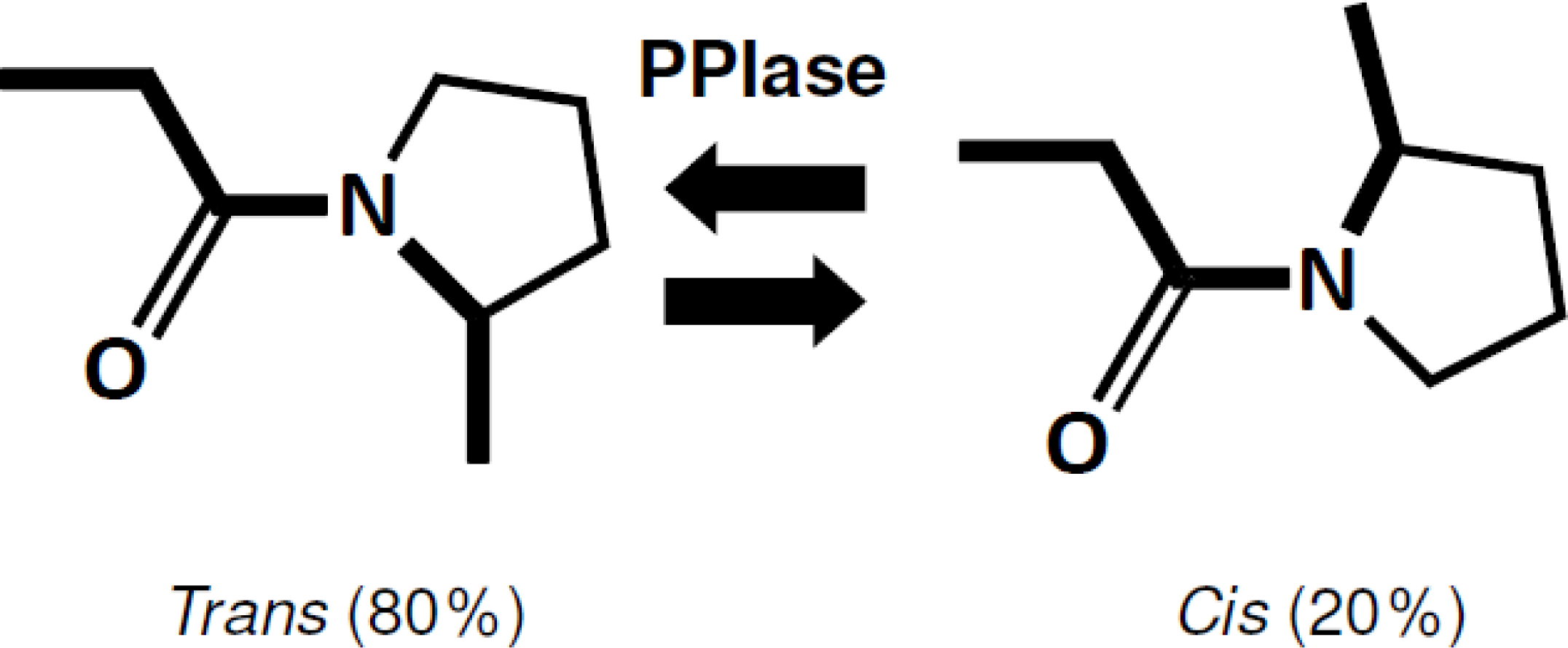
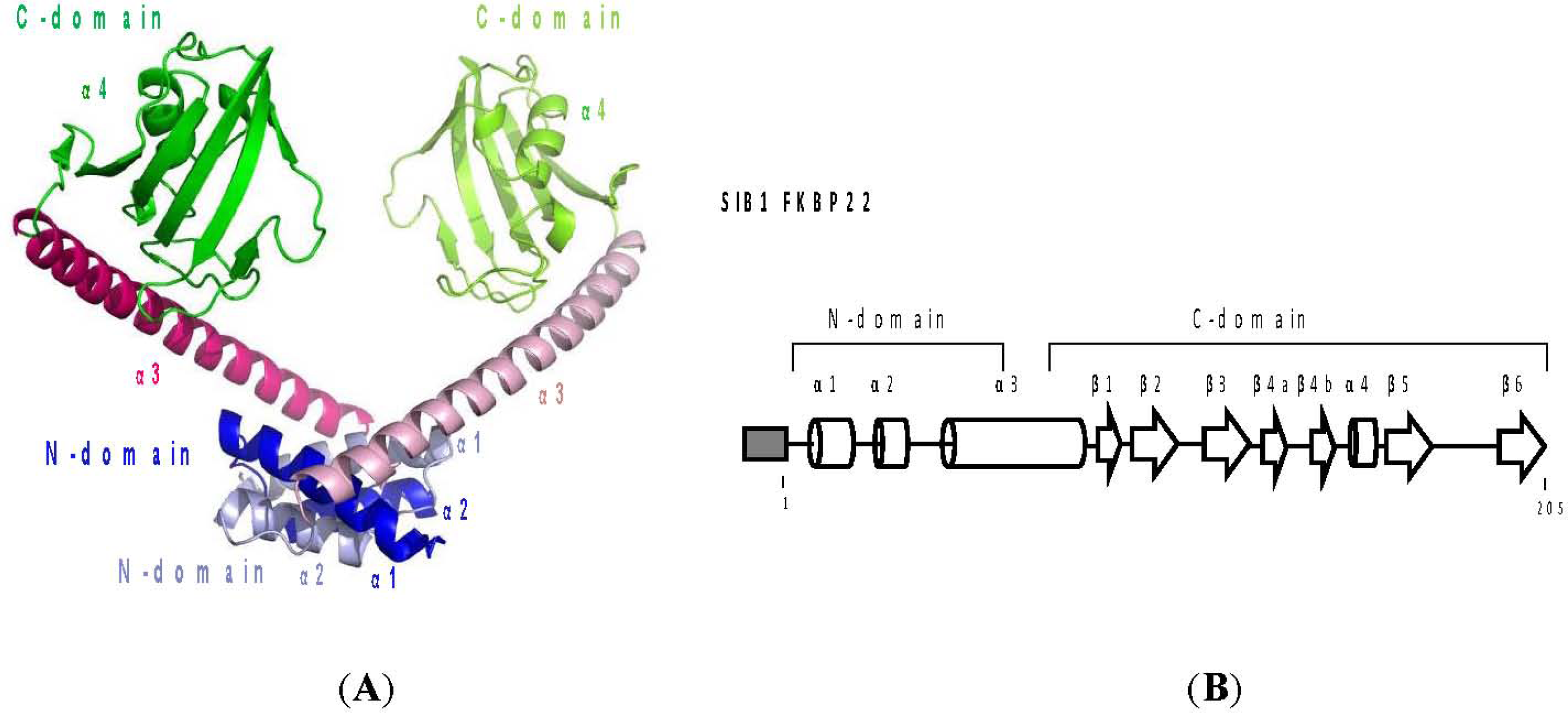
4.1. PPIase Activity of SIB1 FKBP22
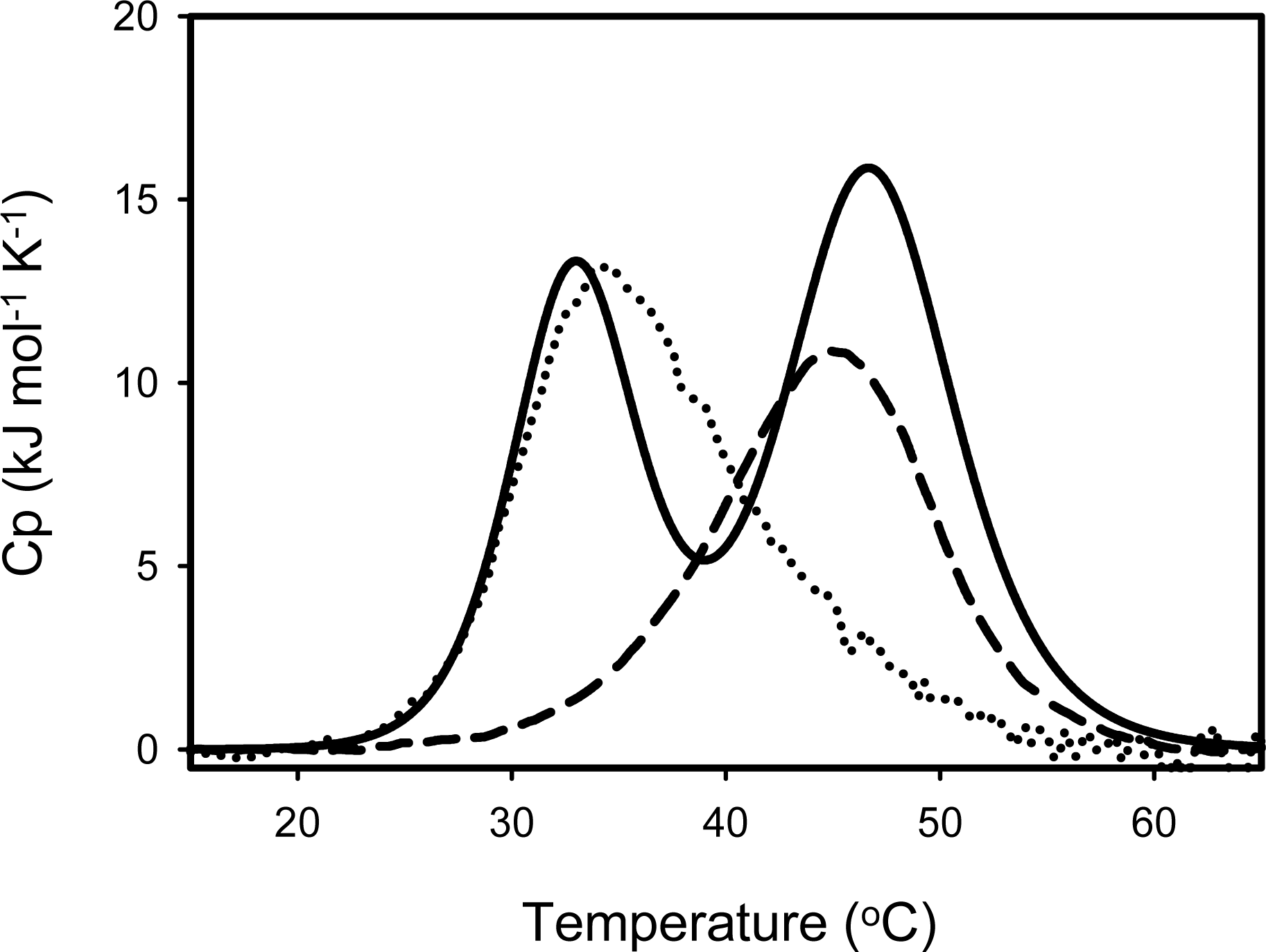
4.2. Stability of SIB1 FKBP22
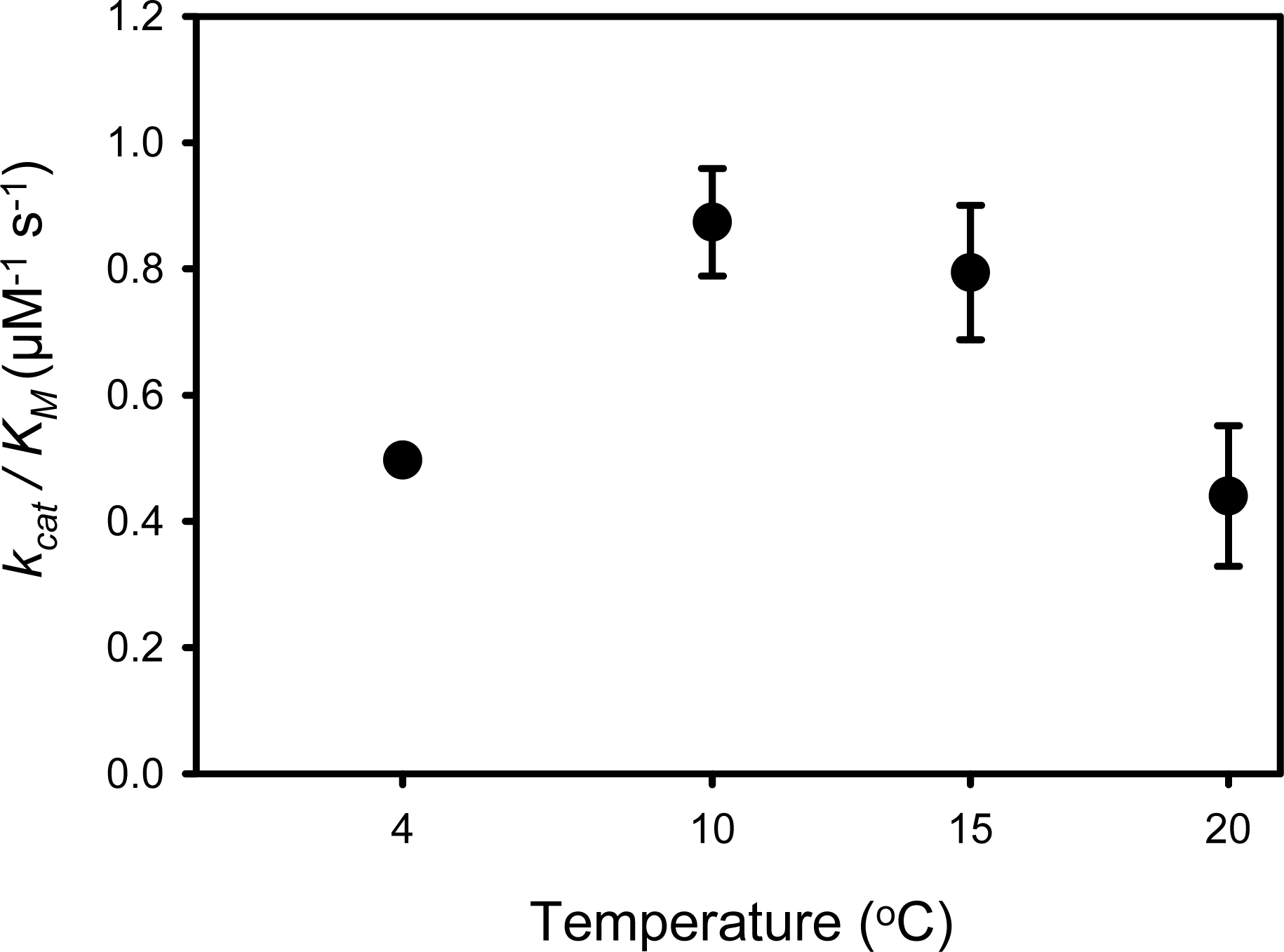
4.3. Relationship between Activity and Stability of SIB1 FKBP22
5. Involvement of SIB1 FKBP22 in Protein Folding
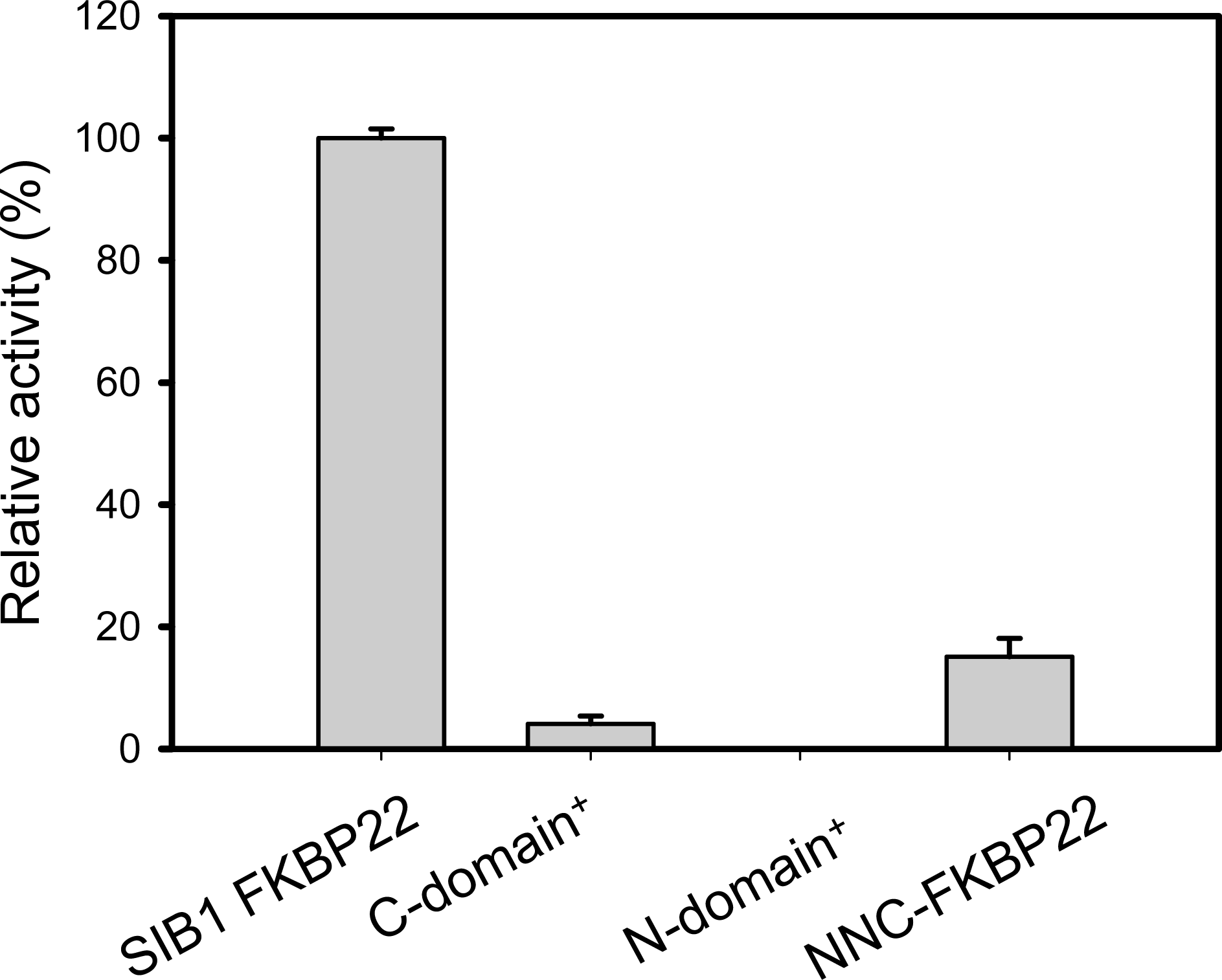
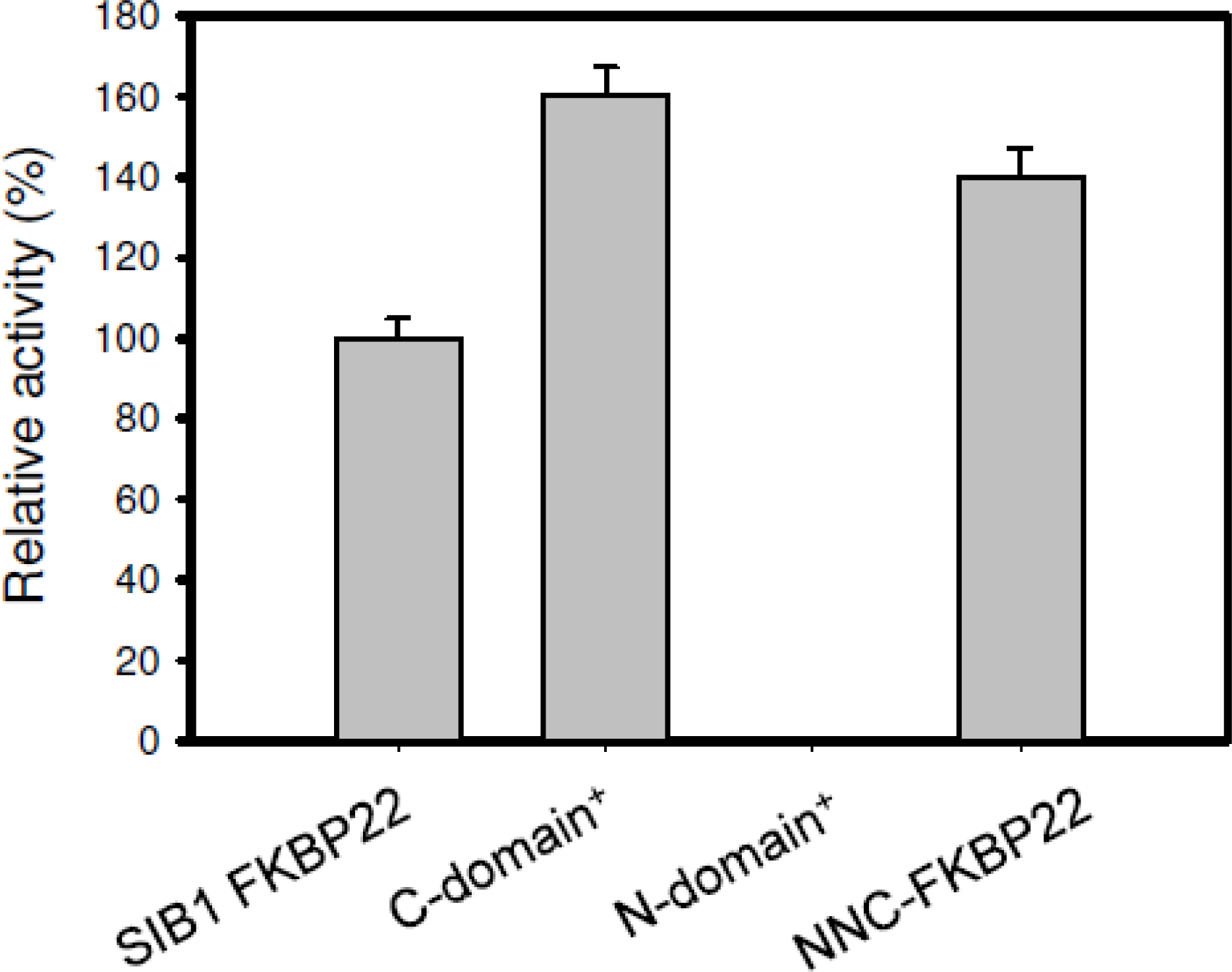
5.1. SIB1 FKBP22 as a Foldase
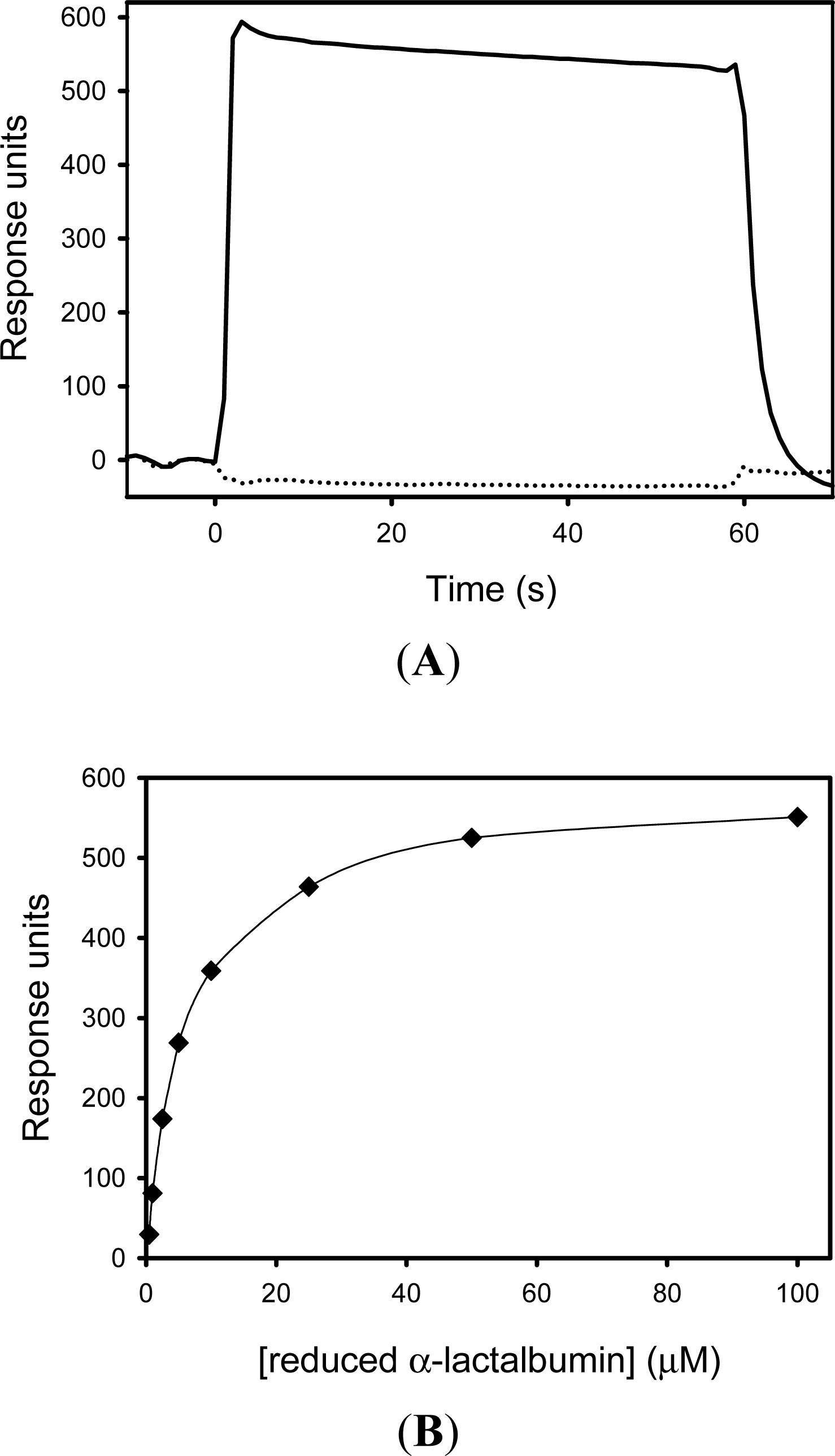
5.2. SIB1 FKBP22 as a Chaperone
5.3. The Relationship between Foldase and Chaperone Activities
6. A Possible Role of SIB1 FKBP22 in Cold Adaptation
7. Other PPIases and Involvement in Cold Adaptation
8. Conclusions
Acknowledgments
References
- Ray, MK. Cold-stress response of low temperature adapted bacteria. Res. Signpost 2006, 37, 1–23. [Google Scholar]
- Hendrik, JP; Hartl, FU. Molecular chaperone functions of heat-shock proteins. Annu. Rev. Biochem 1993, 62, 349–384. [Google Scholar]
- Stetter, KO. Hyperthermophilic prokaryotes. FEMS Microbiol. Rev 1996, 1, 149–158. [Google Scholar]
- Cavicchioli, R; Charlton, T; Ertan, H; Omar, SM; Sidiqui, KS; Williams, TJ. Biotechnological uses of enzymes from psychrophiles. Microb. Biotechnol 2011, 4, 449–460. [Google Scholar]
- Kasana, RC; Gulati, A. Cellullase from psychrophilic microorganism: A review. J. Basic Microbiol 2001, 5, 1–8. [Google Scholar]
- Kasana, RC. Protease from psychrotrophs: An overview. Crit. Rev. Microbiol 2010, 2, 134–145. [Google Scholar]
- Joseph, B; Ramteke, PW; Thomas, G. Cold active microbial lipases: some hot issues and recent developments. Biotechnol. Adv 2008, 5, 457–470. [Google Scholar]
- Suzuki, Y; Mizutani, Y; Tsuji, T; Ohtani, N; Takano, K; Haruki, M; Morikawa, M; Kanaya, S. Gene cloning, overproduction, and characterization of thermolabile alkaline phosphatase from a psychrotrophic bacterium. Biosci. Biotechnol. Biochem 2005, 69, 364–373. [Google Scholar]
- Feller, G; Gerday, C. Psychrophylic enzymes: Hot topics in cold adaptation. Nat. Rev. Microbiol 2003, 1, 200–208. [Google Scholar]
- Piette, F; D’Amico, S; Struvay, C; Mazzucchelli, G; Renaut, J; Tutino, ML; Danchin, A; Leprince, P; Feller, G. Proteomic of life at low temperatures: trigger factor is the primary chaperone in the Antartic bacterium Pseudoalteromonas haloplanktis TAC125. Mol. Microb 2010, 76, 120–132. [Google Scholar]
- Jacob, RP; Schmid, FX. Energetic coupling between native-state prolyl isomerization and conformational protein folding. J. Mol. Biol 2008, 377, 1560–1575. [Google Scholar]
- Kay, JE. Structure-function relationships in the FK5-506-binding protein (FKBP) family of peptidylprolyl cis-trans isomerases. Biochem. J 1996, 314, 361–385. [Google Scholar]
- Wang, CC; Tsou, CL. Enzymes as chaperones and chaperones as enzymes. FEBS Lett 1998, 425, 382–384. [Google Scholar]
- Theuerkorn, M; Fischer, G; Schiene-Fischer, C. Prolyl cis/trans isomerase signaling pathway in cancer. Curr. Opin. Pharmacol 2011, 11, 1–7. [Google Scholar]
- Fanganel, J; Fischer, G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front. Biosci 2004, 9, 3453–3478. [Google Scholar]
- Edlich, F; Fischer, G. Pharmacological targeting of catalyzed protein folding: The example of peptide bond cis/trans isomerases. Handb. Exp. Pharmacol 2006, 172, 359–404. [Google Scholar]
- Scholz, C; Eckert, B; Hagn, F; Schaarschmidt, P; Balbach, J; Schmid, FX. SlyD proteins from different species exhibit high prolyl isomerase and chaperone activities. Biochemistry 1996, 45, 20–33. [Google Scholar]
- Weininger, U; Haupt, C; Schweimer, K; Graubner, W; Kovermann, M; Bruser, T; Szholz, C; Scaarshmidt, P; Zoldak, G; Schmid, FX; et al. NMR solution structure of SlyD from Escherichia coli: Spatial separation of prolyl isomerase and chaperone function. J. Mol. Biol 2009, 387, 295–305. [Google Scholar]
- Saul, FA; Arie, JP; Vulliez-le Normand, B; Kahn, R; Betto, NJM; Bentley, GA. Structure and functional studies of FkpA from Escherichia coli, a cis/trans peptidyl-prolyl isomerase with chaperone activity. J. Mol. Biol 2004, 335, 595–608. [Google Scholar]
- Suzuki, R; Nagata, K; Yumoto, F; Kawakami, M; Nemoto, N; Furutani, M; Adachi, K; Maruyama, T; Tanokura, M. Three-dimensional solution structure of an archaeal FKBP with a dual function of peptidyl prolyl cis-trans isomerase and chaperone-like activities. J. Mol. Biol 2003, 328, 1149–1160. [Google Scholar]
- Li, ZY; Liu, CP; Zhu, LQ; Jing, GZ; Zhou, JM. The chaperone activity of trigger factor is distinct from its isomerase activity during co-expression with adenylate kinase in Escherichia coli. FEBS Lett 2001, 506, 108–112. [Google Scholar]
- Pirkl, F; Buchner, J. Functional analysis of the Hsp90-associated human peptidyl prolyl cis/trans isomerases FKBP51, FKBP52 and Cyp40. J. Mol. Biol 2001, 308, 795–806. [Google Scholar]
- Martinez-Heckert, E; Hendrickson, WA. Structural analysis of protein folding by the long-chain archeal chaperone FKBP26. J. Mol. Biol 2011, 3, 450–464. [Google Scholar]
- Budde, I; Steil, L; Scharf, C; Volker, U; Bremer, E. Adaptation of Bacillus subtilis to growth at low temperature: A combined transcriptomic and proteomic appraisal. Microbiology 2006, 3, 831–853. [Google Scholar]
- Jaremko, L; Jaremko, M; Elfaki, I; Mueller, JW; Ejchart, A; Bayer, P; Zhukov, I. Structure and dynamics of the first archaeal parvulin reveal a new functionally important loop in parvulin-type. J. Biol. Chem 2011, 8, 6554–6565. [Google Scholar]
- Suzuki, Y; Haruki, M; Takano, K; Morikawa, M; Kanaya, S. Possible involvement of an FKBP family member protein from a psychrotrophic bacterium Shewanella sp. SIB1 in cold-adaptation. Eur. J. Biochem 2004, 271, 1372–1381. [Google Scholar]
- Kato, T; Haruki, M; Imanaka, T; Morikawa, M; Kanaya, S. Isolation and characterization of psychotrophic bacteria from oil-reservoir water and oil sands. Appl Microbiol Biotechnol 2001, 55, 794–800. [Google Scholar]
- Kulakova, L; Galkin, A; Kurihara, T; Yoshimura, T; Esaki, N. Cold-active serine alkaline protease from the psychrotrophic bacterium Shewanella strain Ac10: Gene cloning and enzyme purification and characterization. Appl. Environ. Microbiol 1999, 65, 611–617. [Google Scholar]
- Ohtani, N; Haruki, M; Morikawa, M; Kanaya, S. Heat labile ribonuclease HI from a psychrotrophic bacterium: gene cloning, characterization, and site-directed mutagenesis. Protein Eng 2001, 14, 975–982. [Google Scholar]
- Chon, H; Tadokoro, T; Ohtani, N; Koga, Y; Takano, K; Kanaya, S. Identification of RNase HII from psychrotrophic bacterium, Shewanella sp. SIB1 as a high-activity type RNase H. FEBS J 2006, 273, 2264–2275. [Google Scholar]
- Sato, A; Yokotani, S; Tadokoro, T; Tanaka, S; Angkawidjaja, C; Koga, Y; Takano, K; Kanaya, S. Crystal structure of stable protein CutA1 from psychrotrophic bacterium Shewanella sp. SIB1. J. Synchrotron Radiat 2011, 18, 6–10. [Google Scholar]
- Soni, KA; Nannapaneni, R; Tasara, T. The contribution of transcriptomic and proteomic analysis in elucidating stress adaptation responses of Listeria monocytogenes. Foodborne Pathog. Dis 2011, 8, 843–852. [Google Scholar]
- Tasara, T; Stephan, R. Cold stress tolerance of Listeria monocytogenes: A review of molecular adaptive mechanisms and food safety implications. J. Food. Prot 2006, 6, 1473–1484. [Google Scholar]
- Garnier, M; Matamoros, S; Chevret, D; Pilet, MF; Leori, F; Tresse, O. Adaptation to cold and proteomic responses of the psychrotrophic biopreservative Lactococcus piscium strain CNCM I-4031. Appl. Environ. Microbiol 2010, 24, 8011–8018. [Google Scholar]
- Ting, L; Williams, TJ; Cowley, MJ; Lauro, FM; Guilhaus, M; Raftery, MJ; Cavicchioli, R. Cold adaptation in the marine bacterium, Sphingopyxis alaskensis, assesed using quantitative proteomic. Environ. Microbiol 2010, 10, 2658–2676. [Google Scholar]
- Rahfeld, JU; Rucknagel, KP; Stoller, G; Horne, SM; Schierhorn, A; Young, KD; Fischer, G. Isolation and amino acid sequence of a new 22-kDa FKBP-like peptidyl-prolyl cis/transisomerase of Escherichia coli Similarity to Mip-like proteins of pathogenic bacteria. J. Biol. Chem 1996, 271, 22130–22138. [Google Scholar]
- Horne, SM; Young, KD. Escherichia coli and other species of the Enterobacteriaceae encode a protein similar to the family of Mip-like FK506-binding proteins. Arch. Microbiol 1995, 165, 357–365. [Google Scholar]
- Engleberg, NC; Carter, C; Weber, DR; Cianciotto, NP; Eisenstein, BI. DNA sequence of mip, a Legionella pneumophila gene associated with macrophage infectivity. Infect. Immun 1989, 57, 1263–1270. [Google Scholar]
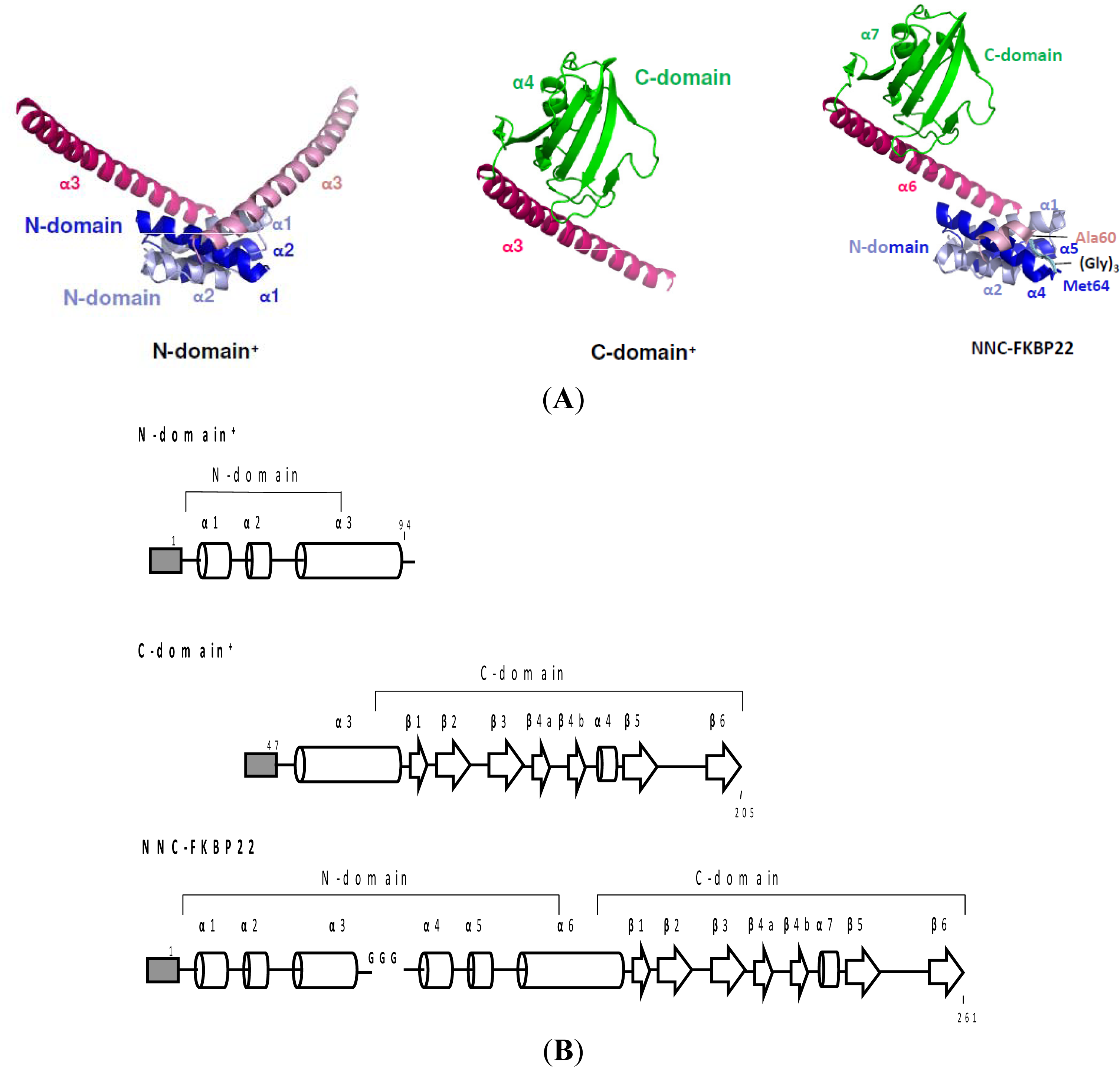
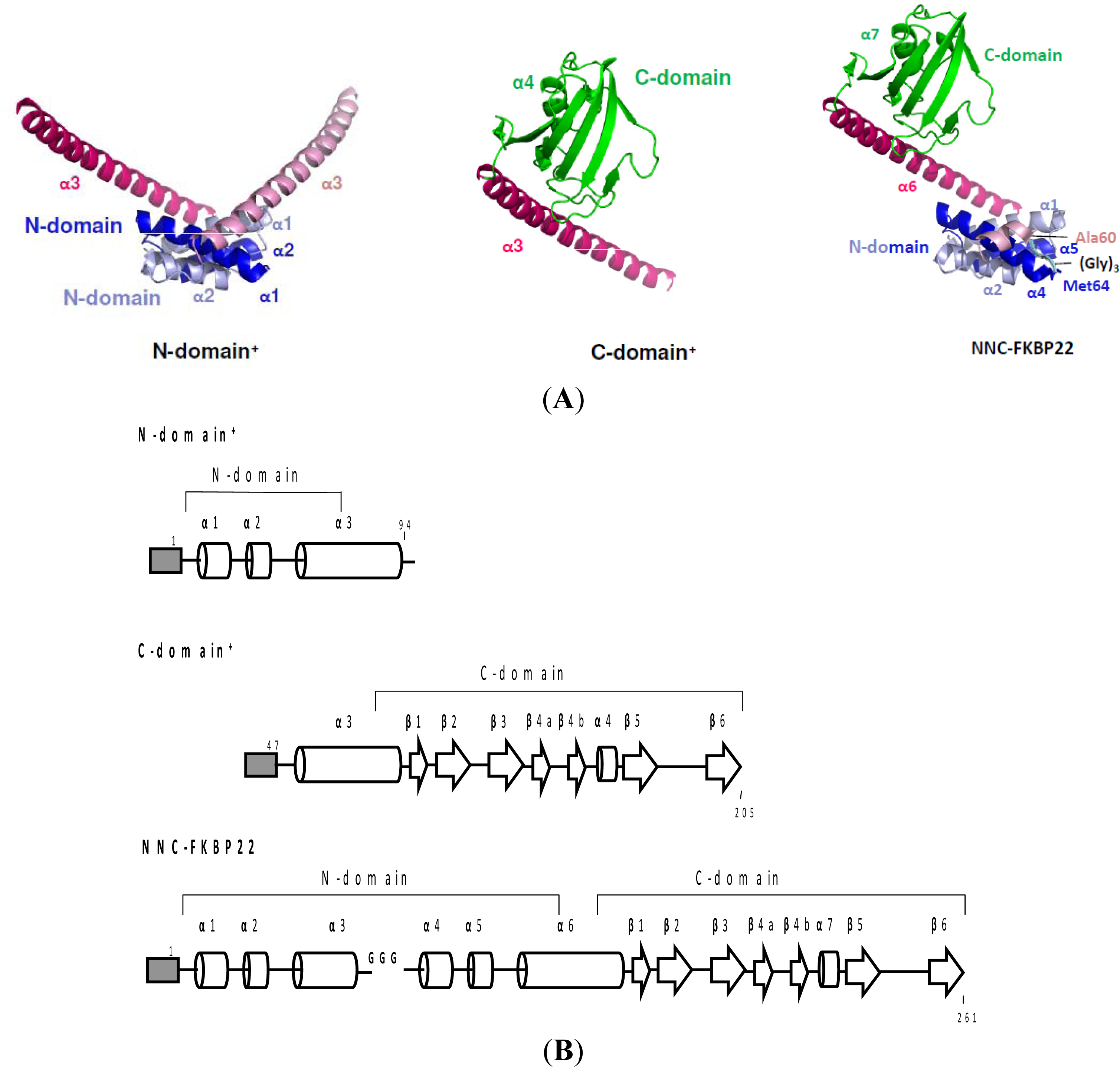
- Suzuki, Y; Takano, K; Kanaya, S. Stabilities and activities of the N- and C-domains of FKBP22 from a psychrotrophic bacterium overproduced in E. coli. FEBS J 2005, 272, 632–642. [Google Scholar]
- Budiman, C; Bando, K; Angkawidjaja, C; Koga, Y; Takano, K; Kanaya, S. Engineering of monomeric FK506-binding protein 22 with peptidyl prolyl cis-trans isomerase: Importance of V-shaped dimeric structure for binding to protein substrate. FEBS J 2009, 276, 4091–4101. [Google Scholar]
- Tradler, T; Stoller, G; Rucknagel, KP; Schierhorn, A; Rahfeld, JU; Fischer, G. Comparative mutational analysis of peptidyl prolyl cis/trans isomerases: active sites of Escherichia coli trigger factor and human FKBP12. FEBS Lett 1997, 40, 184–190. [Google Scholar]
- D’Amico, S; Marx, JC; Gerday, C; Feller, G. Activity-stability relationships in extremophilic enzymes. J. Biol. Chem 2003, 278, 7891–7896. [Google Scholar]
- Collins, T; Meuwis, MA; Gerday, C; Feller, G. Activity, stability and flexibility in glycosidases adapted to extreme thermal environments. J Mol Biol 2003, 328, 419–428. [Google Scholar]
- Fields, PA; Somero, GN. Hot spots in cold adaptation: localized increases in conformational flexibility in lactate dehydrogenase A4 orthologs of Antarctic notothenioid fishes. Proc Natl Acad Sci USA 1998, 95, 11476–11481. [Google Scholar]
- Kiefhaber, T; Quaas, R; Hahn, U; Schmid, FX. Folding of ribonuclease T1. 1. Existence of multiple unfolded states created by proline isomerization. Biochemistry 1990, 29, 3051–3061. [Google Scholar]
- Kiefhaber, T; Quaas, R; Hahn, U; Schmid, FX. Folding of ribonuclease T1. 2. Kinetic models for the folding and unfolding reactions. Biochemistry 1990, 29, 3061–3070. [Google Scholar]
- Schindler, T; Mayr, LM; Landt, O; Hahn, U; Schmid, FX. The role of a trans-proline in the folding mechanism of ribonuclease T1. Eur. J. Biochem 1996, 241, 516–524. [Google Scholar]
- Knappe, TA; Eckert, B; Schaarschmidt, P; Scholz, C; Schmid, FX. Insertion of a chaperone domain converts FKBP12 into a powerful catalyst of protein folding. J. Mol. Biol 2007, 368, 1458–1468. [Google Scholar]
- Hayer-Hartl, MK; Ewbank, JJ; Creighton, TE; Hartl, FU. Conformational specificity of the chaperonin GroEL for the compact folding intermediates of -lactalbumin. EMBO J 1994, 13, 3192–3202. [Google Scholar]
- Okazaki, A; Ikura, T; Nikaido, K; Kuwajima, K. The chaperonin GroEL does not recognize apo-α-lactalbumin in the molten globule state. Nat. Struct. Biol 1994, 1, 439–446. [Google Scholar]
- Scholz, C; Stoller, G; Zarnt, T; Fischer, G; Schmid, FX. Cooperation of enzymatic and chaperone functions of trigger factor in the catalysis of protein folding. EMBO J 1997, 16, 54–58. [Google Scholar]
- Ramm, K; Pluckthun, A. High enzymatic activity and chaperone function are mechanistically related features to the dimeric E. coli peptidyl-prolyl-isomerase FkpA. J. Mol. Biol 2001, 310, 485–498. [Google Scholar]
- Acharya, KR; Stuart, DI; Walker, NP; Lewis, M; Philips, DC. Refined structure of baboon -lactalbumin at 1.7 Å resolution. Comparison with C-type lysozyme. J. Mol. Biol 1989, 1, 99–127. [Google Scholar]
- Acharya, KR; Ren, JS; Sturart, DI; Philips, D; Fenna, RE. Crystal structure of human -lactalbumin at 1.7 Å resolution. J. Mol. Biol 1991, 2, 571–581. [Google Scholar]
- Kuwajima, K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins 1989, 6, 87–103. [Google Scholar]
- Suzuki, Y; Win, OY; Koga, Y; Takano, K; Kanaya, S. Binding analysis of a psychrotrophic FKBP22 to a folding intermediate of protein using surface plasmon resonance. FEBS Lett 2005, 579, 5781–5784. [Google Scholar]
- Arie, JP; Sassoon, N; Betton, JM. Chaperone function of FkpA, a heat shock prolyl isomerase, in the periplasm of Escherichia coli. Mol Microbiol 2001, 39, 199–210. [Google Scholar]
- Hu, K; Galius, V; Pervushin, K. Structural plasticity of peptidyl−prolyl isomerase sFkpA is a key to its chaperone function as revealed by solution NMR. Biochemistry 2006, 45, 11983–11991. [Google Scholar]
- Strocchi, M; Ferrer, M; Timmis, KN; Golyshin, PN. Low temperature-induced system failure in Escherichia coli: insight from rescue by cold-adapted chaperones. Proteomics 2006, 6, 193–206. [Google Scholar]
- Lu, KP; Finn, G; Lee, TH; Nicholson, LL. Prolyl cis-trans isomerization as molecular timer. Nat. Chem. Biol 2007, 3, 619–629. [Google Scholar]
- Ideno, A; Yoshida, T; Iida, T; Furutani, M; Maruyama, T. FK506-binding protein of hyperthermophilic archeum, Thermococcus sp. KS-1, a cold-shock-inducible peptidyl-prolyl cis-trans isomerase with activities to trap and refold denatured proteins. Biochem. J 2001, 357, 465–471. [Google Scholar]
- Kandror, O; Goldberg, AL. Trigger factor is induced upon cold shock and enhances viability of Escherichia coli at low temperatures. Proc. Natl. Acad. Sci. USA 1997, 94, 4978–4981. [Google Scholar]
- Pissavin, C; Hugouvieux-Cotte-Pattat, N. Characterization of a periplasmic peptidyl-prolyl cis-trans isomerase in Erwinia chrysanthemi. FEMS Microbiol. Lett 1997, 157, 59–65. [Google Scholar]
- Missiakas, D; Raina, S. Protein folding in the bacterial periplasm. J. Bacteriol 1997, 179, 2465–2471. [Google Scholar]
- Rouviere, PE; Gross, CA. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev 1996, 10, 3170–3182. [Google Scholar]
- Basak, C; Pathak, SK; Bhattacharyya, A; Pathak, S; Basu, J; Kundu, M. The secreted peptidyl prolyl cis,trans-isomerase HP0175 of Helicobacter pylori induces apoptosis of gastric epithelial cells in a TLR4- and apoptosis signal-regulating kinase 1-dependent manner. J. Immunol 2005, 174, 5672–5680. [Google Scholar]
- DebRoy, S; Aragon, V; Kurtz, S; Cianciotto, NP. Legionella pneumophila Mip, a surface-exposed peptidylproline cis-trans-isomerase, promotes the presence of phospholipase C-like activity in culture supernatants. Infect. Immun 2006, 74, 5152–5160. [Google Scholar]
- Soderberg, MA; Rossier, O; Cianciotto, NP. The type II protein secretion system of Legionella pneumophila promotes growth at low temperatures. J. Bacteriol 2004, 186, 3712–3720. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Transition | Tm (°C) |
|---|---|---|
| SIB1 FKBP22 | First | 32.5 |
| Second | 46.4 | |
| C-domain+ | 35.6 | |
| N-domain+ | 44.4 |
| KD Values (μM) | |
|---|---|
| SIB1 FKBP22 | 2.2 |
| N-domain+ | 3.4 |
| C-domain+ | not detected |
| NNC-FKBP22 | 14.4 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Budiman, C.; Koga, Y.; Takano, K.; Kanaya, S. FK506-Binding Protein 22 from a Psychrophilic Bacterium, a Cold Shock-Inducible Peptidyl Prolyl Isomerase with the Ability to Assist in Protein Folding. Int. J. Mol. Sci. 2011, 12, 5261-5284. https://doi.org/10.3390/ijms12085261
Budiman C, Koga Y, Takano K, Kanaya S. FK506-Binding Protein 22 from a Psychrophilic Bacterium, a Cold Shock-Inducible Peptidyl Prolyl Isomerase with the Ability to Assist in Protein Folding. International Journal of Molecular Sciences. 2011; 12(8):5261-5284. https://doi.org/10.3390/ijms12085261
Chicago/Turabian StyleBudiman, Cahyo, Yuichi Koga, Kazufumi Takano, and Shigenori Kanaya. 2011. "FK506-Binding Protein 22 from a Psychrophilic Bacterium, a Cold Shock-Inducible Peptidyl Prolyl Isomerase with the Ability to Assist in Protein Folding" International Journal of Molecular Sciences 12, no. 8: 5261-5284. https://doi.org/10.3390/ijms12085261