Simultaneous Structural Identification of Natural Products in Fractions of Crude Extract of the Rare Endangered Plant Anoectochilus roxburghii Using 1H NMR/RRLC-MS Parallel Dynamic Spectroscopy


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
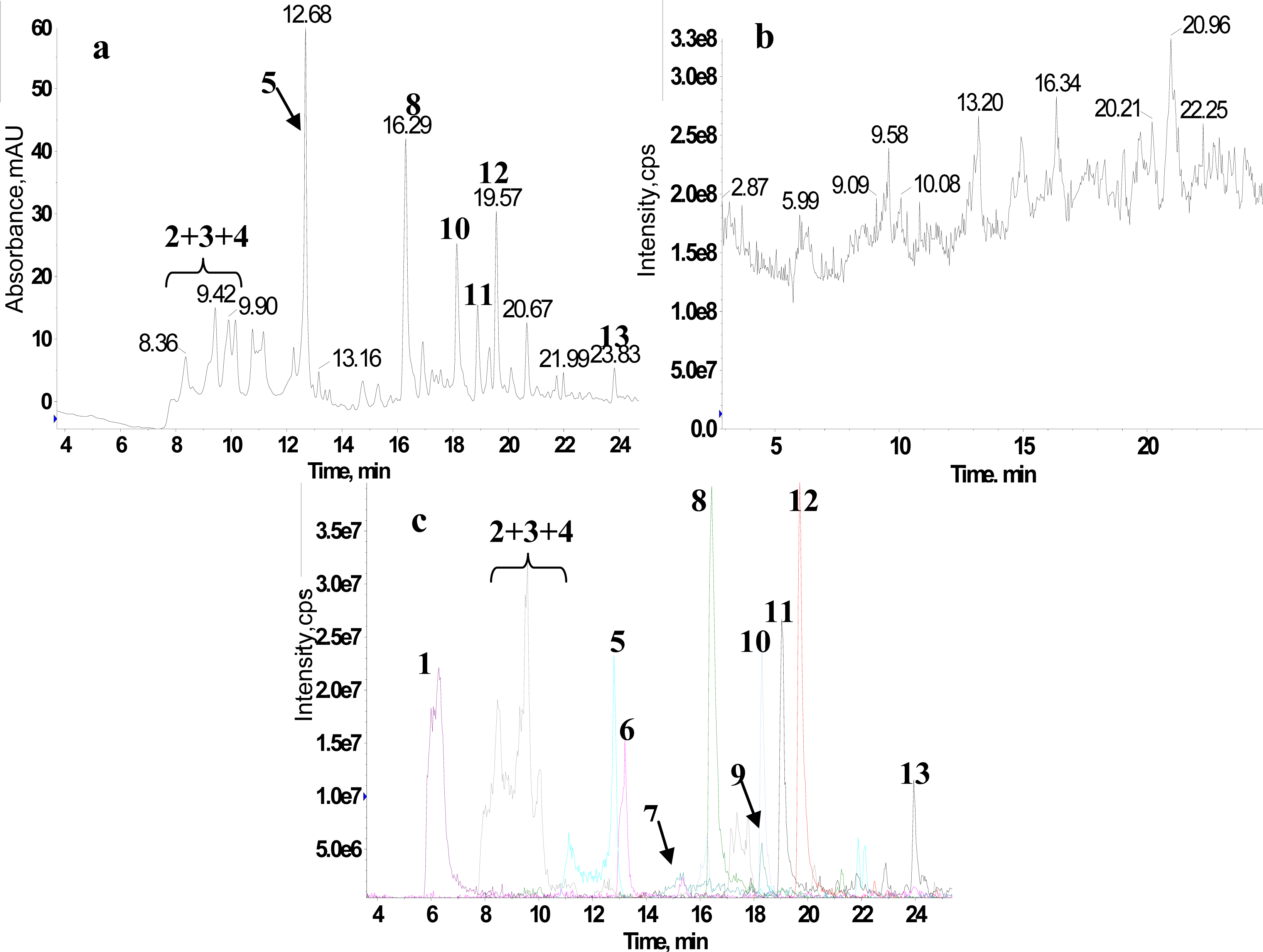
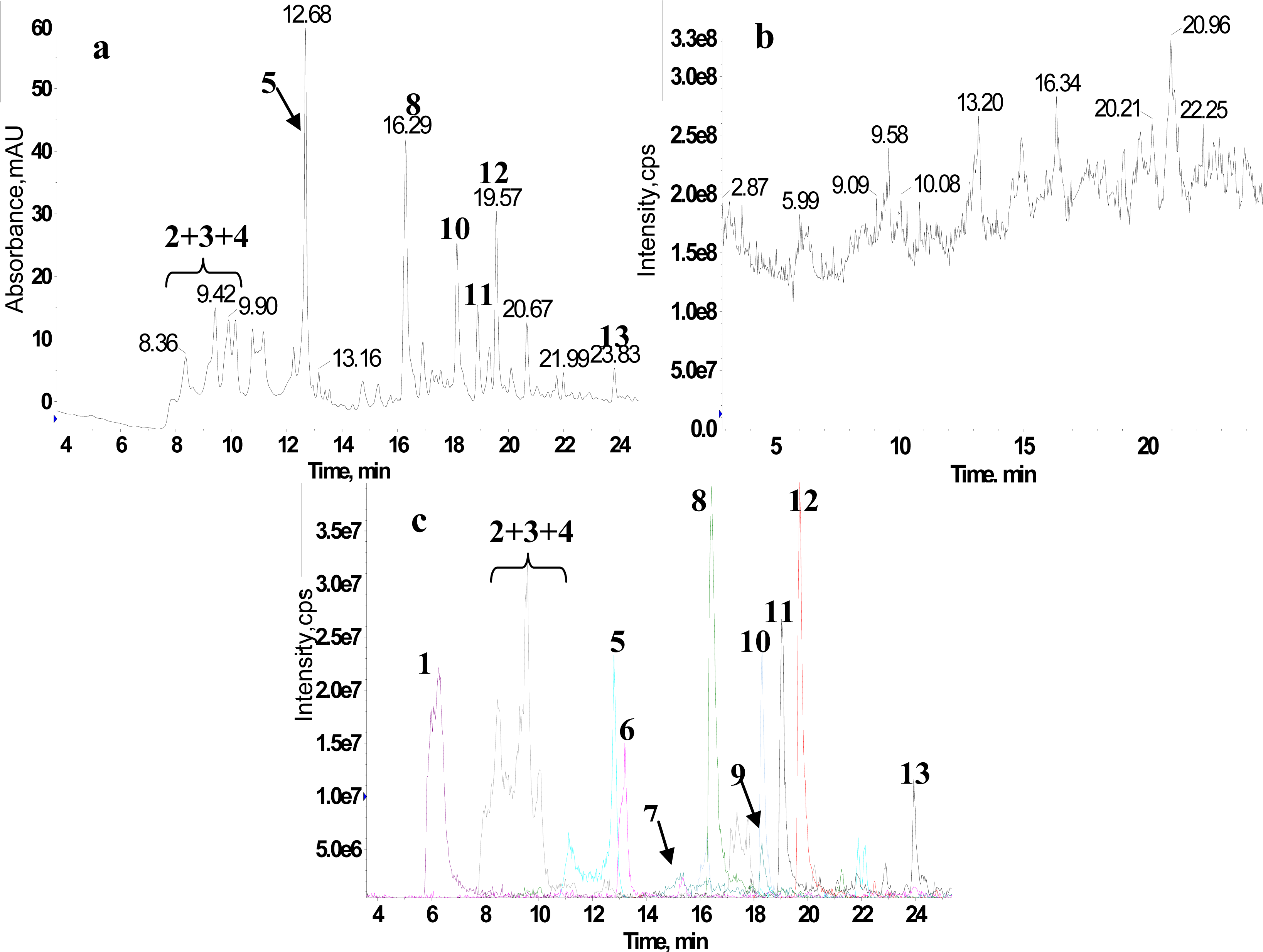
2.1. RRLC-MS Analysis of the Crude Extract
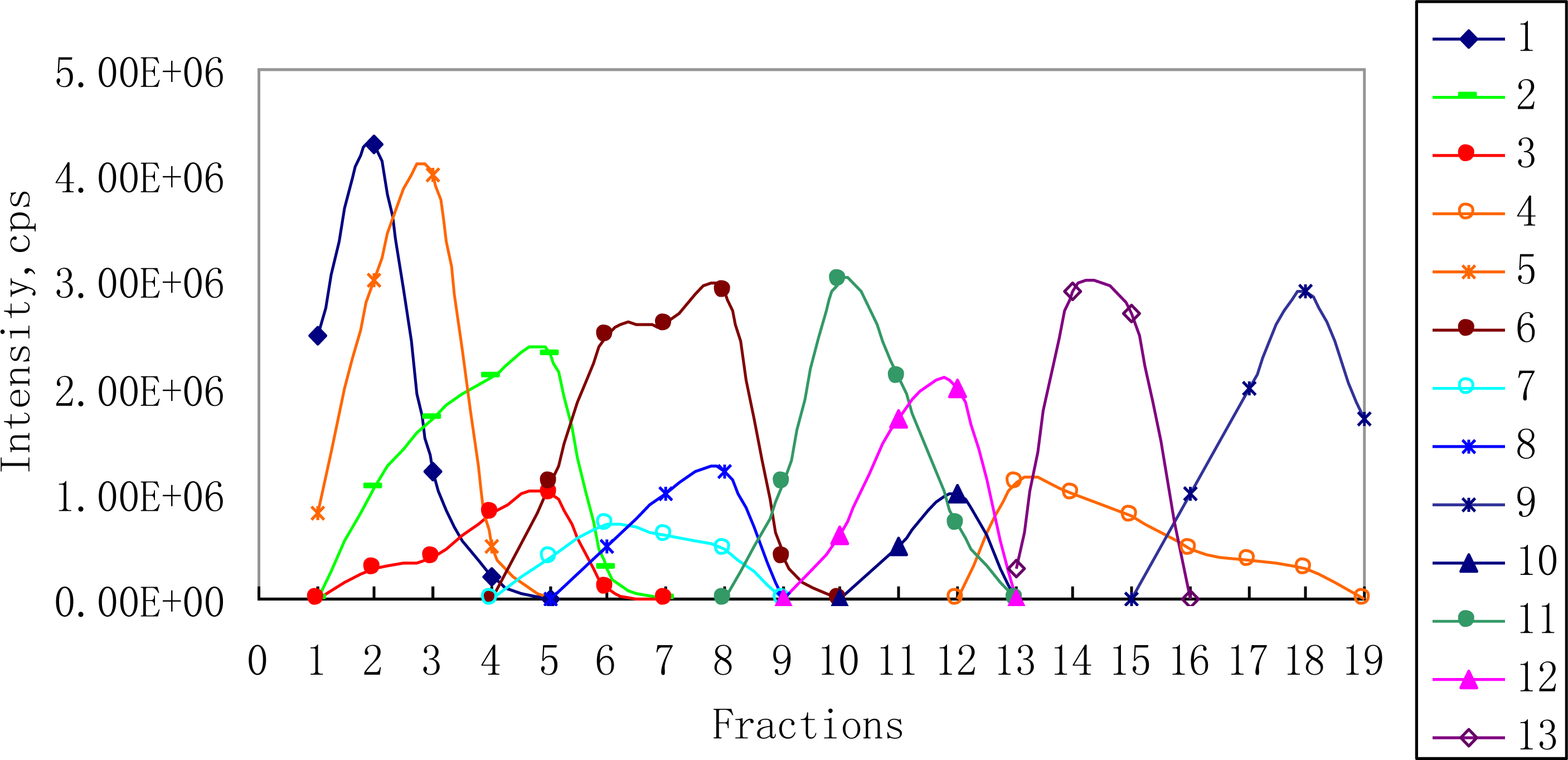
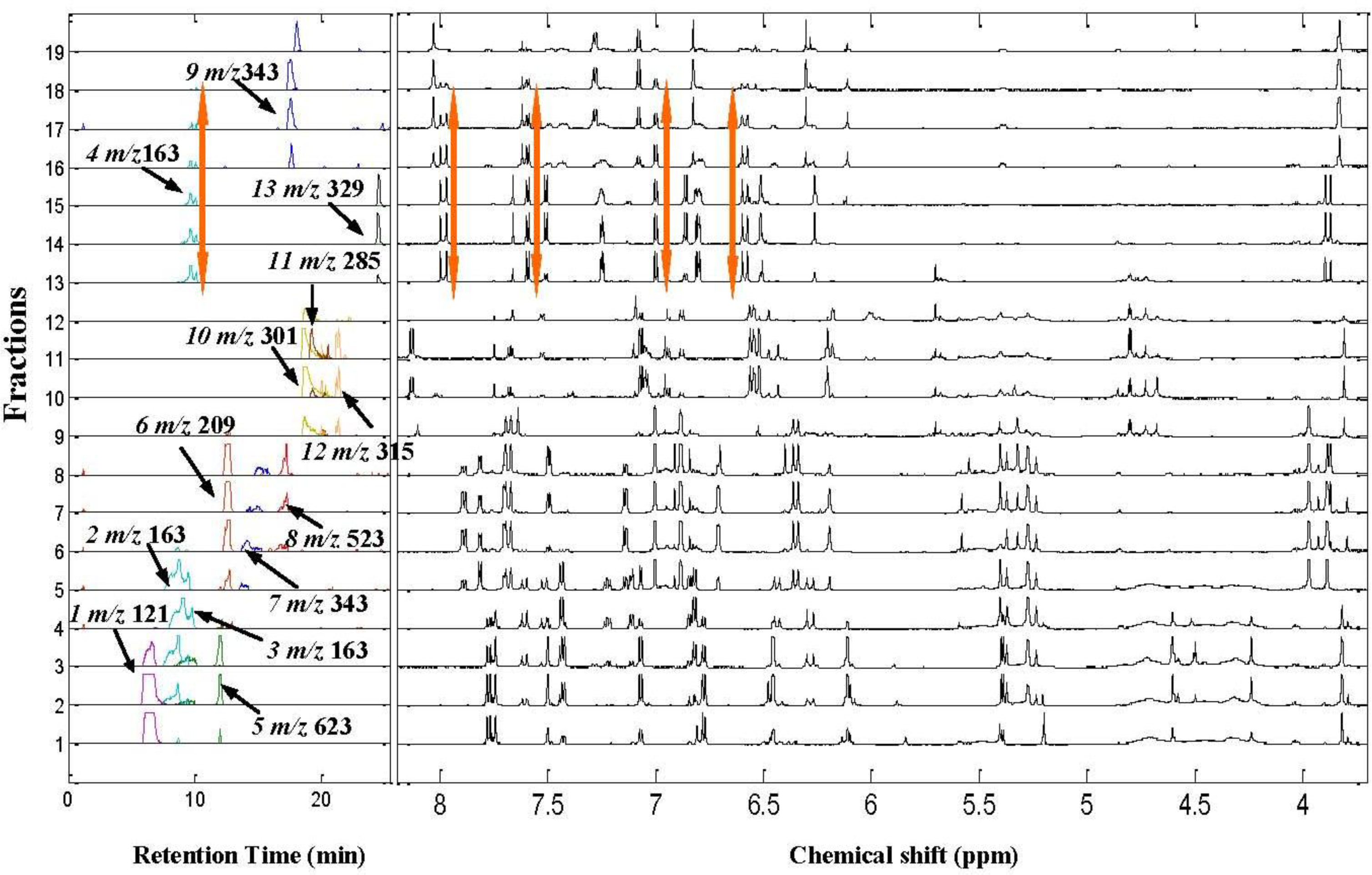
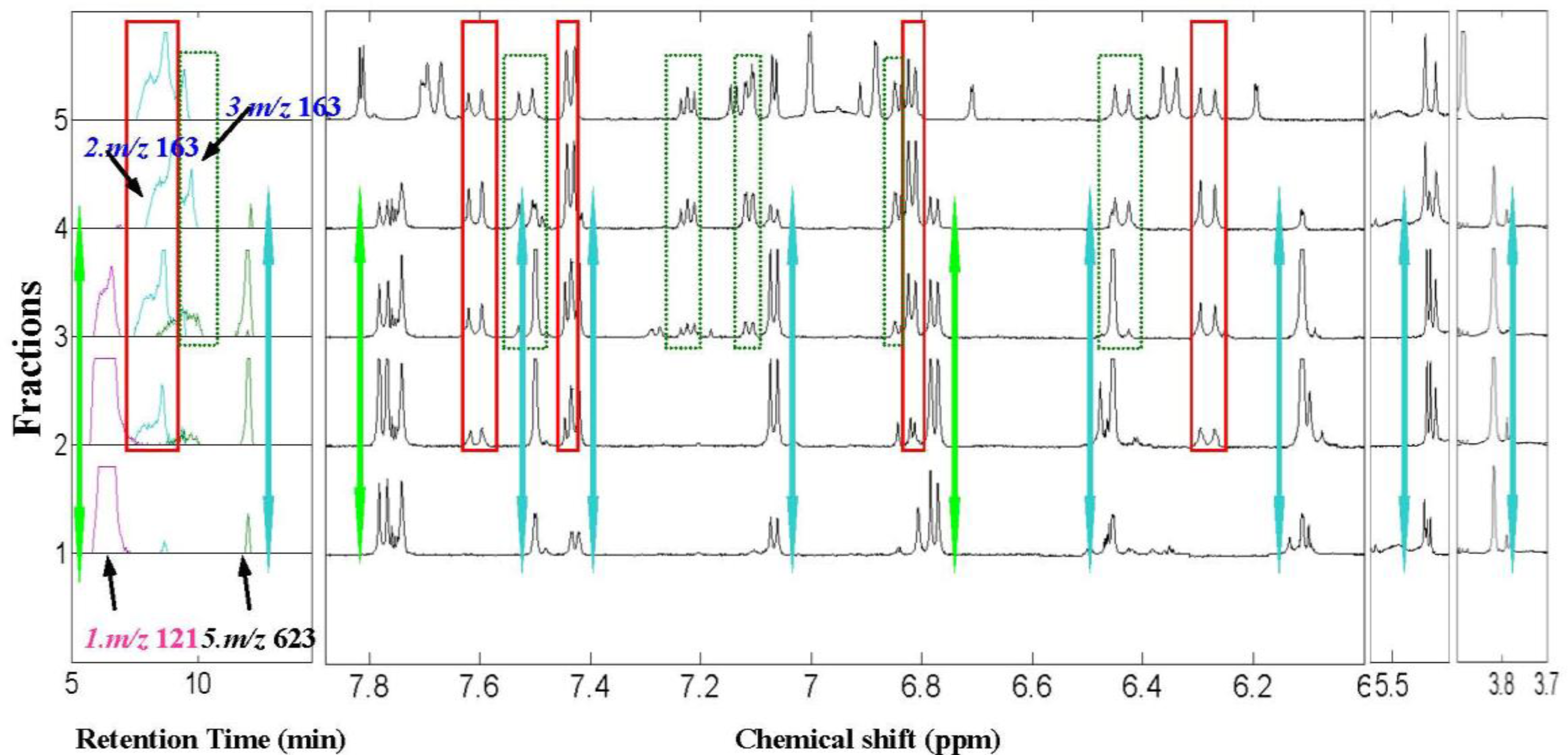
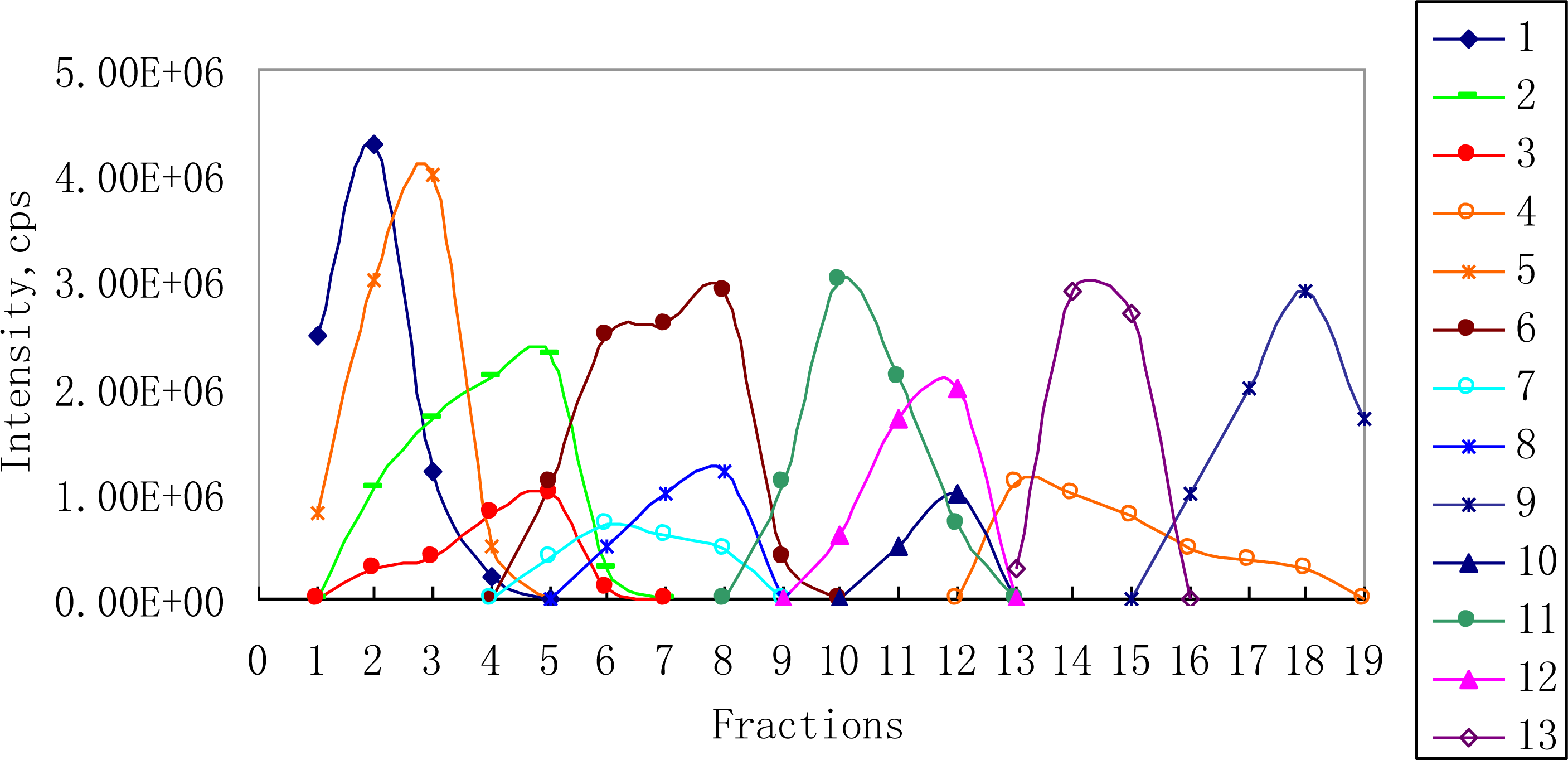
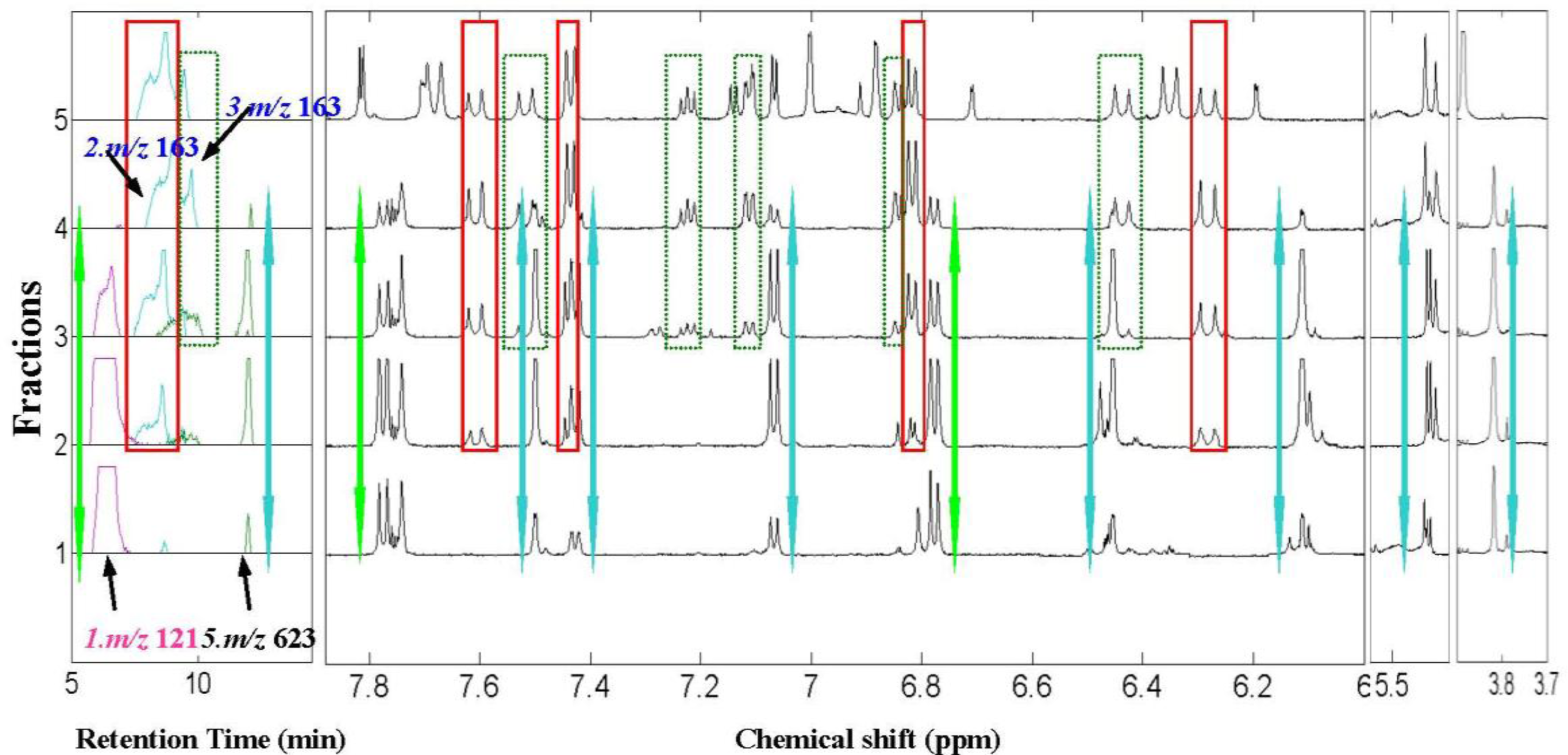
2.2. 1H NMR/RRLC-MS PDS Spectrum
3. Experimental Section
3.1. Reagents
3.2. Fractions Preparation
3.3. Instrumentation and Conditions
3.3.1. RRLC-MS and RRLC-MS/MS Analysis
3.3.2. NMR Samples and Analysis
3.4. Data Processing
4. Conclusions
Acknowledgments
References
- Jonker, N; Kretschmer, A; Kool, J; Fernandez, A; Kloos, D; Krabbe, JG; Lingeman, H; Irth, H. Online magnetic bead dynamic protein-affinity selection coupled to LC-MS for the screening of pharmacologically active compounds. Anal. Chem 2009, 81, 4263–4270. [Google Scholar]
- Liang, F; Li, LJ; Abliz, Z; Yang, YC; Shi, JG. Structural characterization of steroidal saponins by electrospray ionization and fast-atom bombardment tandem mass spectrometry. Rapid Commun. Mass Spectrom 2002, 16, 1168–1173. [Google Scholar]
- Xiang, Y; Abliz, Z; Li, LJ; Huang, X; Yu, SS. Study of structural characteristic features of phenanthriondolizidine alkaloids by fast atom bombardment with tandem mass spectrometry. Rapid Commun. Mass Spectrom 2002, 16, 1668–1674. [Google Scholar]
- Cui, LJ; Abliz, Z; Xia, M; Zhao, LY; Gao, S; He, WY; Xiang, Y; Liang, F; Yu, SS. On-line identification of phenanthroindolizidine alkaloids in a crude extract from Tylophora atrofolliculata by liquid chromatography combined with tandem mass spectrometry. Rapid Commun. Mass Spectrom 2004, 18, 184–190. [Google Scholar]
- Liu, YZ; Liang, F; Cui, LJ; Xia, M; Zhao, LY; Yang, YC; Shi, JG; Abliz, Z. Multi-stage mass spectrometry of furostanol saponins combined with electrospray ionization in positive and negative ion modes. Rapid Commun. Mass Spectrom 2004, 18, 235–238. [Google Scholar]
- Li, B; Abliz, Z; Fu, GM; Tang, MJ; Yu, SS. Characteristic fragmentation behavior of some glucuronide-type triterpenoid saponins using electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom 2005, 19, 381–390. [Google Scholar]
- Ablajan, K; Abliz, Z; Shang, XY; He, JM; Zhang, RP; Shi, JG. Structural characterization of flavonol 3,7-di-O-glycosides and determination of the glycosylation position by using negative ion electrospray ionization tandem mass spectrometry. J. Mass Spectrom 2006, 41, 352–360. [Google Scholar]
- Li, B; Abliz, Z; Tang, MJ; Fu, GM; Yu, SS. Rapid analytical method of triterpenoid saponins in crude extract from Symplocos chinensis using liquid chromatography combined with electrospray ionization tandem mass spectrometry. J. Chromatogr. A 2006, 1101, 53–62. [Google Scholar]
- Geng, P; Zhang, RP; Haji, AA; He, JM; Qu, K; Zhu, HB. Fast profiling of the integral metabolism of flavonols in the active fraction of Gossypium herbaceam L. using liquid chromatography/multi-stage tandem mass spectrometry. Rapid Commun. Mass Spectrom 2007, 21, 1877–1888. [Google Scholar]
- Liu, Y; He, JM; Zhang, RP; Shi, JG; Abliz, Z. Study of the characteristic fragmentation behavior of hydroquinone glycosides by electrospray ionization tandemmass spectrometry with optimization of collision energy. J. Mass Spectrom 2009, 44, 1182–1187. [Google Scholar]
- Xu, KY; Liu, F; Liu, BG; Gao, H; Ning, ZX. Synthesis of 4’,7-diacetoxyapigenin and its apoptotic induction in human hep G2 cells. Int. J. Mol. Sci 2010, 11, 1991–1998. [Google Scholar]
- Shockcor, J; Unger, S; Wilson, ID; Foxall, PJD; Nicholson, JK; Lindon, JC. Combined HPLC, NMR spectroscopy, and ion-trap mass spectrometry with application to the detection and characterization of xenobiotic and endogenous metabolites in human urine. Anal. Chem 1996, 68, 4431–4435. [Google Scholar]
- Salanti, A; Orlandi, M; Tolppa, EL; Zoia, L. Oxidation of isoeugenol by salen complexes with bulky substituents. Int. J. Mol. Sci 2010, 11, 912–926. [Google Scholar]
- Sandvoss, M; Weltring, A; Preiss, A; Levsen, K; Wuensch, G. Combination of matrix solid-phase dispersion extraction and direct on-line liquid chromatography-nuclear magnetic resonance spectroscopy–tandem mass spectrometry as a new efficient approach for the rapid screening of natural products: Application to the total asterosaponin fraction of the starfish Asterias rubens. J. Chromatogr. A 2001, 917, 75–86. [Google Scholar]
- Exarchou, V; Godejohann, M; van Beek, TA; Gerothanassis, IP; Vervoort, J. LC-UV-solid-phase extraction-NMR-MS combined with a cryogenic flow probe and its application to the identification of compounds present in Greek oregano. Anal. Chem 2003, 75, 6288–6294. [Google Scholar]
- Spraul, M; Freund, AS; Nast, RE; Withers, RS; Maas, WE; Corcoran, O. Advancing NMR sensitivity for LC-NMR-MS using a cryoflow probe: Application to the analysis of acetaminophen metabolites in urine. Anal. Chem 2003, 75, 1536–1541. [Google Scholar]
- Godejohann, M; Tseng, LH; Braumann, U; Fuchser, J; Spraul, M. Characterization of a paracetamol metabolite using on-line LC-SPE-NMR-MS and a cryogenic NMR probe. J. Chromatogr. A 2004, 1058, 191–196. [Google Scholar]
- Fritsche, J; Angoelal, R; Dachtler, M. On-line liquid-chromatography–nuclear magnetic resonance spectroscopy-mass spectrometry coupling for the separation and characterization of secoisolariciresinol diglucoside isomers in flaxseed. J. Chromatogr. A 2002, 972, 195–203. [Google Scholar]
- Lin, Y; Schiavo, S; Orjala, J; Vouros, P; Kautz, R. Microscale LC-MS-NMR platform applied to the identification of active cyanobacterial metabolites. Anal. Chem 2008, 80, 8045–8054. [Google Scholar]
- Schrader, W; Geiger, J; Godejohann, M. Studies of complex reactions using modern hyphenated methods: α-Pinene ozonolysis as a model reaction. J. Chromatogr. A 2005, 1075, 185–196. [Google Scholar]
- Elipe, MVS. Advantages and disadvantages of nuclear magnetic resonance spectroscopy as a hyphenated technique. Anal. Chim. Acta 2003, 497, 1–25. [Google Scholar]
- Jaroszewski, JW. Hyphenated NMR methods in natural products research. Planta Med 2005, 71, 691–700. [Google Scholar]
- Cloarec, O; Dumas, ME; Craig, A; Barton, RH; Trygg, J; Hudson, J; Blancher, C; Gauguier, D; Lindon, JC; Holmes, E; Nicholson, JK. Statistical total correlation spectroscopy: An exploratory approach for latent biomarker identification from metabolic 1H NMR data sets. Anal. Chem 2005, 77, 1282–1289. [Google Scholar]
- Crockford, DJ; Holmes, E; Lindon, JC; Plumb, RS; Zirah, S; Bruce, SJ; Rainville, P; Stumpf, CL; Nicholson, JK. Statistical heterospectroscopy, an approach to the integrated analysis of NMR and UPLC-MS data sets: Application in metabonomic toxicology studies. Anal. Chem 2006, 78, 363–371. [Google Scholar]
- Dai, DM; He, JM; Sun, RX; Zhang, RP; Aisa, HA; Abliz, Z. Nuclear magnetic resonance and liquid chromatography–mass spectrometry combined with an incompleted separation strategy for identifying the natural products in crude extract. Anal. Chim. Acta 2009, 632, 221–228. [Google Scholar]
- Asahi Encyclopedia. In The World of Plants; Asahi Shinbun: Tokyo, Japan, 1997; Volume 9, pp. 243–244.
- Zhang, YH; Cai, JY; Pi, HF. Antihyperglycemic activity of kinsenoside, a high yielding constituent from Anoectochilus roxburghii in streptozotocin diabetic rats. Ethnopharmacology 2007, 114, 141–145. [Google Scholar]
- Shyur, LF; Chenb, CH; Lo, CP; Wang, SY; Kang, PL; Sun, SJ; Chang, CA; Tzeng, CM; Yang, NS. Induction of apoptosis in MCF-7 human breast cancer by phytochemicals from Anoectochilus formosanus. J. Biomed. Sci 2004, 11, 928–939. [Google Scholar]
- Chang, CJ; Wu, SM; Yang, PW. High-pressure carbon dioxide and co-solvent extractions of crude oils from plant materials. Innovat. Food Sci. Emerg. Tech 2001, 1, 187–191. [Google Scholar]
- Lin, CC; Huang, PC; Lin, JM. Antioxidant and hepatoprotective effects of Anoectochilus formosanus and Gynostemma pentaphyllum. Am. J. Chin. Med 2000, 28, 87–96. [Google Scholar]
- Shih, CC; Wu, YW; Lin, WC. Scavenging of reactive oxygen species and inhibition of the oxidation of low density lipoprotein by the aqueous extraction of Anoectochilus formosanus. Am. J. Chin. Med 2003, 31, 25–36. [Google Scholar]
- He, CN; Wang, CL; Guo, SX; Xiao, PG. Research on chemical constituents and activities of Anoectochilus. J. Chin. Phom 2004, 39, 81–84. [Google Scholar]
- Koop, CE. The future of medicine. Science 2002, 295, 233. [Google Scholar]
- He, CN; Wang, CL; Guo, SX; Yang, JS; Xiao, PG. Study on chemical constituents of Anoectochilus roxburghii. Chin. Pharm. J 2005, 40, 581–583. [Google Scholar]
- He, CN; Wang, CL; Guo, SX; Yang, JS; Xiao, PG. Study on chemical constituents in herbs of Anoectochilus roxburghii II. Chin. J. Chin. Mater. Med 2005, 30, 761–763. [Google Scholar]
- Zhang, HY; Pan, X. Research on the constituents and activities of Anoectochilus roxburghii. Strait Pharmaceut. J 2009, 21, 82–84. [Google Scholar]
- Zhao, FF. Study on rapid breeding of Anoectochilus roxburghii. Forest Invent. Plann 2009, 34, 71–72. [Google Scholar]
- Liu, Y; He, JM; Sun, RX; Liu, C; Zhang, RP; Shi, JG; Abliz, Z. Simultaneous structural identification of constituents in active herbal extract of Forsythia Suspensa using NMR/LC-MS parallel dynamic spectroscopy. Chin J AnalChem 2011, 39. (in press).. [Google Scholar]
- Suárez, M; Macia, A; Romero, MP; Motilva, MJ. Improved liquid chromatography tandem mass spectrometry method for the determination of phenolic compounds in virgin olive oil. J. Chromatogr. A 2008, 1214, 90–99. [Google Scholar]
- Ablajan, K. Study on the Mass Spectrometric Analytical Method of Flavonoid Glycosides. Ph.D. Thesis; Peking Union Medical College: Beijing, China. 2007; 73–75. [Google Scholar]
- Guan, J; Wang, CL; Guo, SX. Isolation and structural elucidation of flavonoids from Anoecotochilus roxburhii. Chin. Trad. Herb. Drug 2005, 36, 1450–1453. [Google Scholar]
- Charisiadis, P; Exarchou, V; Troganis, AT; Gerothanassis, IP. Exploring the “fogrotten” –OH NMR spectral region in natural products. Chem. Commun 2010, 46, 3589–3591. [Google Scholar]
- Nerantzaki, AA; Tsiafoulis, CG; Charisiadis, P; Kontogianni, VG; Gerothanassis, IP. Novel determination of the total phenolic content in the crude plant extracts by the use of 1H NMR of the –OH spectral region. Anal. Chim. Acta 2011, 688, 54–60. [Google Scholar]
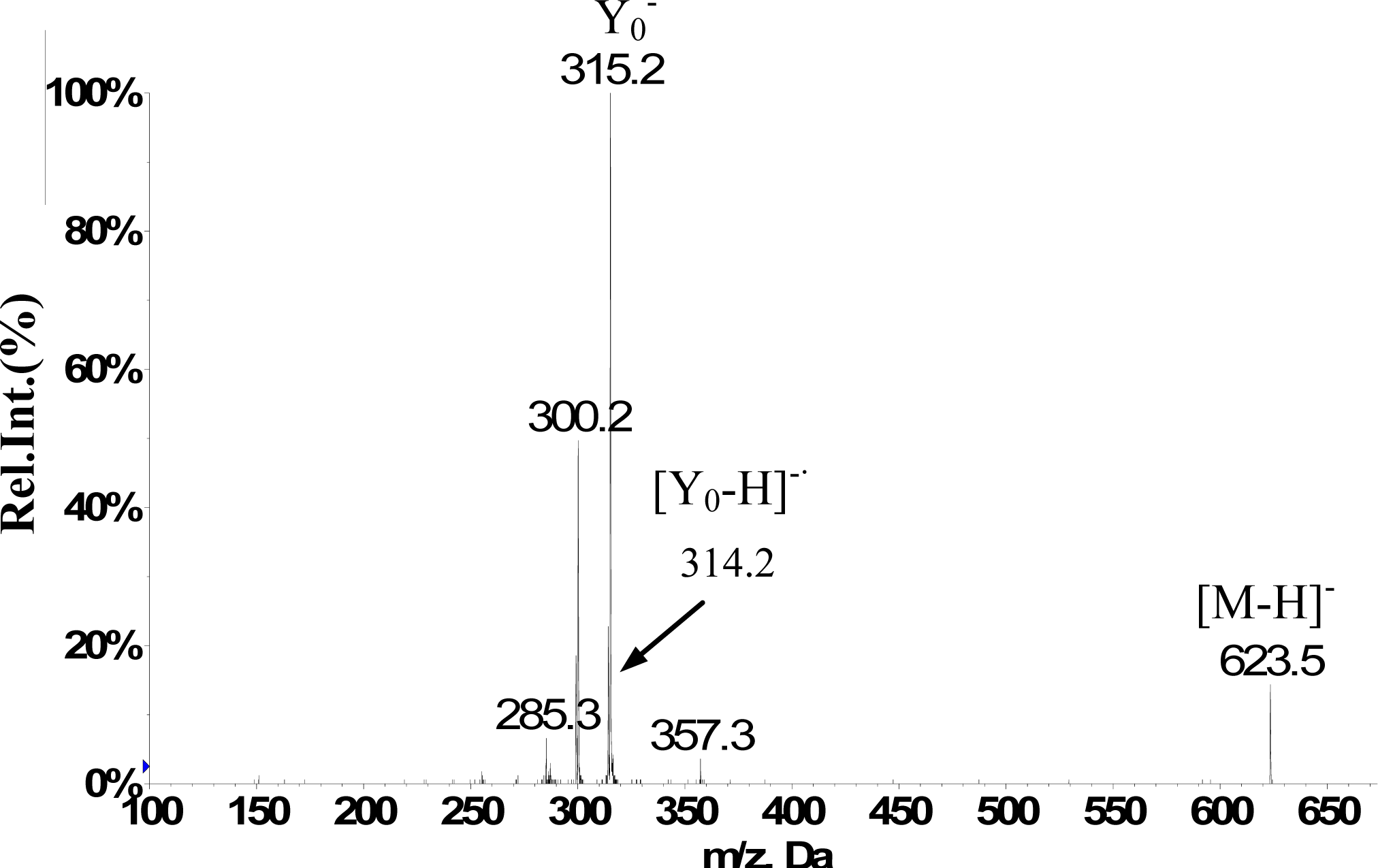


{kind=link}
{kind=link}
{kind=link}
{kind=link}
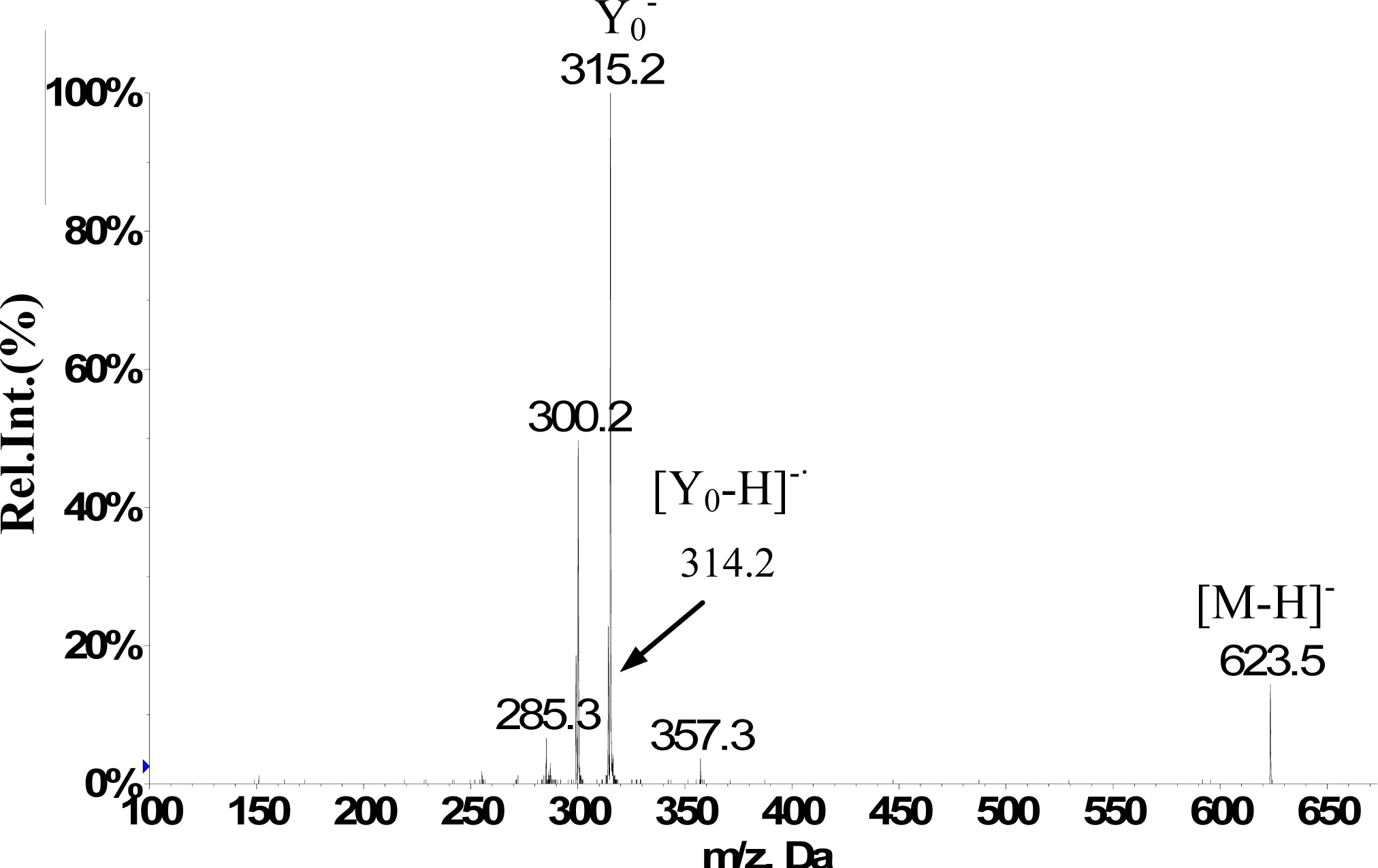{kind=link}
{kind=link}
| Compound | Rt (min) | [M-H]− (m/z) | 1H NMR data (δ, ppm; J, Hz) | LC-MS/MS data (m/z, relative abundances, ℅) |
|---|---|---|---|---|
| p-hydroxybenzaldehyde (1) | 6.8 | 121 | H–CHO 9.87s, H-2,6 7.76d (2H, 8.5Hz), H-3,5 6.78d (2H, 8.5Hz) | 121(100), 92(97), 65(10) |
| trans-4-hydroxycinnamic acid (2) | 8.5 | 163 | H–OH 9.78brs, H-2,6 7.44d (2H, 8.6 Hz), H-3,5 6.80d (2H, 8.6 Hz), H-7 7.59 d (16.0 Hz), H-8 6.27d (16.0 Hz) | 163(10), 119(100), 93(20) |
| trans-3-hydroxycinnamic acid (3)* | 9.2 | 163 | H-2 7.03a, H-4 6.85dd (2.0, 8.5Hz), H-5 7.23t (8.5,8.5Hz), H-6 7.10a(8.5,2.0Hz), H-7 6.43d(15.9Hz), H-8 7.52d (15.9Hz) | 163(10), 119(100), 93(20) |
| trans-2-hydroxycinnamic acid (4)* | 9.7 | 163 | H-3 7.00d (8.1Hz), H-4 6.88 m, H-5 7.24 m, H-6 7.59d (7.7Hz), H-7 7.99 d (16Hz), H-8 6.59d (16Hz) | 163(10), 119(100), 93(30) |
| Isorhamnetin-3-O-[6″-rhamnosyl(1→6)] glucopyranoside(5) | 12.1 | 623 | H-2’ 7.50 d (2.0Hz), H-5’ 7.06 d (8.5Hz), H-6’ 7.41dd (2.0,8.5Hz), H-6 6.11 d(2.0Hz), H-8 6.45d (2.0Hz), H-1″ 5.39a, H–OCH3 3.83s (3H) | 623(25), 357(10), 315(100), 300(50), 285(10) |
| 5-hydroxyferulic acid (6)* | 13.0 | 209 | H-2 7.00d(1.5Hz), H-6 6.88d(1.5Hz), H-7 7.69d (15.5Hz), H-8 6.33d(15.5Hz), H–OCH3 3.97s (3H) | 209(70), 165(40), 141(100) |
| 3,5-dihydroxy-3’,4’,7-trimethoflavone (7) | 15.0 | 343 | H-6’7.85dd (2.0,8.5Hz), H-2’7.77 d (2.0Hz), H–OCH3 3.84 s (9H), H-5’7.14 d (8.5Hz), H-8 6.71 d (2.0Hz), H-6 6.19 d (2.0Hz) | 313(90), 285(15), 254(50), 242(100), 198(30),151(10) |
| 5,6,3’,4’-tetrahydroxy-7,5’-trimethoflavonol-3’-O-glucoside (8)* | 16.2 | 523 | H-2’ 7.50d(1.5Hz), H-6’ 7.48d (1.5Hz), H-8 6.91s, H–OCH3 3.89s (3H), 3.87s (3H) | 523(70), 361(100), 329(30), 315(40), 299(20), 179(10) |
| 5,4’-dihydroxy-6,7,3’-trimethoflavone (9) | 18.4 | 343 | H-2’8.03d (1.5HZ), H-6’7.29dd (1.5,8.0Hz), H 5’7.08d (8.0Hz), H-8 6.82s, H-3 6.30s H–OCH3 3.84s (9H) | 343(25), 328(49), 313(100), 298(10), 242(37), 214(8) |
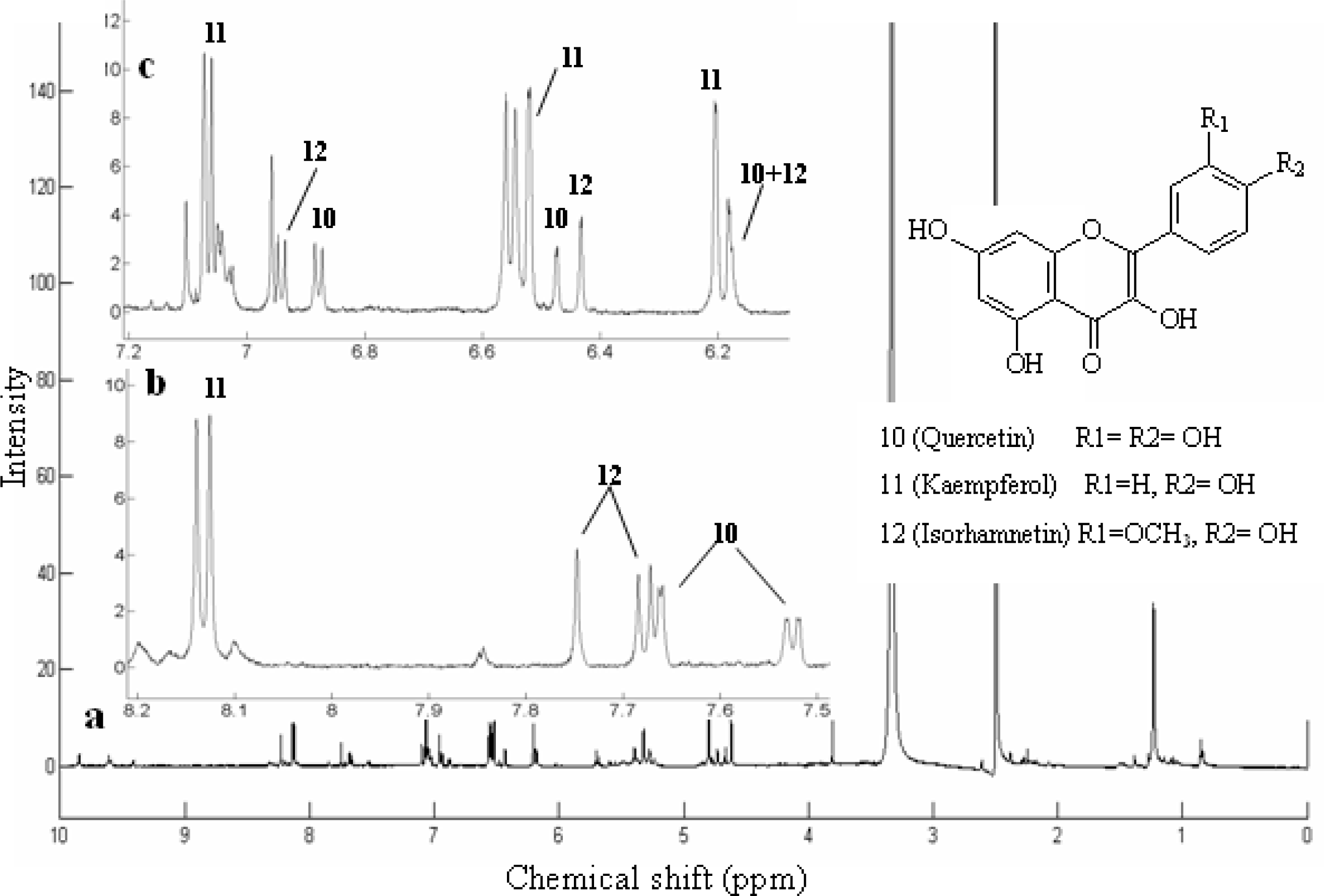
| Quercetin (10) | 18.4 | 301 | H-2’7.65d (2.0Hz), H-5’6.88d (8.5Hz), H-8 6.47d (2.0Hz), H-6’7.53dd (2.0,8.5Hz), H-6 6.18 d (2.0Hz), H-3’-OH 9.35brs, H-3-OH 9.40 brs, H-4’-OH 9.65 brs | 301(84), 273(7), 179(40), 151(100), 121(26), 107(43) |
| Kaempferol (11)* | 18.9 | 285 | H-2 ‘,6’ 8.15 d (2H, 8.8 Hz), H-3’,5’7.07 d (2H, 8.8 Hz), H-8 6.53 d (2.0 Hz), H-6 6.27d(2.0 Hz) | 285(100), 229(6), 211(7),185(8) |
| Isorhamnetin (12) | 19.6 | 315 | H-2’ 7.74s, H-6’ 7.68d (8.5Hz), H-5’ 6.93d (8.5Hz), H-6 6.18d (2.0Hz),H-8 6.43 d (2.0Hz), H–OCH33.81s (3H) | 315(90), 300(100), 271(12), 255(10), 227(15), 164(20), 151(35) |
| Quercetin 3,4’-dimethyl ether (13) | 24.6 | 329 | H-2’7.66d (2.0Hz), H-6’7.51dd (2.0, 8.5Hz), H-5’6.87d (8.5Hz), H-8 6.51d (1.8Hz), H-6 6.26d (1.8Hz), H–OCH3 3.89s (3H), 3.87s (3H) | 329(70), 314(64), 299(45), 285(58), 271(95), 243(100), 199(22) |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, X.-X.; He, J.-M.; Wang, C.-L.; Zhang, R.-P.; He, W.-Y.; Guo, S.-X.; Sun, R.-X.; Abliz, Z. Simultaneous Structural Identification of Natural Products in Fractions of Crude Extract of the Rare Endangered Plant Anoectochilus roxburghii Using 1H NMR/RRLC-MS Parallel Dynamic Spectroscopy. Int. J. Mol. Sci. 2011, 12, 2556-2571. https://doi.org/10.3390/ijms12042556
Wang X-X, He J-M, Wang C-L, Zhang R-P, He W-Y, Guo S-X, Sun R-X, Abliz Z. Simultaneous Structural Identification of Natural Products in Fractions of Crude Extract of the Rare Endangered Plant Anoectochilus roxburghii Using 1H NMR/RRLC-MS Parallel Dynamic Spectroscopy. International Journal of Molecular Sciences. 2011; 12(4):2556-2571. https://doi.org/10.3390/ijms12042556
Chicago/Turabian StyleWang, Xiao-Xue, Jiu-Ming He, Chun-Lan Wang, Rui-Ping Zhang, Wen-Yi He, Shun-Xing Guo, Rui-Xiang Sun, and Zeper Abliz. 2011. "Simultaneous Structural Identification of Natural Products in Fractions of Crude Extract of the Rare Endangered Plant Anoectochilus roxburghii Using 1H NMR/RRLC-MS Parallel Dynamic Spectroscopy" International Journal of Molecular Sciences 12, no. 4: 2556-2571. https://doi.org/10.3390/ijms12042556