A Newly Isolated Thermostable Lipase from Bacillus sp.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
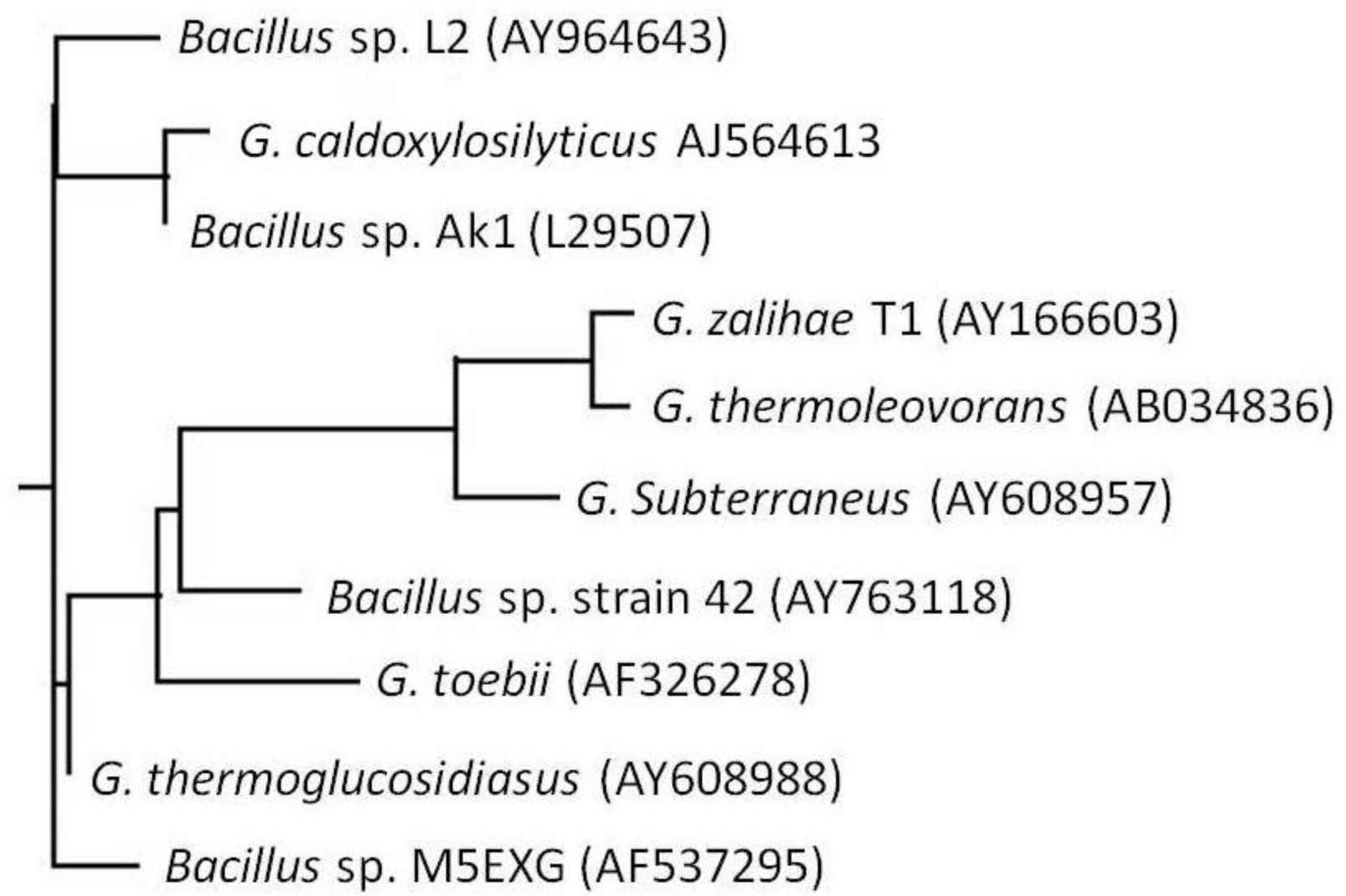
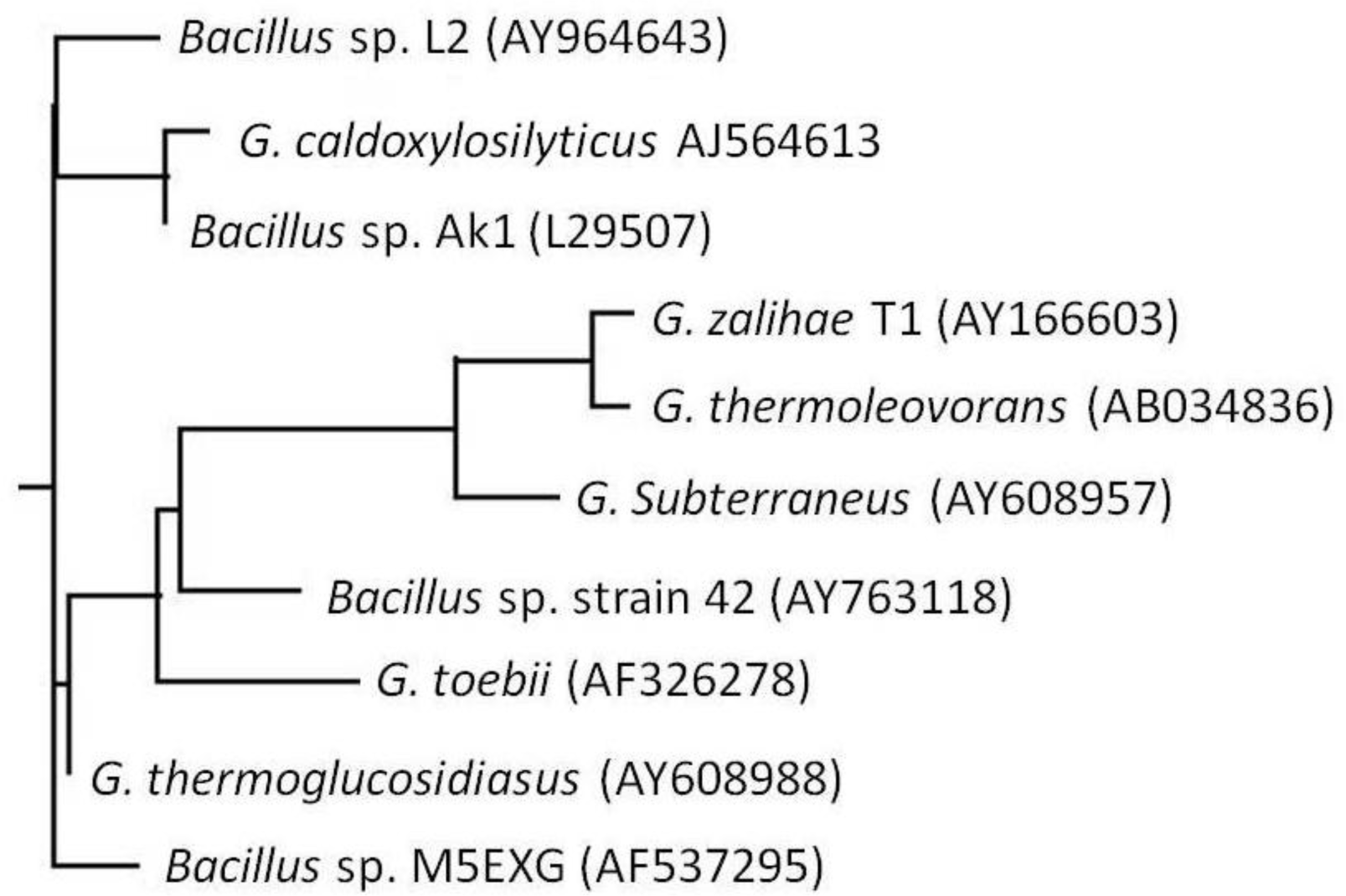
2.1. Bacterial Identification
2.2. Isolation and PCR Cloning of the Thermostable L2 Lipase Gene
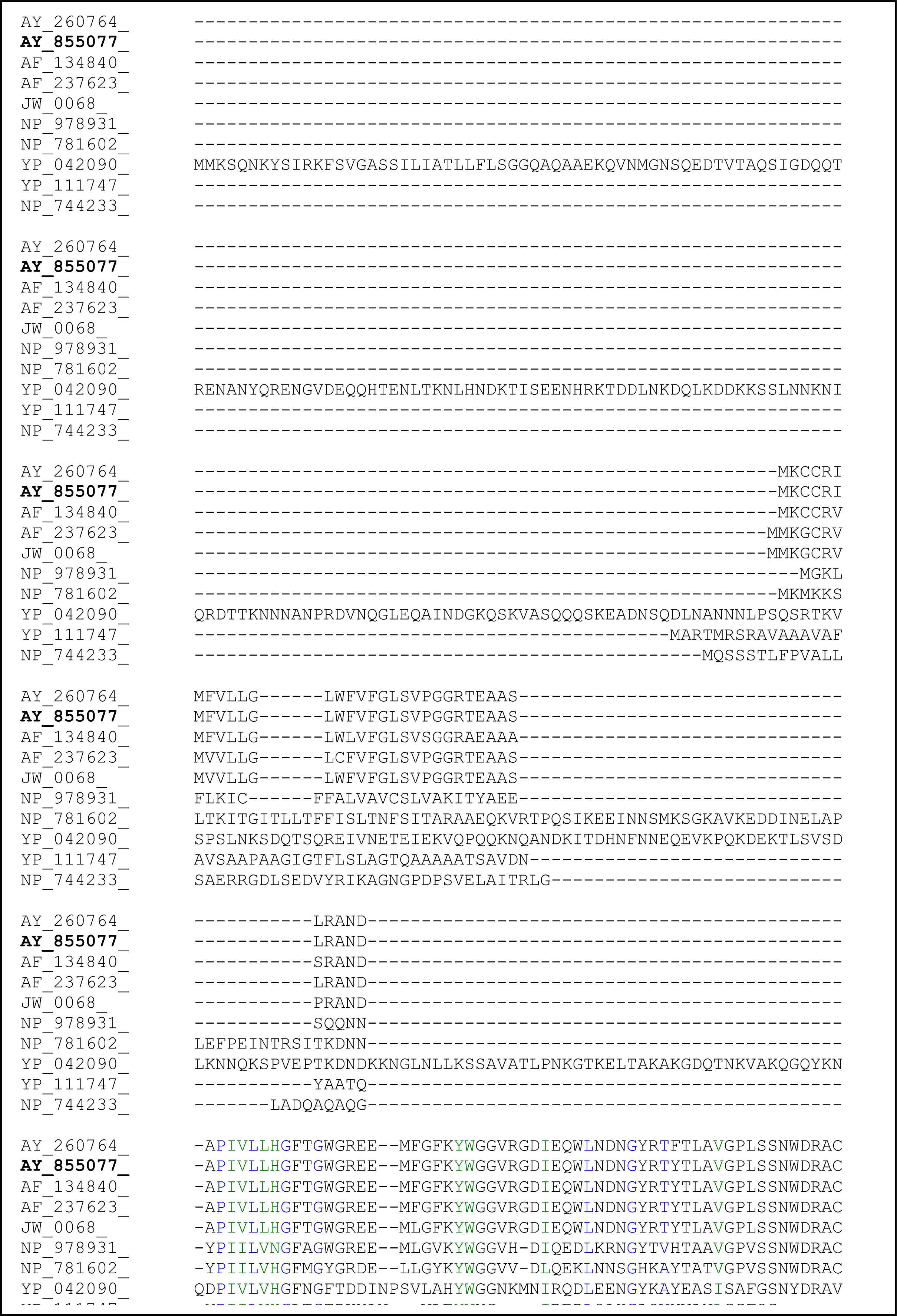
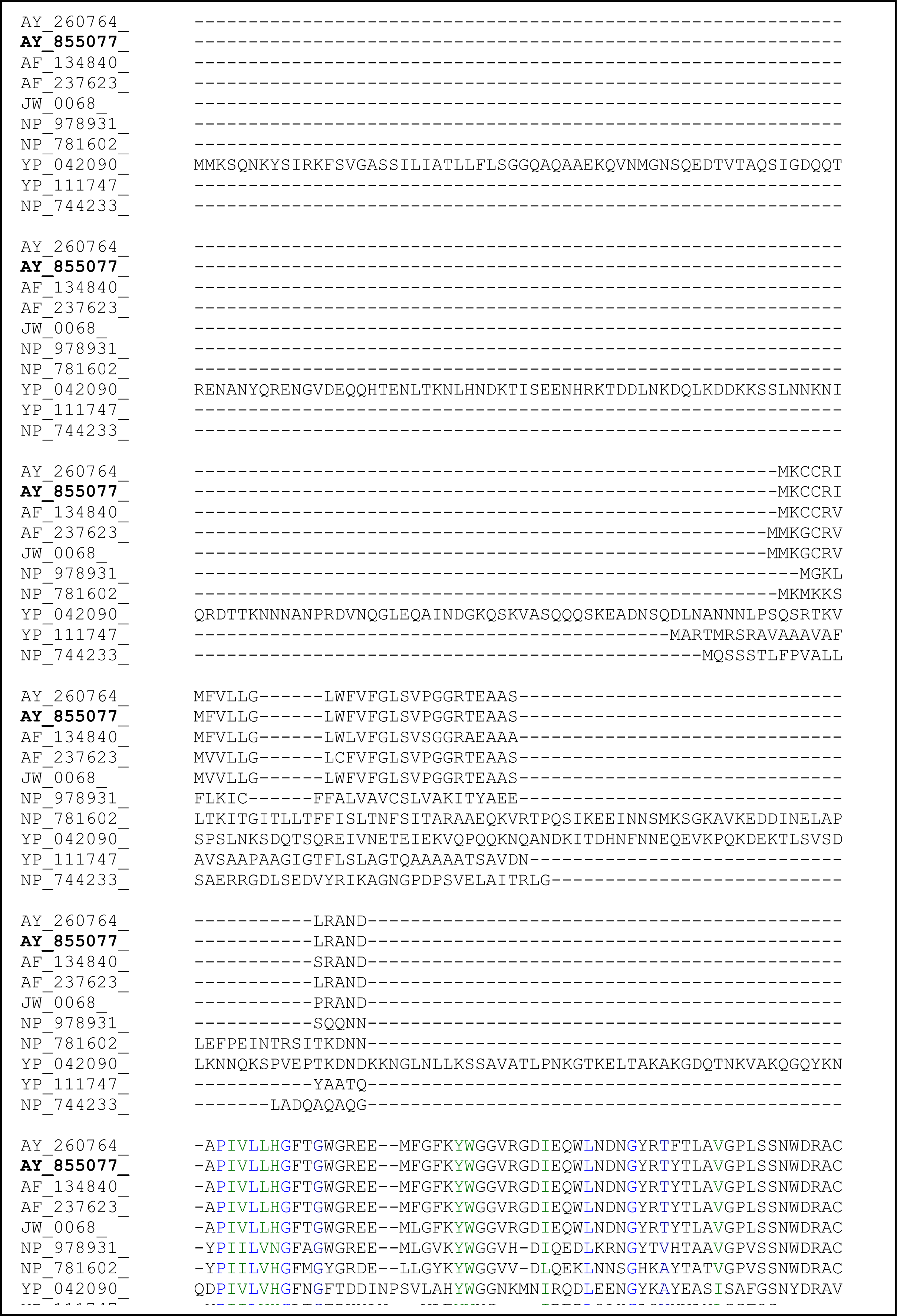
2.3. Sequence Analysis of the Thermostable L2 Lipase
2.4. Overexpression of the L2 Lipase Gene
2.5. Purification of Recombinant L2 Lipase
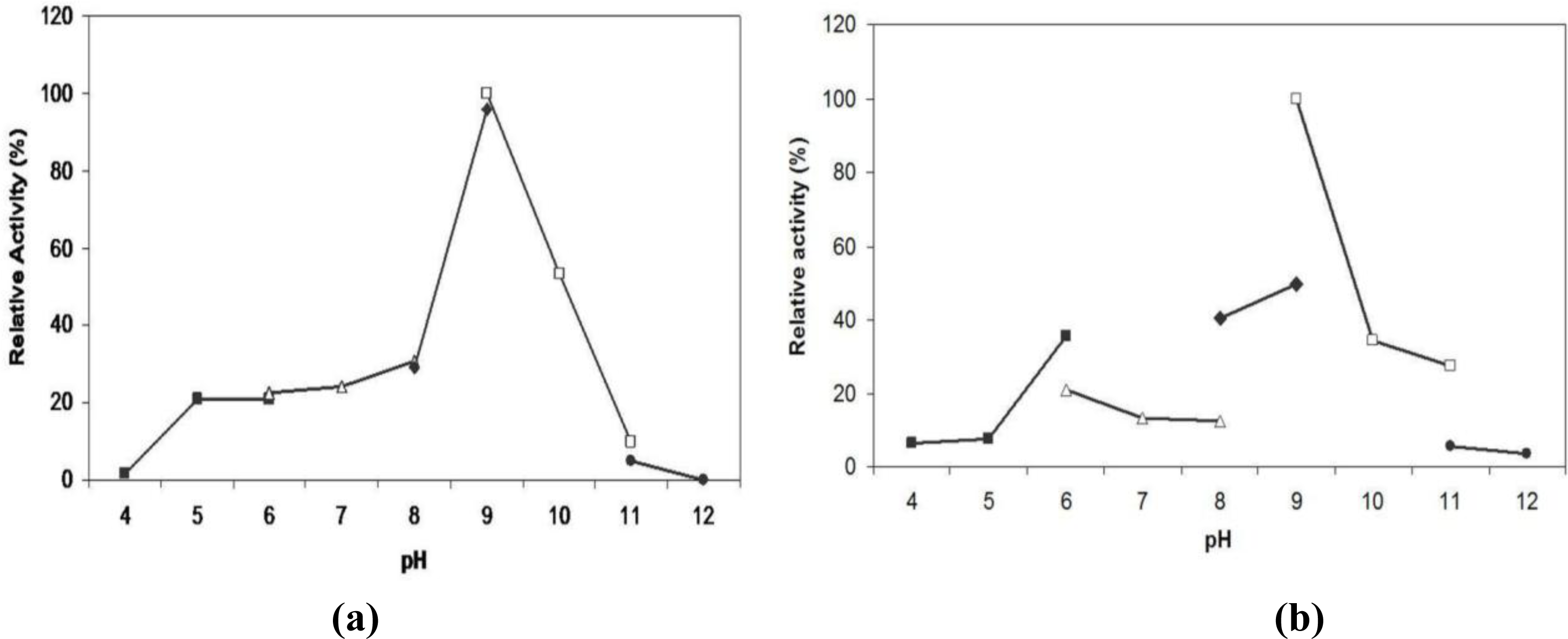
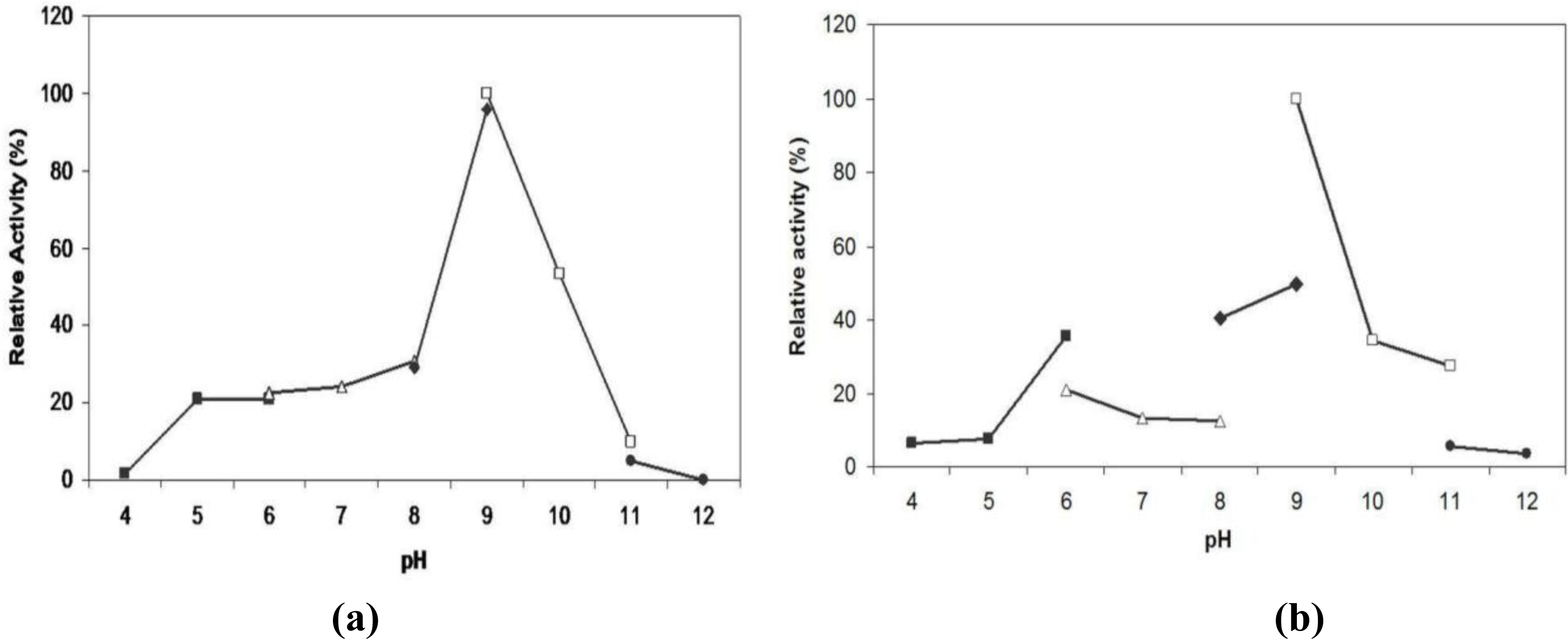
2.6. Effect of pH on Lipase Activity and Stability
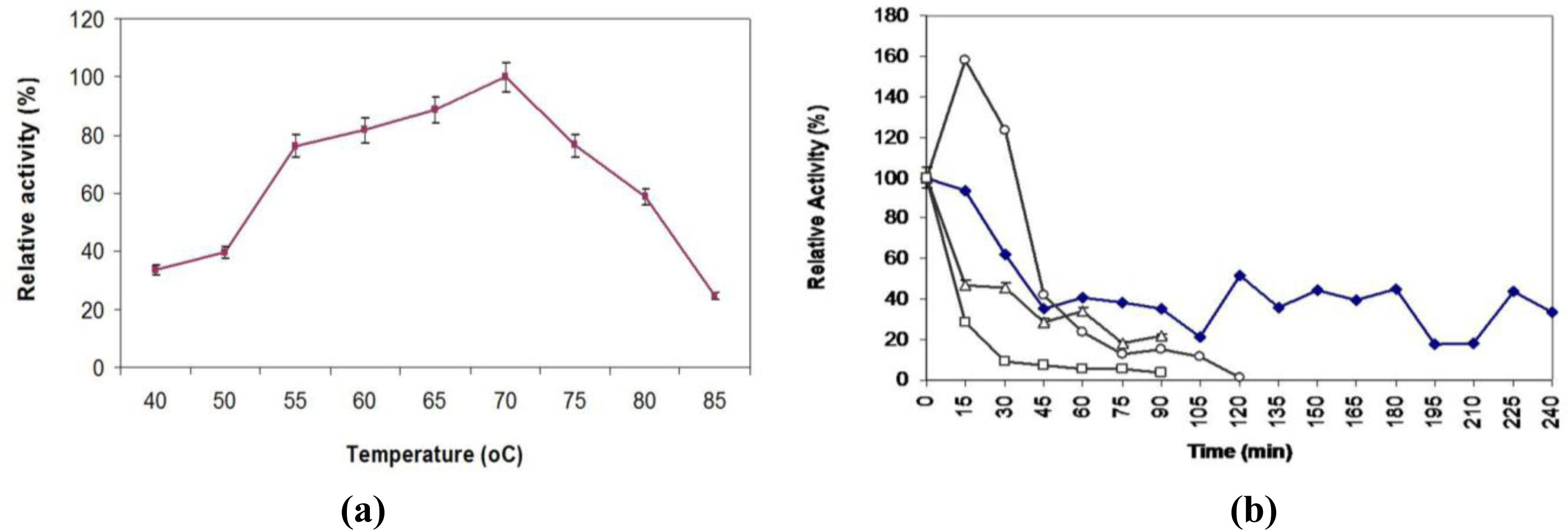
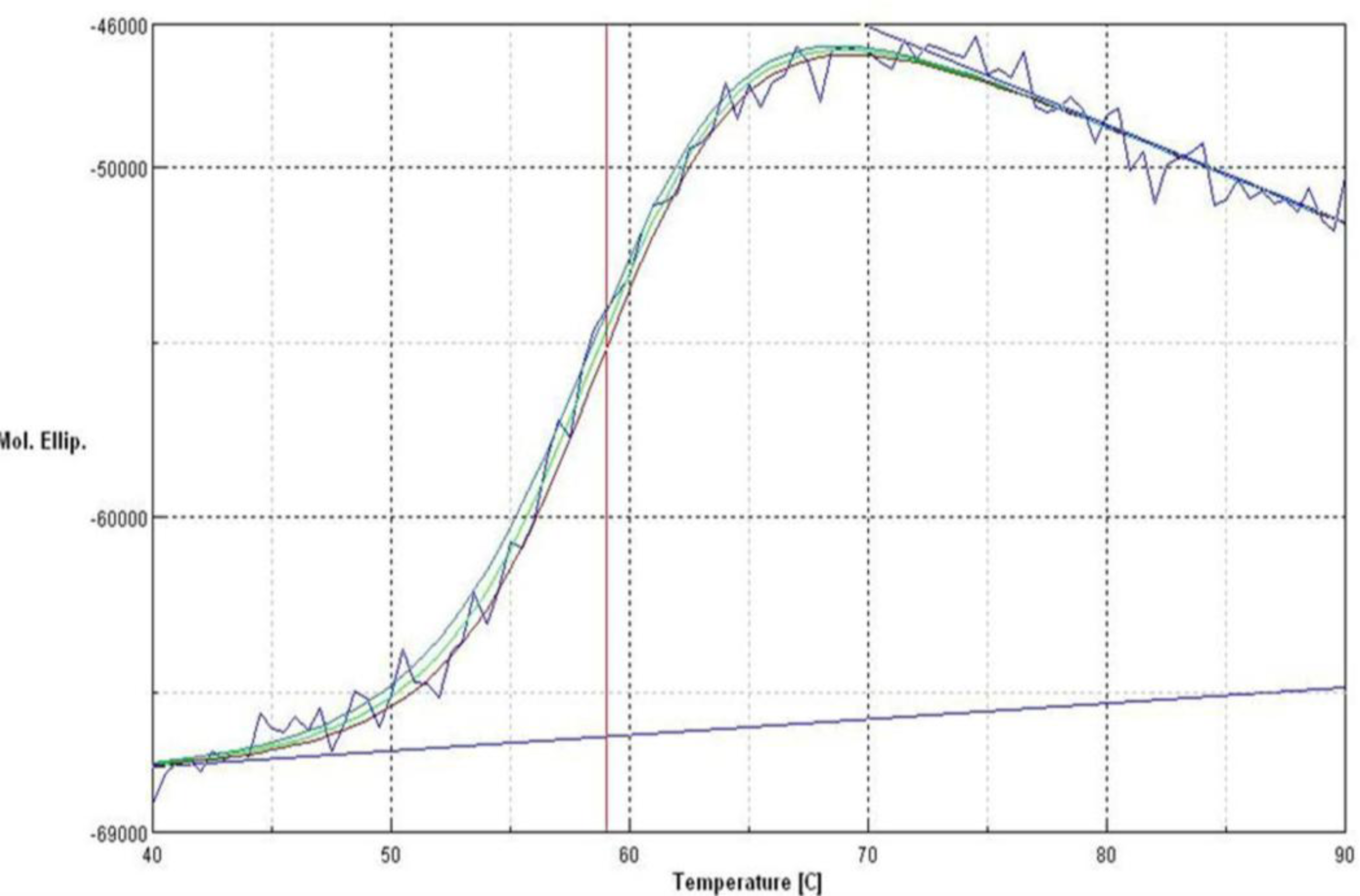
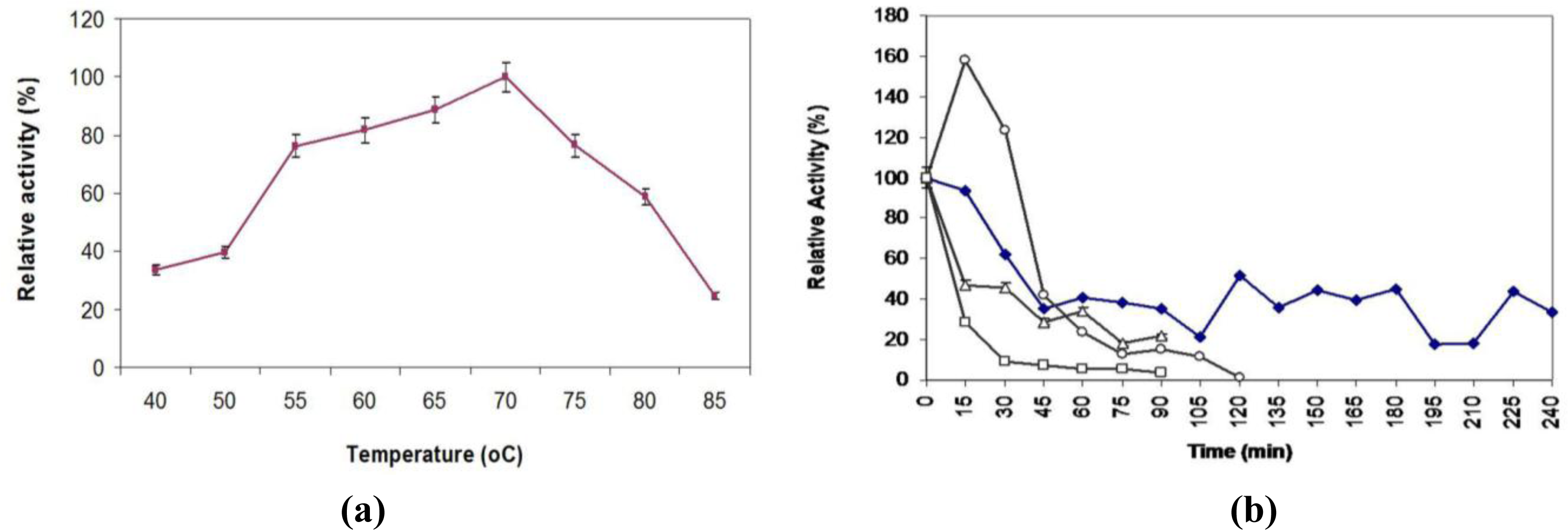
2.7. Effect of Temperature on Activity and Thermostability Profile
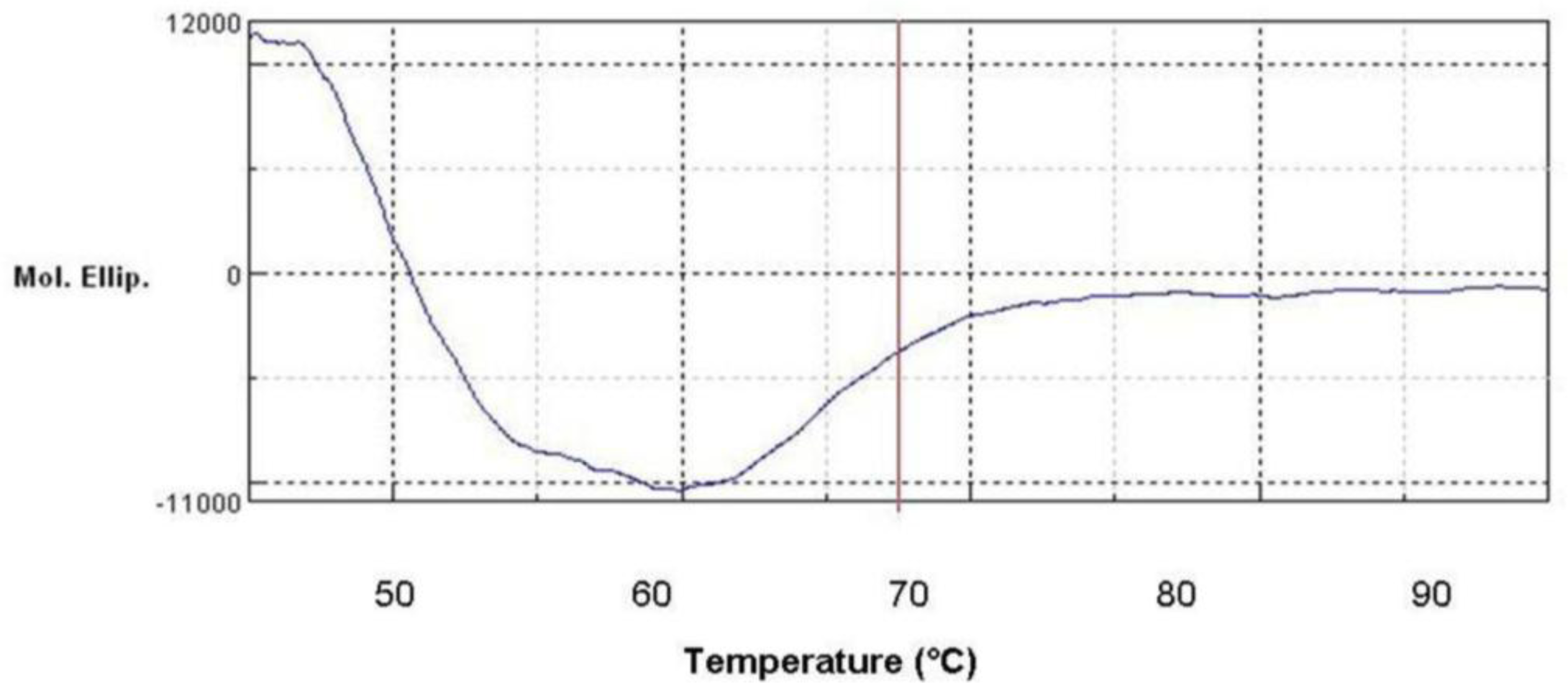
2.8. Denatured Protein Analysis of L2 Lipase
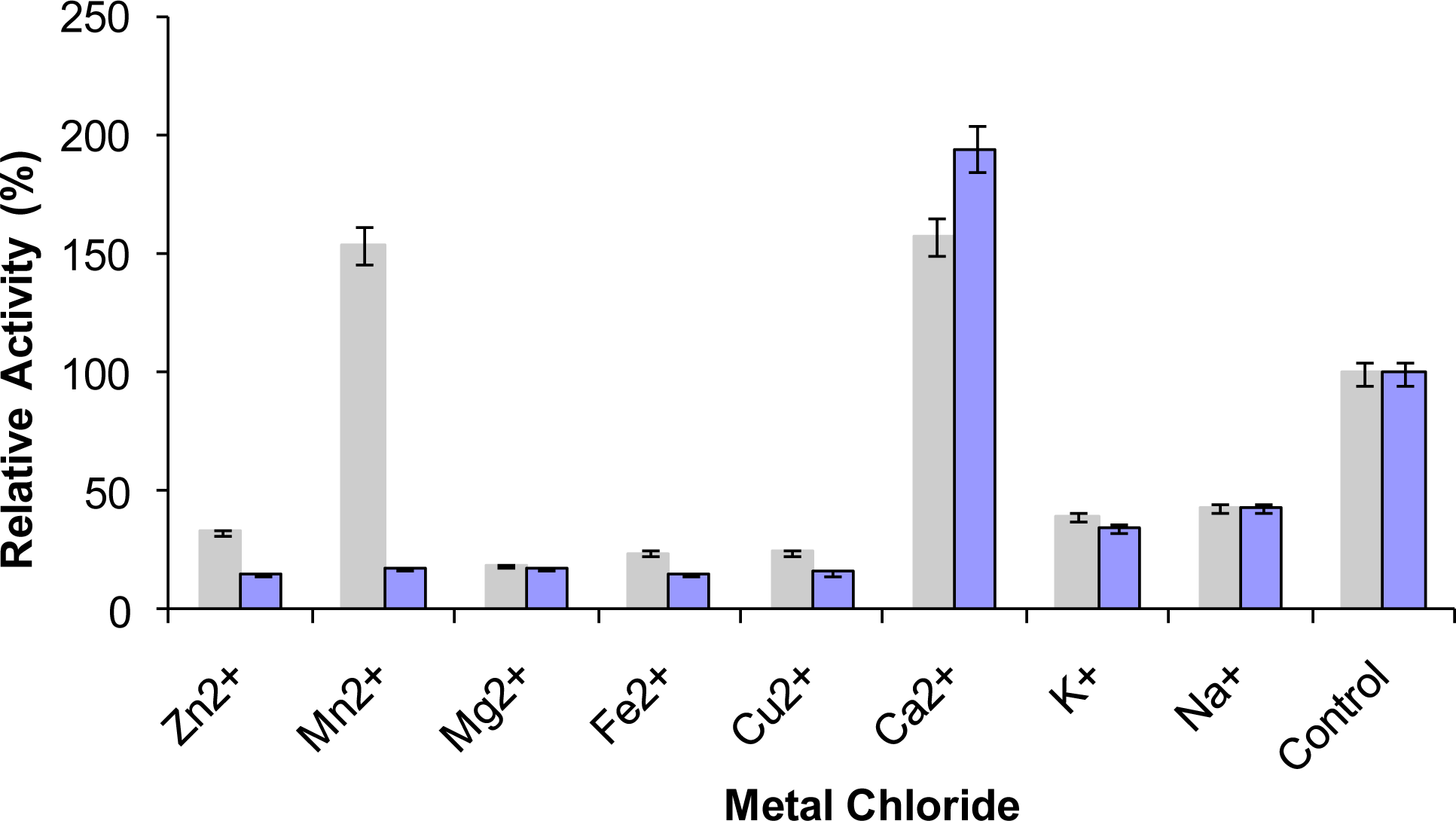
2.9. Effect of Metal Ions on Lipase Activity
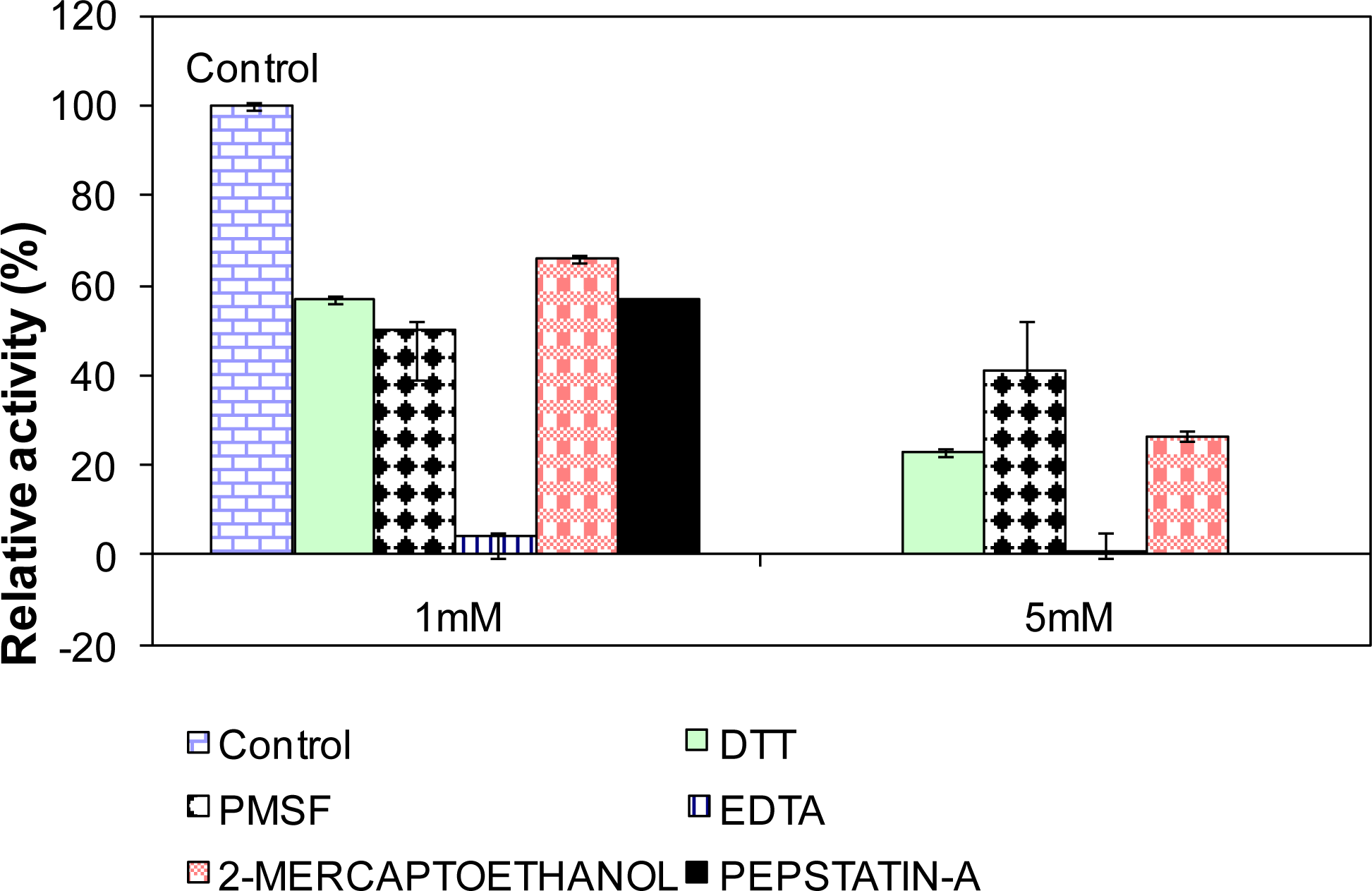
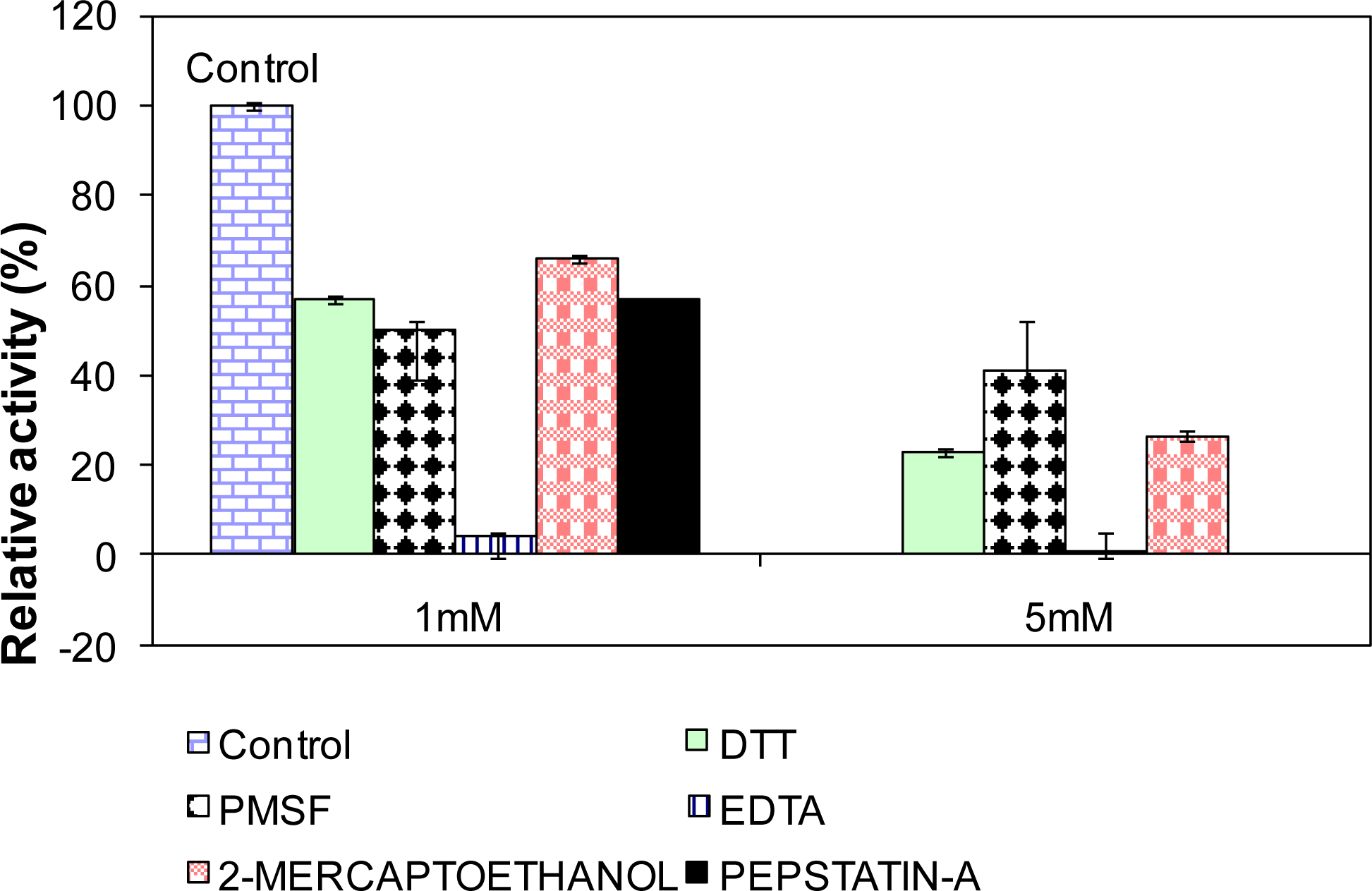
2.10. Influence of Various Effectors on L2 Lipase Activity
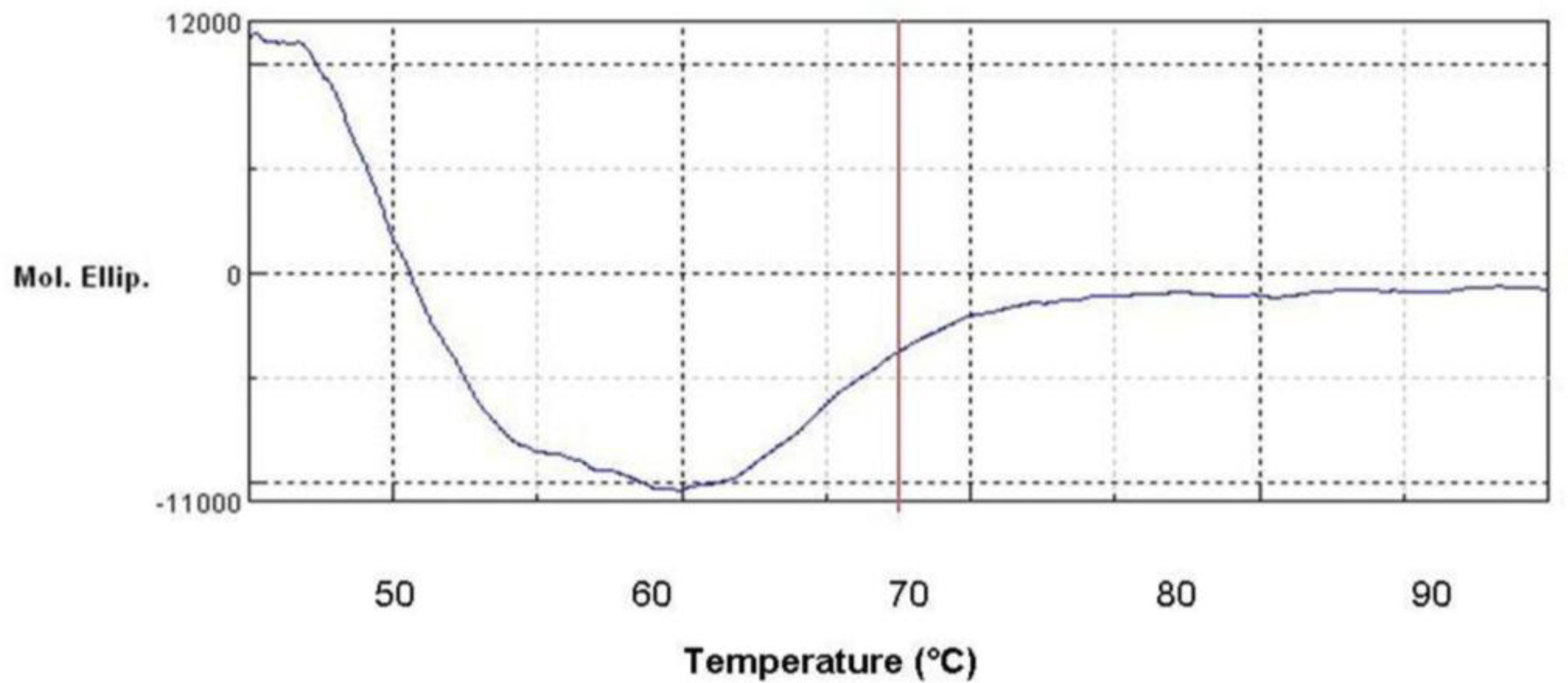
2.11. Secondary Structure Prediction of L2 Lipase by Circular Dichroism (CD) Spectral Analysis
3. Experimental Section
3.1. Strains, Plasmids and Growth Conditions
3.2. Nucleic Acid Manipulation
3.3. 16S rDNA Gene Sequence Amplification
3.4. Phylogenetic Tree Analysis
3.5. Isolation of the Lipase Gene
- Lip F2: 5′-TAG AGA ACG GAA GCC AAG AAG A-3′
- Lip R2: 5′-GAG CCG TTC AAA ATA ATG GTC G-3′
- P1R1: 5′-TTA AGG CTG CAA GCT CGC CAACTG-3′
- F: 5′-CAG AAA ACC CGA CAA TTG CCG-3′
- Mature: 5′-GCA TCC CTA CGC GCC CAT GAT-3′
3.6. Cloning of PCR Product
3.7. Sequencing of the Thermostable Lipase Gene
3.8. Expression of the L2 Lipase Gene
3.9. Assay of Lipase Activity
3.10. Effect of Different Concentrations of Inducer (IPTG) on Crude Lipase Expression
3.11. Time-Course Analysis of Crude Lipase Expression
3.12. Purification of Recombinant Lipase L2
3.13. SDS-PAGE Analysis of Bacterial Protein
3.14. Effect of pH on Lipase Activity and Stability
3.15. Effect of Temperature on Lipase Activity and Stability
3.16. Denatured Protein Analysis
3.17. Effect of Metal Ions on Lipase Activity
3.18. Influence of Various Effectors on Lipase Activity
3.19. Circular Dichroism (CD) Spectral Analysis of L2 Lipase
4. Conclusions
Acknowledgments
References
- Arpigny, JL; Jaeger, K-E. Bacterial lipolytic enzymes: Classification and properties. Biochem. J 1999, 343, 177–183. [Google Scholar]
- Jaeger, K; Eggert, T. Lipases for biotechnology. Curr. Opin. Biotechnol 2002, 13, 390–397. [Google Scholar]
- Sharma, R; Chisti, Y; Banerjee, UC. Production, purification, characterization, and applications of lipases. Biotech. Adv 2002, 19, 627–662. [Google Scholar]
- Coolbear, T; Daniel, RM; Morgan, HW. The enzymes from extreme thermophiles: Bacterial sources, thermostabilities and industrial relevance. Adv. Biochem. Eng. Biotechnol 1992, 45, 57–98. [Google Scholar]
- Nazina, TN; Tourova, TP; Poltaraus, AB; Novikova, EV; Grigoryan, AA; Ivanova, AE; Lysenko, AM; Petrunyaka, VV; Osipov, GA; Belyaev, SS; Ivanov, MV. Taxonomic study of aerobic thermophilic bacilli: Descriptions of Geobacillus subterraneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermoglucosidasius and Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G. thermocatenulatus, G. thermoleovorans, G. kaustophilus, G. thermoglucosidasius and G. thermodenitrificans. Int. J. Syst. Evol. Micr 2001, 51, 433–446. [Google Scholar]
- ExPASy Tools. ExPASy Proteomics Server. Swiss Institute of Bioinformatics: Lausanne, Switzerland, 2011. Available online: http://www.expasy.org/tool (accessed on 14 April 2011).
- SignalP Server. SignalP V2.0.b2. Center for Biological Sequence Analysis (CBS): Lyngby, Denmark, 2011. Available online: http://www.cbs.dtu.dk/services/SignalP-2.0/ (accessed on 14 April 2011).
- Bornscheuer, UT; Bessler, C; Srinivas, R; Krishna, SH. Optimizing lipase and related enzymes for efficient application. Trends Biotech 2002, 20, 433–437. [Google Scholar]
- Hamid, AS. Cloning and Expression of Alkaline Protease Gene from Bacillus Stearothermophilus Strain F1; MS Thesis; Universiti Putra Malaysia: Serdang, Malaysia, 2000. [Google Scholar]
- Sinchaikul, S; Sookkheo, B; Phutrakul, S; Pan, FM; Chen, ST. Optimization of a thermostable lipase from Bacillus stearothermophilus P1: Overexpression, purification, and characterization. Protein Expr. Purif 2001, 22, 388–398. [Google Scholar]
- Leow, TC; Rahman, RNZRA; Basri, M; Salleh, AB. High level expression of thermostable lipase from Geobacillus sp. Strain T1. Biosci. Biotech. Biochem 2004, 68, 96–103. [Google Scholar]
- Nthangeni, MB; Patterton, HG; van Tonder, A; Vergeer, WP. Over-expression and properties of a purified recombinant Bacillus licheniformis lipase: A comparative report on Bacillus lipases. Enzyme Microbial. Technol 2001, 28, 705–712. [Google Scholar]
- Mosbah, H; Sayari, A; Bezzine, S; Gargouri, Y. Expression, purification, and characterization of His-tagged Staphylococcus xylosus lipase wild-type and its mutant Asp 290 Ala. Protein Expr. Purif 2006, 47, 516–523. [Google Scholar]
- Dharmsthiti, S; Luchai, S. Production, purification and characterization of thermophilic lipase from Bacillus sp. THL027. FEMS Microbiol. Lett 1999, 179, 241–246. [Google Scholar]
- Jaeger, KE; Ransac, S; Dijkstra, BW; Clson, C; Heuvel, MV; Misset, O. Bacterial lipases. FEMS Microbiol. Rev 1994, 15, 29–63. [Google Scholar]
- Nawani, N; Dosanjh, NS; Kaur, J. A novel thermostable lipase from a thermophilic Bacillus sp.: Characterization and esterification studies. Biotechnol. Lett 1998, 20, 997–1000. [Google Scholar]
- Sharma, R; Chisti, Y; Banerjee, UC. Production, purification, characterization, and applications of lipases. Biotech. Adv 2002, 19, 627–662. [Google Scholar]
- Wang, Y; Srivastava, KC; Shen, GJ; Wang, HY. Thermostable alkaline lipase from a newly isolated themrophilic Bacillus strain, A30–1 (ATCC 53841). J. Ferment. Bioengineer 1995, 79, 433–438. [Google Scholar]
- Lee, DW; Kim, HW; Lee, KW; Kim, BC; Choe, EA; Lee, HS; Kim, DS; Pyun, YR. Purification and characterization of two distinct thermostable lipases from the gram-posistive thermophilic bacterium Bacillus thermoleovorans ID-1. Enzyme Microbial. Technol 2001, 29, 363–371. [Google Scholar]
- Kim, H-K; Park, S-Y; Lee, J-K; Oh, T-K. Gene cloning and characterisation of thermostable lipase from Bacillus stearothermophilus. Biosci. Biotech. Biochem 1998, 62, 66–71. [Google Scholar]
- Gary, CJ. Stabilization of enzymes with soluble additives. In Thermostability of Enzyme; Gupta, MN, Ed.; Narosa: New Delhi, India, 1995; pp. 124–143. [Google Scholar]
- Sreerama, N; Venyaminov, SY; Woody, RW. Estimation of protein secondary structure from circular dichroism spectra: Inclusion of denatured proteins with native proteins in the analysis. Anal. Biochem 2000, 287, 243–251. [Google Scholar]
- Schröder, O; Tippner, D; Wagner, R. Towards the three-dimensional structure of the Escherichia coli DNA-binding protein H-NS: A CD and fluorescence study. Biochem. Biop. Res. Co 2001, 282, 219–227. [Google Scholar]
- Yang, JT; Wu, CSC; Martinez, HM. Calculation of protein conformation from circular dichroism. Meth. Enzymol 1986, 130, 208–269. [Google Scholar]
- Shariff, FM; Leow, TC; Mukred, AD; Salleh, AB; Basri, M; Rahman, RNZRA. Production of L2 lipase by Bacillus sp. strain L2: Nutritional and physical factors. J. Basic Microbiol 2007, 47, 406–412. [Google Scholar]
- National Center for Biotechnology Information (NCBI) Home Page, Bethesda, MD, USA, 2011. Available online: http://www.nlm.ncbi.nih.gov (accessed on 14 April 2011).
- Thompson, JD; Higgins, DG; Gibson, TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl. Acids Res 1994, 22, 4673–4680. [Google Scholar]
- SDSC Biology Workbench. San Diego Supercomputer Center: San Diego, USA, 2011. Available online: http://workbench.sdsc.edu/ (accessed on 14 April 2011).
- NCBI/BLAST-Basic Logic Alignment Search Tool. National Center for Biotechnology Information (NCBI): Bethesda, MD, USA, 2011. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 14 April 2011).
- Kwon, DK; Rhee, JS. A simple and rapid colorimetric method for determination of free fatty acids for lipase assay. J. Am. Oil. Chem. Soc 1986, 63, 89–92. [Google Scholar]
- Laemmli, UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 380–685. [Google Scholar]



 ) and 10 mM (
) and 10 mM (
 ) prior to lipase assay. The remaining activity was determined at 70 °C using olive oil emulsion (1:1, v/v in Glycine-NaOH buffer pH 9) and expressed as a percentage of the activity without the metal chlorides.
) and 10 mM (
) prior to lipase assay. The remaining activity was determined at 70 °C using olive oil emulsion (1:1, v/v in Glycine-NaOH buffer pH 9) and expressed as a percentage of the activity without the metal chlorides.
) prior to lipase assay. The remaining activity was determined at 70 °C using olive oil emulsion (1:1, v/v in Glycine-NaOH buffer pH 9) and expressed as a percentage of the activity without the metal chlorides.
) and 10 mM (
) prior to lipase assay. The remaining activity was determined at 70 °C using olive oil emulsion (1:1, v/v in Glycine-NaOH buffer pH 9) and expressed as a percentage of the activity without the metal chlorides.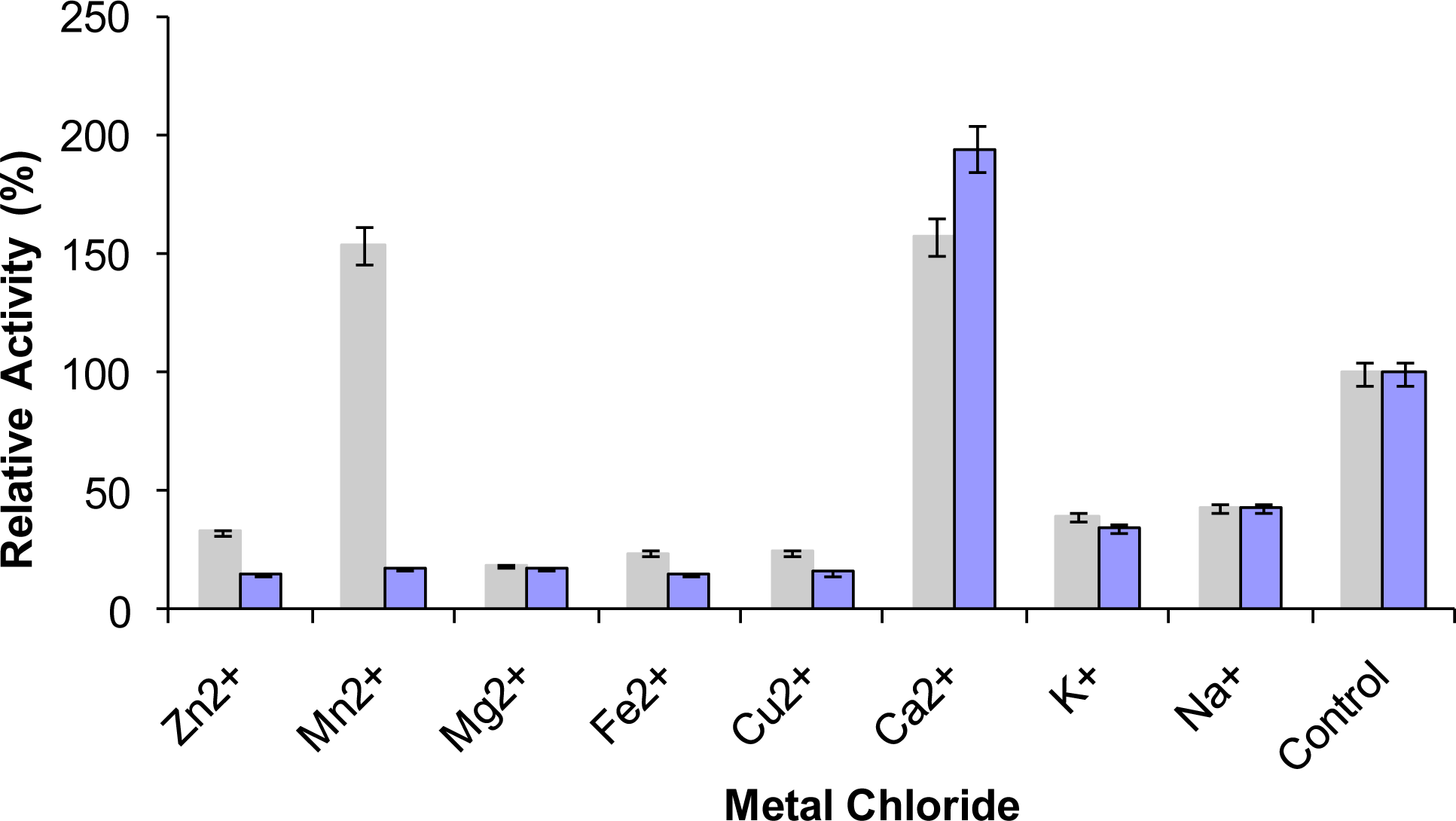
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shariff, F.M.; Rahman, R.N.Z.R.A.; Basri, M.; Salleh, A.B. A Newly Isolated Thermostable Lipase from Bacillus sp. Int. J. Mol. Sci. 2011, 12, 2917-2934. https://doi.org/10.3390/ijms12052917
Shariff FM, Rahman RNZRA, Basri M, Salleh AB. A Newly Isolated Thermostable Lipase from Bacillus sp. International Journal of Molecular Sciences. 2011; 12(5):2917-2934. https://doi.org/10.3390/ijms12052917
Chicago/Turabian StyleShariff, Fairolniza Mohd, Raja Noor Zaliha Raja Abd. Rahman, Mahiran Basri, and Abu Bakar Salleh. 2011. "A Newly Isolated Thermostable Lipase from Bacillus sp." International Journal of Molecular Sciences 12, no. 5: 2917-2934. https://doi.org/10.3390/ijms12052917