Functioning Nanomachines Seen in Real-Time in Living Bacteria Using Single-Molecule and Super-Resolution Fluorescence Imaging

Abstract
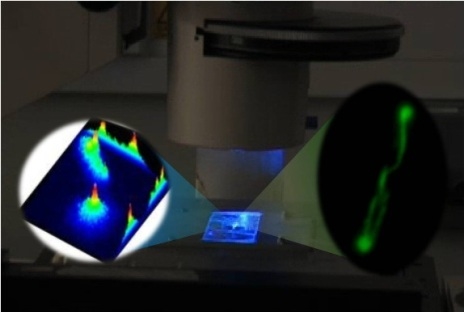
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
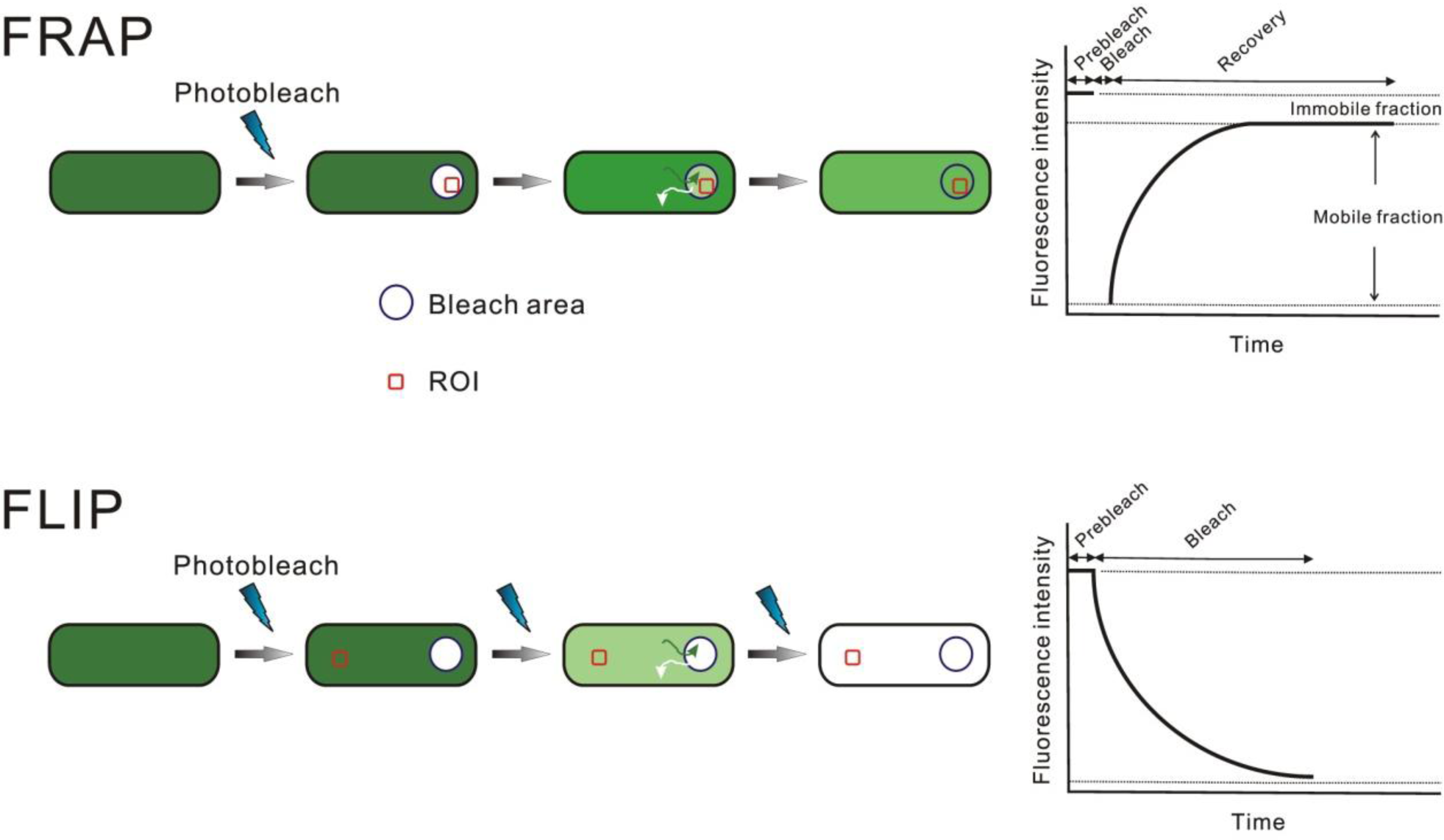
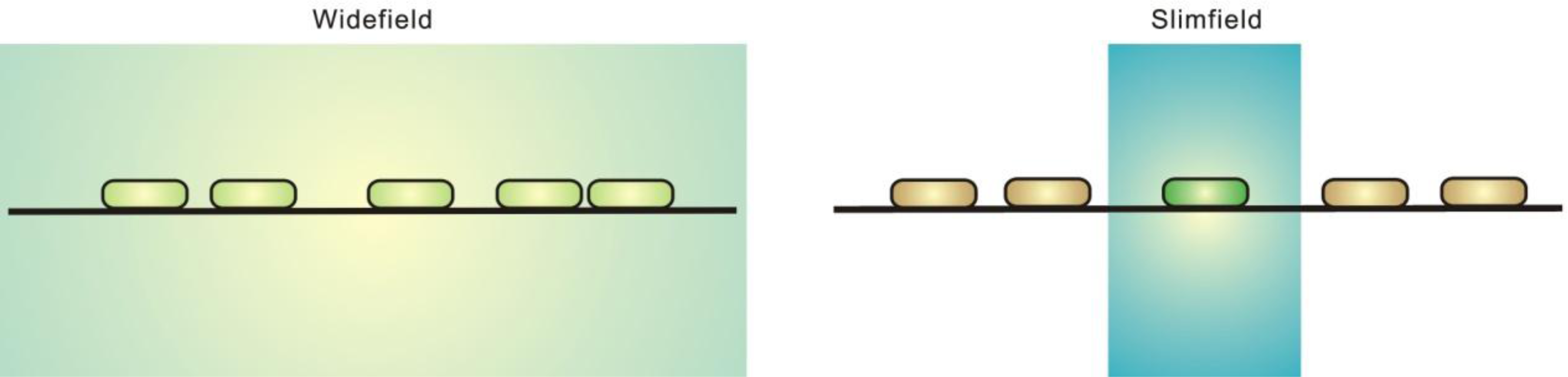{kind=link}
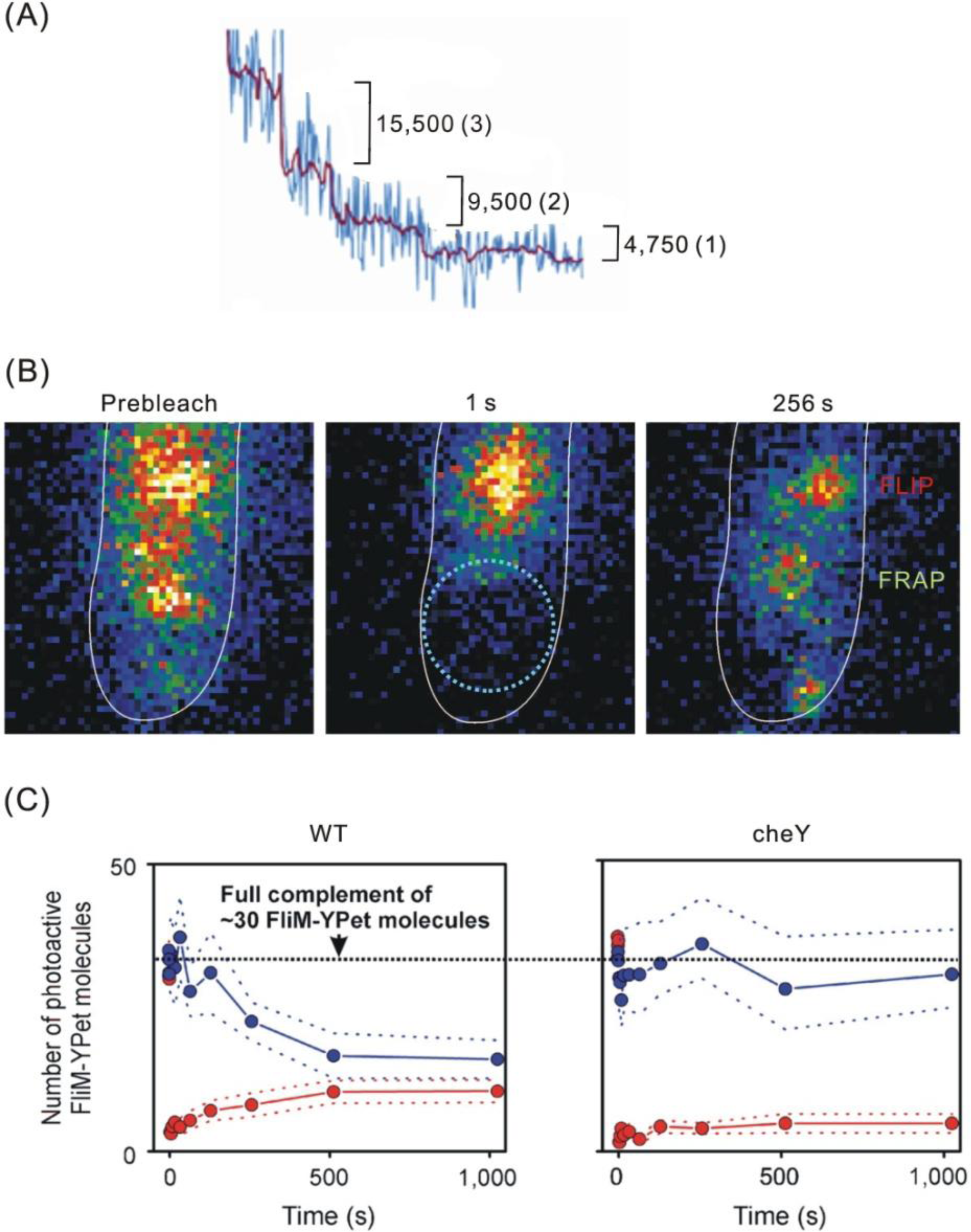{kind=link}
{kind=link}
1. Introduction
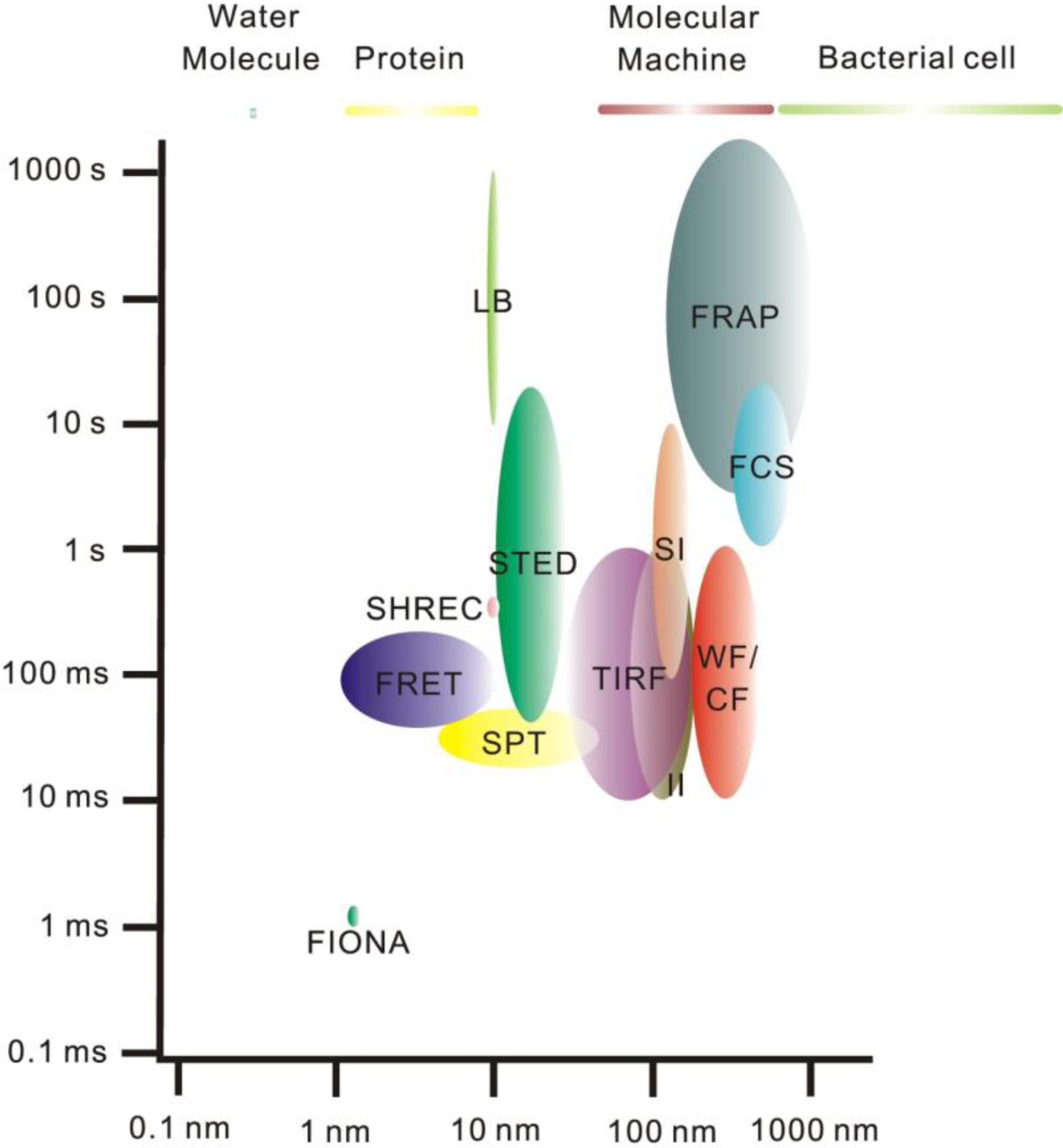
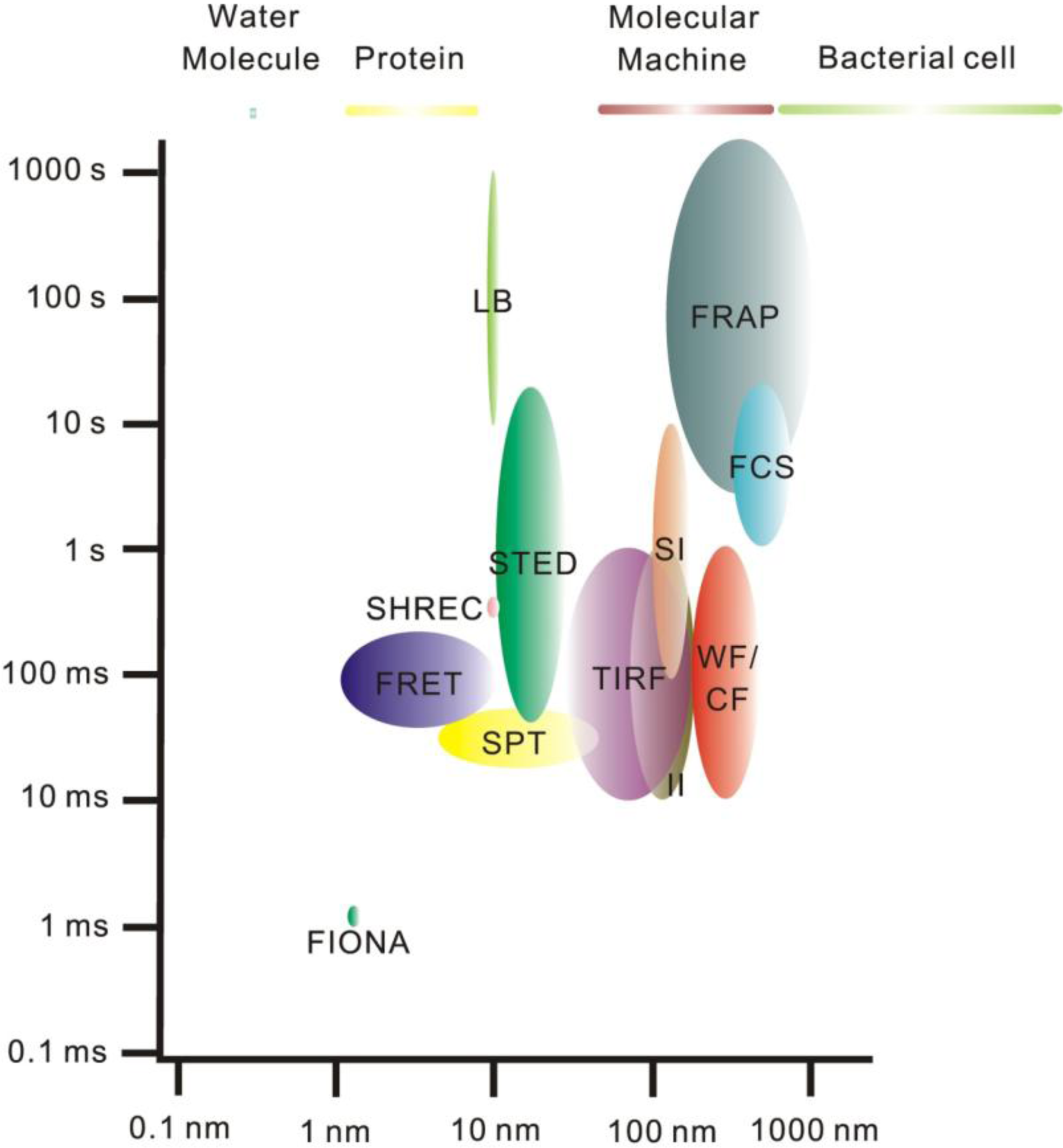
2. Single-Molecule Fluorescence Microscopy
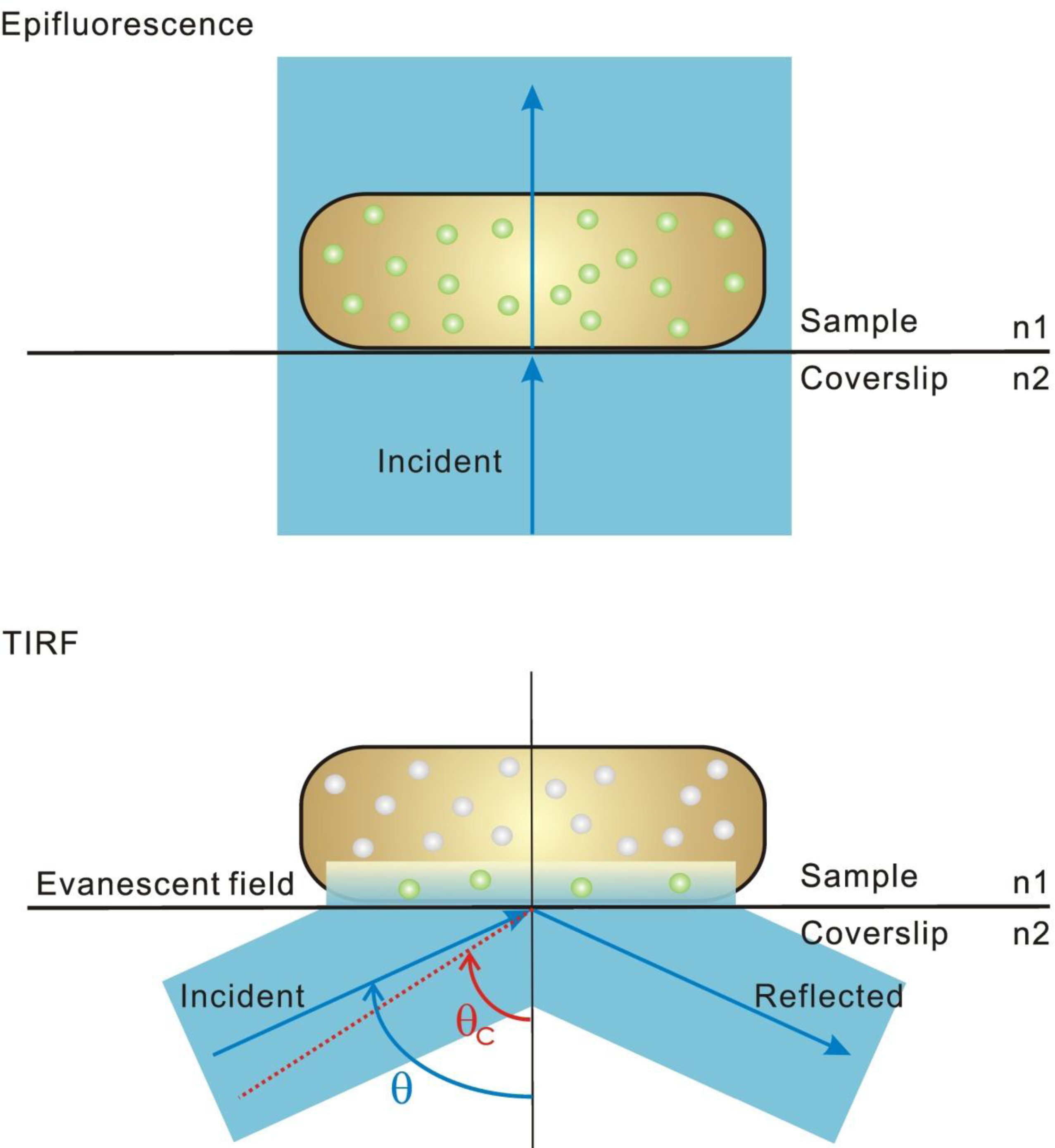
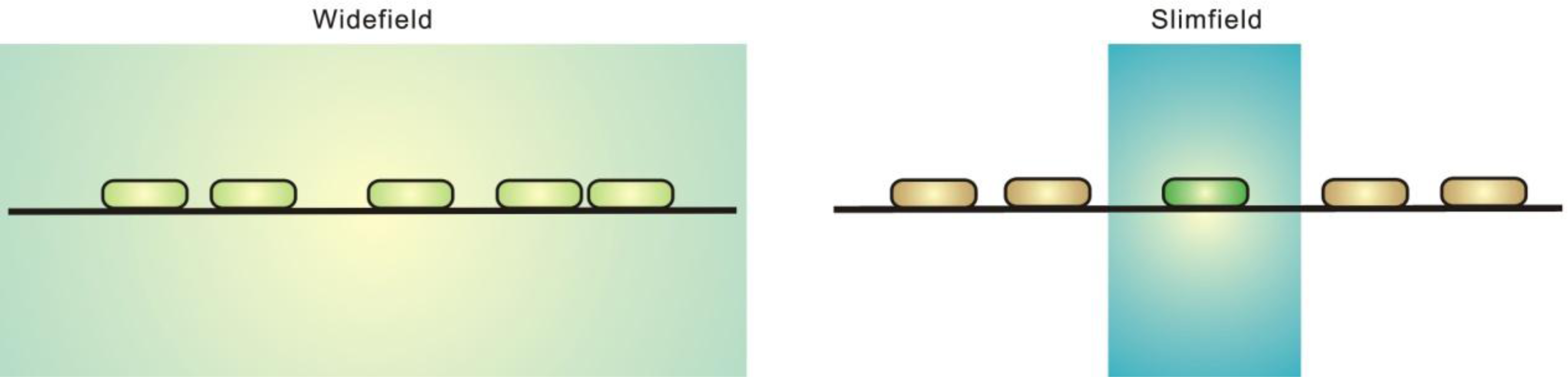
2.1. Fluorescence Imaging: A Brief Introduction
2.2. Fluorescent Proteins and Their Applications
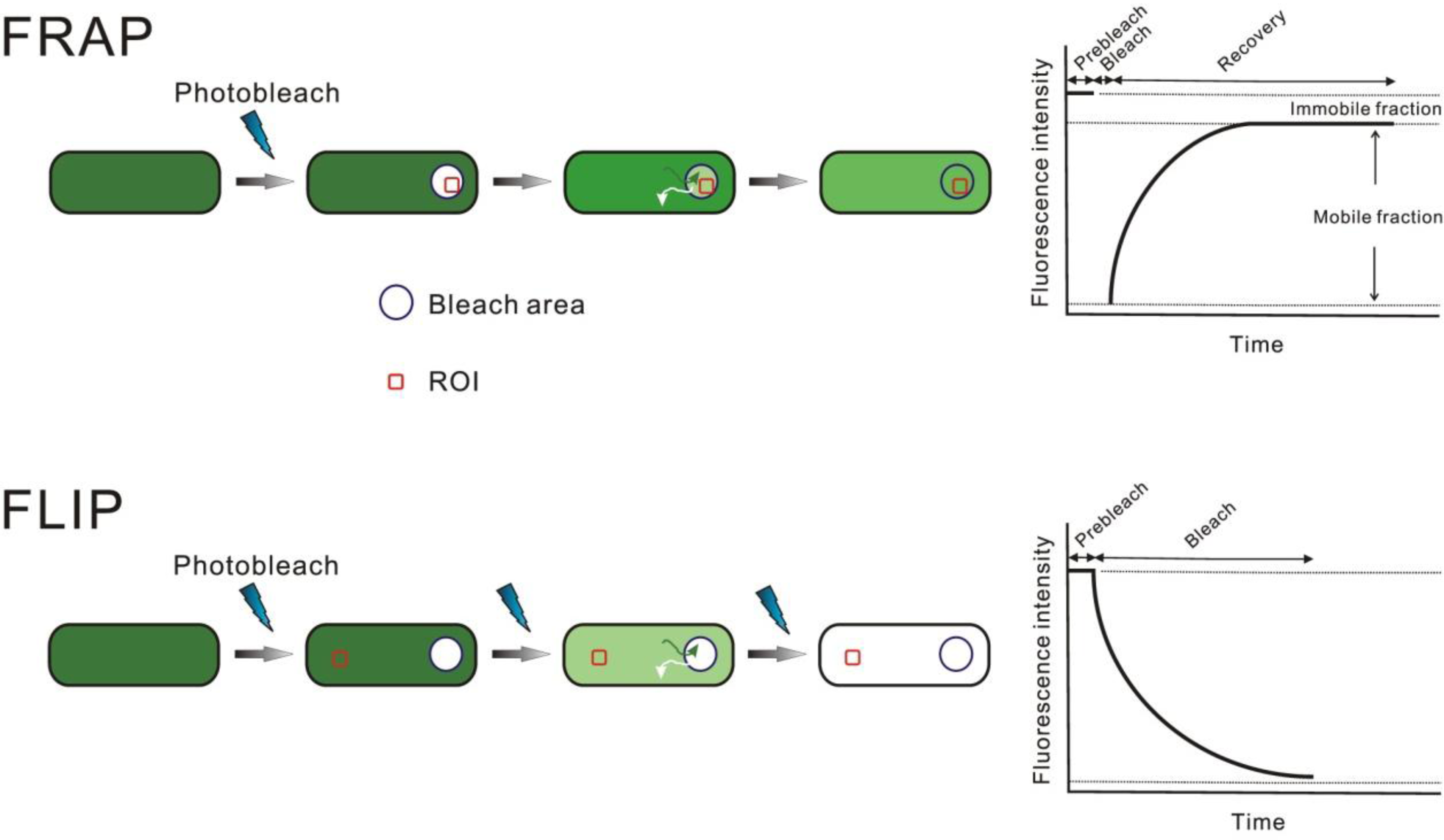
Photobleaching of Fluorescent Proteins
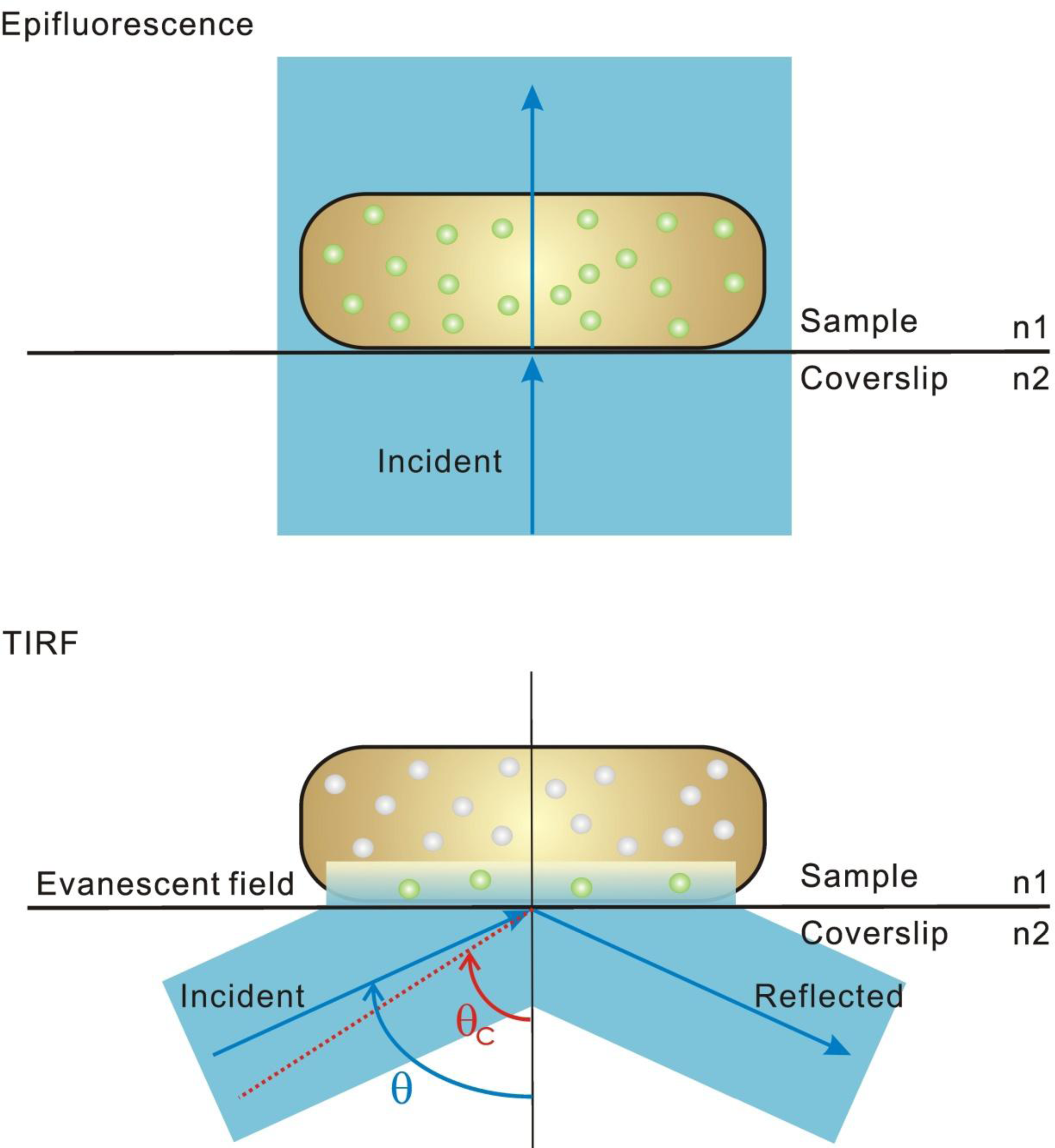
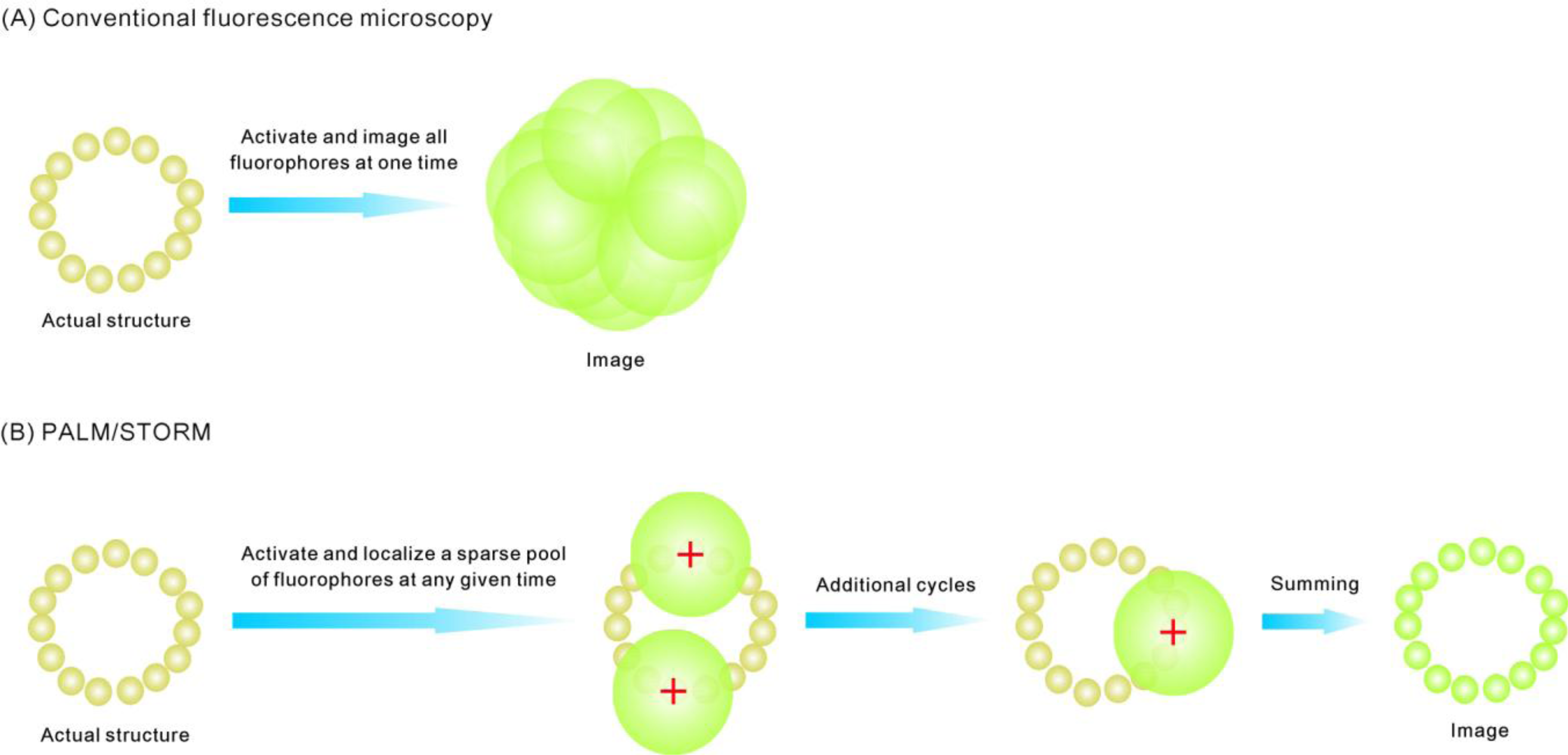
3. Super-Resolution and Single-Molecule Fluorescence Microscopy
4. Experimental Studies
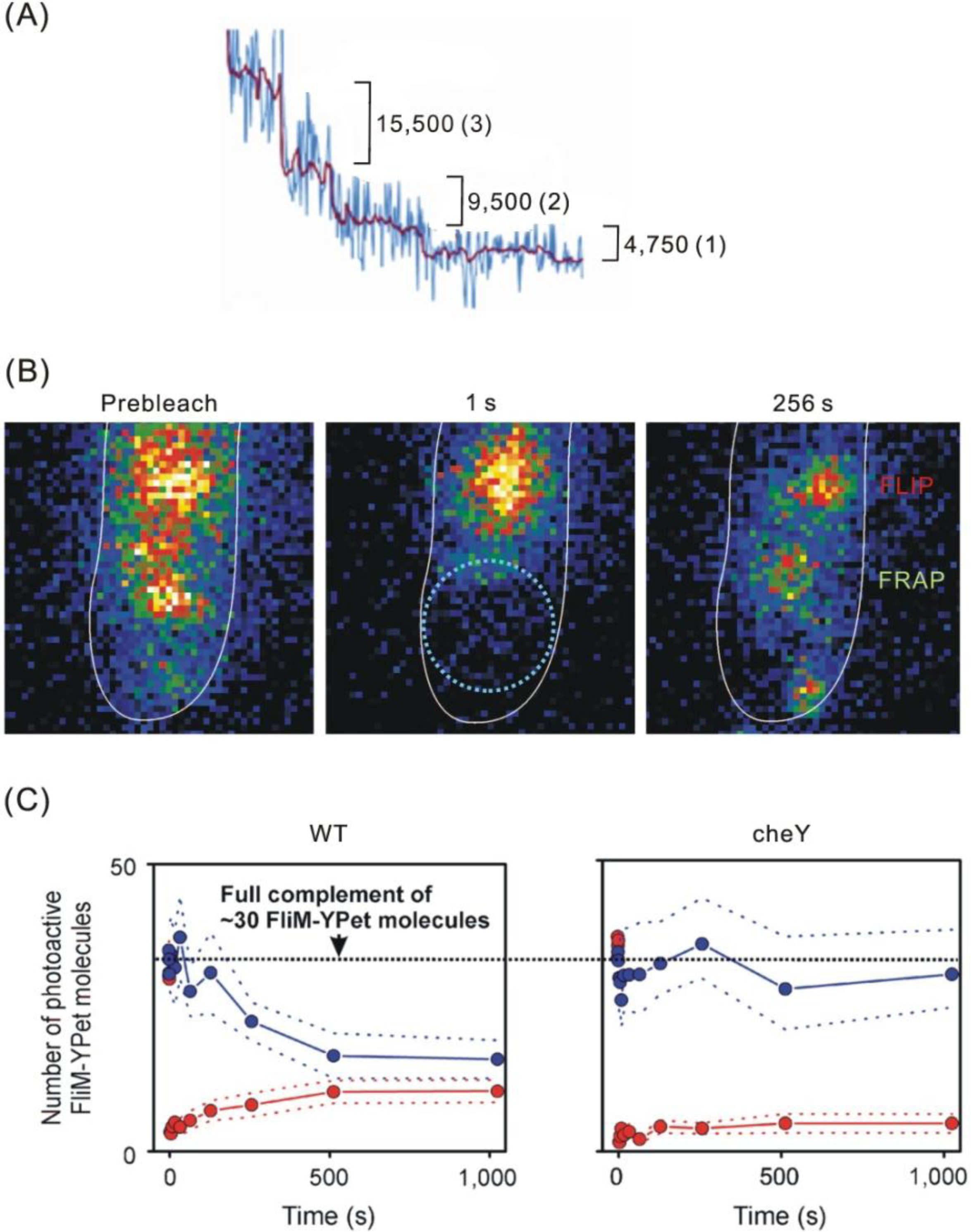
4.1. Flagellar Motors
4.2. Membrane Transporters and Energetic Complexes
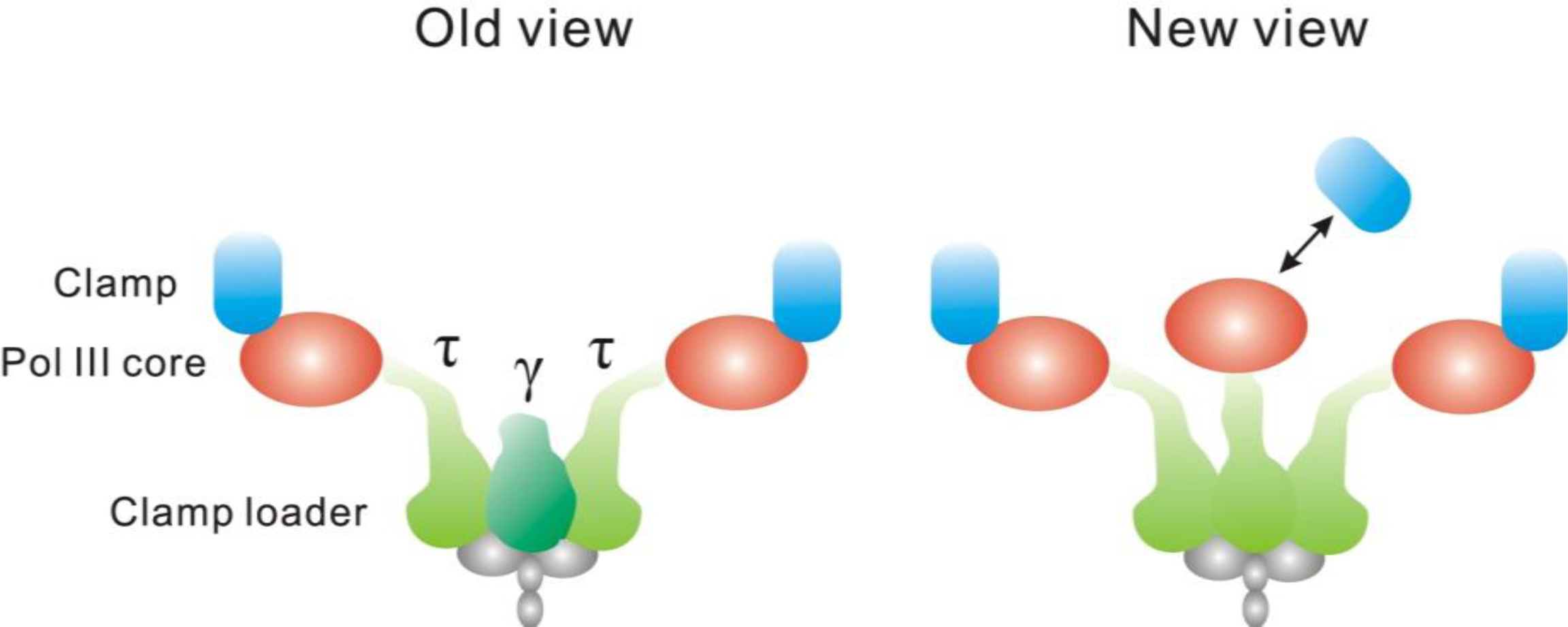
4.3. The Replisome
5. Cytoskeletons
5.1. MreB
5.2. FtsZ
5.3. ParA
6. Conclusions
Acknowledgments
References and Notes
- Alberts, B. The Cell as a Collection of Protein Machines: Preparing the Next Generation of Molecular Biologists. Cell 1998, 92, 291–294. [Google Scholar]
- Kinbara, K; Aida, T. Toward Intelligent Molecular Machines: Directed Motions of Biological and Artificial Molecules and Assemblies. Chem. Rev 2005, 105, 1377–1400. [Google Scholar]
- Mavroidis, C; Dubey, A; Yarmush, ML. Molecular Machines. Annu. Rev. Biomed. Eng 2004, 6, 363–395. [Google Scholar]
- Piccolino, M. Biological Machines: From Mills to Molecules. Nat. Rev. Mol. Cell Biol 2000, 1, 149–152. [Google Scholar]
- van den Heuvel, MGL; Dekker, C. Motor Proteins at Work for Nanotechnology. Science 2007, 317, 333–336. [Google Scholar]
- Cabeen, MT; Jacobs-Wagner, C. The Bacterial Cytoskeleton. Annu. Rev. Genet 2010, 44, 365–392. [Google Scholar]
- Erhardt, M; Namba, K; Hughes, KT. Bacterial Nanomachines: The Flagellum and Type III Injectisome. Cold Spring Harb. Perspect. Biol 2010, 2, a000299. [Google Scholar]
- Erickson, HP; Anderson, DE; Osawa, M. FtsZ in Bacterial Cytokinesis: Cytoskeleton and Force Generator All in One. Microbiol. Mol. Biol. Rev 2010, 74, 504–528. [Google Scholar]
- Gerdes, K; Howard, M; Szardenings, F. Pushing and Pulling in Prokaryotic DNA Segregation. Cell 2010, 141, 927–942. [Google Scholar]
- Hayes, F; Barilla, D. The Bacterial Segrosome: A Dynamic Nucleoprotein Machine for DNA Trafficking and Segregation. Nat. Rev. Microbiol 2006, 4, 133–143. [Google Scholar]
- Löwe, J; Amos, LA. Evolution of Cytomotive Filaments: The Cytoskeleton from Prokaryotes to Eukaryotes. Int. J. Biochem. Cell Biol 2009, 41, 323–329. [Google Scholar]
- Allemand, JF; Maier, B. Bacterial Translocation Motors Investigated by Single Molecule Techniques. FEMS Microbiol. Rev 2009, 33, 593–610. [Google Scholar]
- Lenn, T; Leake, MC; Mullineaux, CW. Are Escherichia coli OXPHOS Complexes Concentrated in Specialized Zones within the Plasma Membrane? Biochem. Soc. Trans 2008, 36, 1032–1036. [Google Scholar]
- Pomerantz, RT; O’Donnell, M. Replisome Mechanics: Insights into a Twin DNA Polymerase Machine. Trends Microbiol 2007, 15, 156–164. [Google Scholar]
- Robinson, C; Matos, CFRO; Beck, D; Ren, C; Lawrence, J; Vasisht, N; Mendel, S. Transport and Proofreading of Proteins by the Twin-Arginine Translocation (Tat) System in Bacteria. Biochim. Biophys. Acta 2011, 1808, 876–884. [Google Scholar]
- Gitai, Z. New Fluorescence Microscopy Methods for Microbiology: Sharper, Faster, and Quantitative. Curr. Opin. Microbiol 2009, 12, 341–346. [Google Scholar]
- Chiu, W; Baker, ML; Almo, SC. Structural Biology of Cellular Machines. Trends Cell Biol 2006, 16, 144–150. [Google Scholar]
- Nogales, E; Grigorieff, N. Molecular Machines. J. Cell Biol 2001, 152, F1–F10. [Google Scholar]
- Peters, R. Single-Molecule Fluorescence Analysis of Cellular Nanomachinery Components. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 371–394. [Google Scholar]
- Hohlbein, J; Gryte, K; Heilemann, M; Kapanidis, AN. Surfing on a New Wave of Single-Molecule Fluorescence Methods. Phys. Biol 2010, 7, 031001. [Google Scholar]
- Kapanidis, AN; Strick, T. Biology, One Molecule at a Time. Trends Biochem. Sci 2009, 34, 234–243. [Google Scholar]
- Leake, MC. Shining the Spotlight on Functional Molecular Complexes: The New Science of Single-Molecule Cell Biology. Commun. Integr. Biol 2010, 3, 415–418. [Google Scholar]
- Vale, RD. Microscopes for Fluorimeters: The Era of Single Molecule Measurements. Cell 2008, 135, 779–785. [Google Scholar]
- Walter, NG; Huang, C-Y; Manzo, AJ; Sobhy, MA. Do-It-Yourself Guide: How to Use the Modern Single-Molecule Toolkit. Nat. Meth 2008, 5, 475–489. [Google Scholar]
- Wennmalm, S; Simon, SM. Studying Individual Events in Biology. Annu. Rev. Biochem 2007, 76, 419–446. [Google Scholar]
- Zlatanova, J; van Holde, K. Single-Molecule Biology: What Is It and How Does It Work? Mol. Cell 2006, 24, 317–329. [Google Scholar]
- Xie, XS; Yu, J; Yang, WY. Living Cells as Test Tubes. Science 2006, 312, 228–230. [Google Scholar]
- Gierasch, LM; Gershenson, A. Post-Reductionist Protein Science, or Putting Humpty Dumpty Back Together Again. Nat. Chem. Biol 2009, 5, 774–777. [Google Scholar]
- Sung, M-H; McNally, JG. Live Cell Imaging and Systems Biology. Wiley Interdiscip. Rev. Syst. Biol. Med 2011, 3, 167–182. [Google Scholar]
- Sako, Y. Imaging Single Molecules in Living Cells for Systems Biology. Mol. Syst. Biol 2006, 2, 56. [Google Scholar]
- Xie, XS; Choi, PJ; Li, G-W; Lee, NK; Lia, G. Single-Molecule Approach to Molecular Biology in Living Bacterial Cells. Annu. Rev. Biophys 2008, 37, 417–444. [Google Scholar]
- Lichtman, JW; Conchello, JA. Fluorescence Microscopy. Nat. Meth 2005, 2, 910–919. [Google Scholar]
- Schermelleh, L; Heintzmann, R; Leonhardt, H. A Guide to Super-Resolution Fluorescence Microscopy. J. Cell Biol 2010, 190, 165–175. [Google Scholar]
- Thompson, MA; Biteen, JS; Lord, SJ; Conley, NR; Moerner, WE. Molecules and Methods for Super-Resolution Imaging. In Methods in Enzymology; Nils, GW, Ed.; Academic Press: Burlington, MD, USA, 2010. [Google Scholar]
- Waters, JC. Accuracy and Precision in Quantitative Fluorescence Microscopy. J. Cell Biol 2009, 185, 1135–1148. [Google Scholar]
- Toomre, D; Bewersdorf, J. A New Wave of Cellular Imaging. Annu. Rev. Cell Dev. Biol 2010, 26, 285–314. [Google Scholar]
- North, AJ. Seeing Is Believing? A Beginners’ Guide to Practical Pitfalls in Image Acquisition. J. Cell Biol 2006, 172, 9–18. [Google Scholar]
- Brown, CM. Fluorescence Microscopy—Avoiding the Pitfalls. J. Cell Sci 2007, 120, 1703–1705. [Google Scholar]
- Xiao, J. Single-Molecule Imaging in Live Cells. In Handbook of Single-Molecule Biophysics; Hinterdorfer, P, Oijen, A, Eds.; Springer: New York, NY, USA, 2009; pp. 43–93. [Google Scholar]
- Giepmans, BNG; Adams, SR; Ellisman, MH; Tsien, RY. The Fluorescent Toolbox for Assessing Protein Location and Function. Science 2006, 312, 217–224. [Google Scholar]
- Fernandez-Suarez, M; Ting, AY. Fluorescent Probes for Super-Resolution Imaging in Living Cells. Nat. Rev. Mol. Cell Biol 2008, 9, 929–943. [Google Scholar]
- Chudakov, DM; Matz, MV; Lukyanov, S; Lukyanov, KA. Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol. Rev 2010, 90, 1103–1163. [Google Scholar]
- Kremers, G-J; Gilbert, SG; Cranfill, PJ; Davidson, MW; Piston, DW. Fluorescent Proteins at a Glance. J. Cell Sci 2011, 124, 157–160. [Google Scholar]
- Wiedenmann, J; Oswald, F; Nienhaus, GU. Fluorescent Proteins for Live Cell Imaging: Opportunities, Limitations, and Challenges. IUBMB Life 2009, 61, 1029–1042. [Google Scholar]
- Patterson, G; Davidson, M; Manley, S; Lippincott-Schwartz, J. Superresolution Imaging Using Single-Molecule Localization. Annu. Rev. Phys. Chem 2010, 61, 345–367. [Google Scholar]
- Phillips, GJ. Green Fluorescent Protein—A Bright Idea for the Study of Bacterial Protein Localization. FEMS Microbiol. Lett 2001, 204, 9–18. [Google Scholar]
- Lee, H-LD; Lord, SJ; Iwanaga, S; Zhan, K; Xie, H; Williams, JC; Wang, H; Bowman, GR; Goley, ED; Shapiro, L; Twieg, RJ; Rao, J; Moerner, WE. Superresolution Imaging of Targeted Proteins in Fixed and Living Cells Using Photoactivatable Organic Fluorophores. J. Am. Chem. Soc 2010, 132, 15099–15101. [Google Scholar]
- Lippincott-Schwar, J; Altan-Bonnet, N; Patterson, GH. Photobleaching and Photoactivation: Following Protein Dynamics in Living Cells. Nat Cell Biol 2003, (Suppl), S7–S14. [Google Scholar]
- Huang, B; Babcock, H; Zhuang, X. Breaking the Diffraction Barrier: Super-Resolution Imaging of Cells. Cell 2010, 143, 1047–1058. [Google Scholar]
- Huang, B. Super-Resolution Optical Microscopy: Multiple Choices. Curr. Opin. Chem. Biol 2010, 14, 10–14. [Google Scholar]
- Huang, B; Bates, M; Zhuang, X. Super-Resolution Fluorescence Microscopy. Annu. Rev. Biochem 2009, 78, 993–1016. [Google Scholar]
- Toprak, E; Kural, C; Selvin, PR. Super-Accuracy and Super-Resolution: Getting Around the Diffraction Limit. In Methods in Enzymology; Nils, GW, Ed.; Academic Press: Burlington, MD, USA, 2010. [Google Scholar]
- Lord, SJ; Lee, H-LD; Moerner, WE. Single-Molecule Spectroscopy and Imaging of Biomolecules in Living Cells. Anal. Chem 2010, 82, 2192–2203. [Google Scholar]
- Leake, MC; Dobbie, IM; Robson, A; Delalez, N. Visualizing Single Molecular Complexes in Vivo Using Advanced Fluorescence Microscopy. J. Vis. Exp 2009, 31, e1508. [Google Scholar]
- Mattheyses, AL; Simon, SM; Rappoport, JZ. Imaging with Total Internal Reflection Fluorescence Microscopy for the Cell Biologist. J. Cell Sci 2010, 123, 3621–3628. [Google Scholar]
- Lidke, DS; Wilson, BS. Caught in the Act: Quantifying Protein Behaviour in Living Cells. Trends Cell Biol 2009, 19, 566–574. [Google Scholar]
- McEvoy, A; Greenfield, D; Bates, M; Liphardt, J. Q&A: Single-Molecule Localization Microscopy for Biological Imaging. BMC Biol 2010, 8, 106. [Google Scholar]
- Yildiz, A; Selvin, PR. Fluorescence Imaging with One Nanometer Accuracy: Application to Molecular Motors. Acc. Chem. Res 2005, 38, 574–582. [Google Scholar]
- Biteen, JS; Moerner, WE. Single-Molecule and Superresolution Imaging in Live Bacteria Cells. Cold Spring Harb Perspect Biol 2010. [Google Scholar] [CrossRef]
- Park, H; Toprak, E; Selvin, PR. Single-Molecule Fluorescence to Study Molecular Motors. Q. Rev. Biophys 2007, 40, 87–111. [Google Scholar]
- Kural, C; Kim, H; Syed, S; Goshima, G; Gelfand, VI; Selvin, PR. Kinesin and Dynein Move a Peroxisome in Vivo: A Tug-of-War or Coordinated Movement? Science 2005, 308, 1469–1472. [Google Scholar]
- A good review of the technique is found in Ref. 52, but it was invented as a technology in Ref. 63 with a name for the technique added in Ref. 64, both studies using walking of myosin V as the biological test system.
- Warshaw, DM; Kennedy, GG; Work, SS; Krementsova, EB; Beck, S; Trybus, KM. Differential Labeling of Myosin V Heads with Quantum Dots Allows Direct Visualization of Hand-over-Hand Processivity. Biophys. J 2005, 88, L30–L32. [Google Scholar]
- Churchman, LS; Okten, Z; Rock, RS; Dawson, JF; Spudich, JA. Single Molecule High-Resolution Colocalization of Cy3 and Cy5 Attached to Macromolecules Measures Intramolecular Distances through Time. Proc. Natl. Acad. Sci. USA 2005, 102, 1419–1423. [Google Scholar]
- Joglekar, AP; Bloom, K; Salmon, ED. In Vivo Protein Architecture of the Eukaryotic Kinetochore with Nanometer Scale Accuracy. Curr. Biol 2009, 19, 694–699. [Google Scholar]
- Wan, X; O’Quinn, RP; Pierce, HL; Joglekar, AP; DeLuca, JG; Desai, A; Yen, TJ; Salmon, ED. Protein Architecture of the Human Kinetochore Microtubule Attachment Site. Cell 2009, 137, 672–684. [Google Scholar]
- Gordon, MP; Ha, T; Selvin, PR. Single-Molecule High-Resolution Imaging with Photobleaching. Proc. Natl. Acad. Sci. USA 2004, 101, 6462–6465. [Google Scholar]
- Qu, X; Wu, D; Mets, L; Scherer, NF. Nanometer-Localized Multiple Single-Molecule Fluorescence Microscopy. Proc. Natl. Acad. Sci. USA 2004, 101, 11298–11303. [Google Scholar]
- Balci, H; Ha, T; Sweeney, HL; Selvin, PR. Interhead Distance Measurements in Myosin VI via SHRImP Support a Simplified Hand-over-Hand Model. Biophys. J 2005, 89, 413–417. [Google Scholar]
- Pertsinidis, A; Zhang, Y; Chu, S. Subnanometre Single-Molecule Localization, Registration and Distance Measurements. Nature 2010, 466, 647–651. [Google Scholar]
- Manley, S; Gillette, JM; Lippincott-Schwartz, J. Single-Particle Tracking Photoactivated Localization Microscopy for Mapping Single-Molecule Dynamics. In Methods in Enzymology; Nils, GW, Ed.; Academic Press: Burlington, MD, USA, 2010; Volume 475, pp. 109–120. [Google Scholar]
- Bowman, G; Comolli, L; Zhu, J; Eckart, M; Koenig, M; Downing, K; Moerner, W; Earnest, T; Shapiro, L. A Polymeric Protein Anchors the Chromosomal Origin/ParB Complex at a Bacterial Cell Pole. Cell 2008, 134, 945–955. [Google Scholar]
- Deich, J; Judd, EM; McAdams, HH; Moerner, WE. Visualization of the Movement of Single Histidine Kinase Molecules in Live Caulobacter Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 15921–15926. [Google Scholar]
- Plank, M; Wadhams, GH; Leake, MC. Millisecond Timescale Slimfield Imaging and Automated Quantification of Single Fluorescent Protein Molecules for Use in Probing Complex Biological Processes. Integr. Biol 2009, 1, 602–612. [Google Scholar]
- Sowa, Y; Berry, RM. Bacterial Flagellar Motor. Q. Rev. Biophys 2008, 41, 103–132. [Google Scholar]
- Leake, MC; Chandler, JH; Wadhams, GH; Bai, F; Berry, RM; Armitage, JP. Stoichiometry and Turnover in Single, Functioning Membrane Protein Complexes. Nature 2006, 443, 355–358. [Google Scholar]
- Delalez, NJ; Wadhams, GH; Rosser, G; Xue, Q; Brown, MT; Dobbie, IM; Berry, RM; Leake, MC; Armitage, JP. Signal-Dependent Turnover of the Bacterial Flagellar Switch Potein FliM. Proc. Natl. Acad. Sci. USA 2010, 107, 11347–11351. [Google Scholar]
- Brown, PN; Terrazas, M; Paul, K; Blair, DF. Mutational Analysis of the Flagellar Protein FliG: Sites of Interaction with FliM and Implications for Organization of the Switch Complex. J. Bacteriol 2007, 189, 305–312. [Google Scholar]
- Manson, MD. How 34 Pegs Fit into 26 + 8 Holes in the Flagellar Motor. J. Bacteriol 2007, 189, 291–293. [Google Scholar]
- Manson, MD. Dynamic Motors for Bacterial Flagella. Proc. Natl. Acad. Sci. USA 2010, 107, 11151–11152. [Google Scholar]
- Yuan, J; Zweers, J; van Dijl, J; Dalbey, R. Protein Transport across and into Cell Membranes in Bacteria and Archaea. Cell. Mol. Life Sci 2010, 67, 179–199. [Google Scholar]
- Natale, P; Brüser, T; Driessen, AJM. Sec- and Tat-Mediated Protein Secretion across the Bacterial Cytoplasmic Membrane—Distinct Translocases and Mechanisms. Biochim. Biophys. Acta 2008, 1778, 1735–1756. [Google Scholar]
- Leake, MC; Greene, NP; Godun, RM; Granjon, T; Buchanan, G; Chen, S; Berry, RM; Palmer, T; Berks, BC. Variable Stoichiometry of the TatA Component of the Twin-Arginine Protein Transport System Observed by in Vivo Single-Molecule Imaging. Proc. Natl. Acad. Sci. USA 2008, 105, 15376–15381. [Google Scholar]
- Dudkina, NV; Kouril, R; Peters, K; Braun, H-P; Boekema, EJ. Structure and Function of Mitochondrial Supercomplexes. Biochim. Biophys. Acta 2010, 1797, 664–670. [Google Scholar]
- Johnson, AS; van Horck, S; Lewis, PJ. Dynamic Localization of Membrane Proteins in Bacillus subtilis. Microbiology 2004, 150, 2815–2824. [Google Scholar]
- Lenn, T; Leake, MC; Mullineaux, CW. Clustering and Dynamics of Cytochrome bd-I Complexes in the Escherichia coli Plasma Membrane in Vivo. Mol. Microbiol 2008, 70, 1397–1407. [Google Scholar]
- Nan, B; Chen, J; Neu, JC; Berry, RM; Oster, G; Zusman, DR. Myxobacteria gliding Motility Requires Cytoskeleton Rotation Powered by Proton Motive Force. Proc. Natl. Acad. Sci. USA 2011, 108, 2498–2503. [Google Scholar]
- Kobayashi, R; Suzuki, T; Yoshida, M. Escherichia coli Phage-Shock Protein A (PspA) Binds to Membrane Phospholipids and Repairs Proton Leakage of the Damaged Membranes. Mol. Microbiol 2007, 66, 100–109. [Google Scholar]
- Joly, N; Engl, C; Jovanovic, G; Huvet, M; Toni, T; Sheng, X; Stumpf, MPH; Buck, M. Managing Membrane Stress: the Phage Shock Protein (Psp) Response, from Molecular Mechanisms to Physiology. FEMS Microbiol. Rev 2010, 34, 797–827. [Google Scholar]
- Lenn, T; Gkekas, CN; Bernard, L; Engl, C; Jovanovic, G; Buck, M; Ying, L. Measuring the Stoichiometry of Functional PspA Complexes in Living Bacterial Cells by Single Molecule Photobleaching. Chem. Commun 2011, 47, 400–402. [Google Scholar]
- Engl, C; Jovanovic, G; Lloyd, LJ; Murray, H; Spitaler, M; Ying, L; Errington, J; Buck, M. In Vivo Localizations of Membrane Stress Controllers PspA and PspG in Escherichia coli. Mol. Microbiol 2009, 73, 382–396. [Google Scholar]
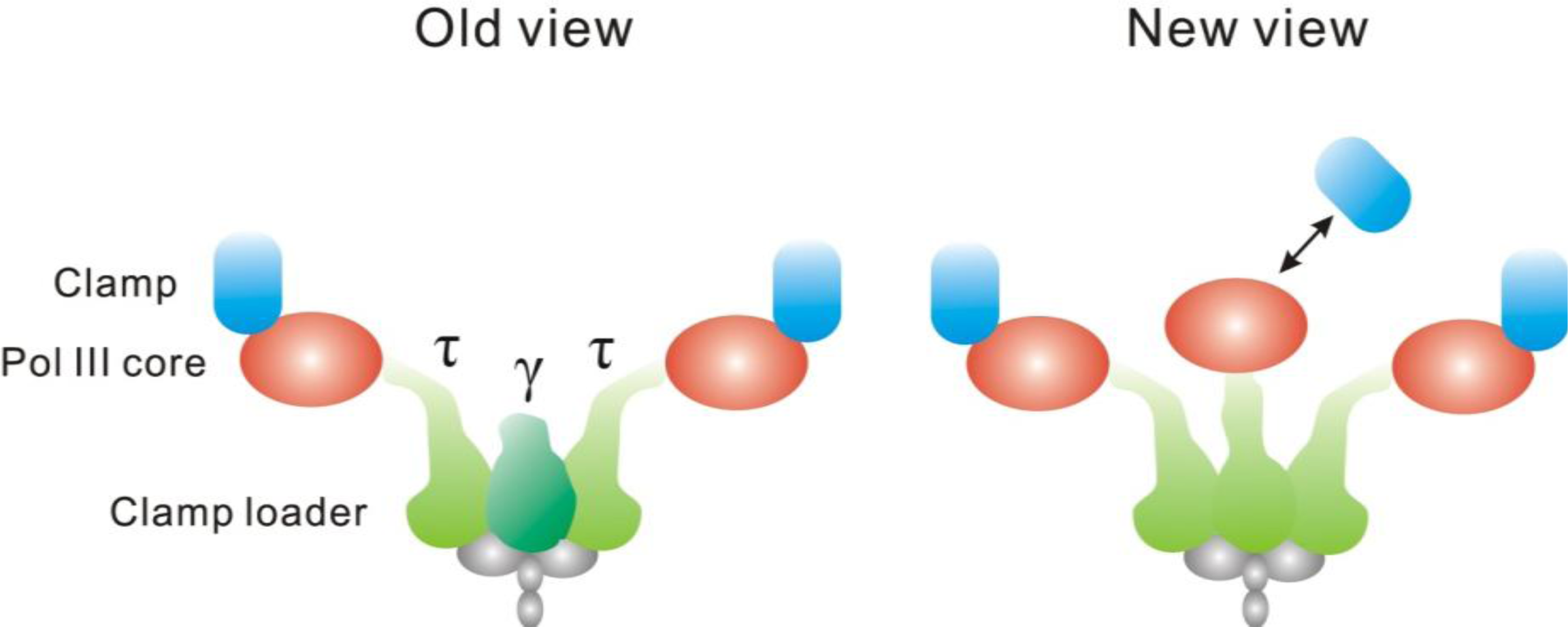
- van Oijen, AM; Loparo, JJ. Single-Molecule Studies of the Replisome. Annu. Rev. Biophys 2010, 39, 429–448. [Google Scholar]
- Perumal, SK; Yue, H; Hu, Z; Spiering, MM; Benkovic, SJ. Single-Molecule Studies of DNA Replisome Function. Biochim. Biophys. Acta 2010, 1804, 1094–1112. [Google Scholar]
- Reyes-Lamothe, R; Possoz, C; Danilova, O; Sherratt, DJ. Independent Positioning and Action of Escherichia coli Replisomes in Live Cells. Cell 2008, 133, 90–102. [Google Scholar]
- Reyes-Lamothe, R; Sherratt, DJ; Leake, MC. Stoichiometry and Architecture of Active DNA Replication Machinery in Escherichia coli. Science 2010, 328, 498–501. [Google Scholar]
- McInerney, P; Johnson, A; Katz, F; O’Donnell, M. Characterization of a Triple DNA Polymerase Replisome. Mol. Cell 2007, 27, 527–538. [Google Scholar]
- Shaevitz, JW; Gitai, Z. The Structure and Function of Bacterial Actin Homologs. Cold Spring Harb Perspect Biol 2010. [Google Scholar] [CrossRef]
- Kim, SY; Gitai, Z; Kinkhabwala, A; Shapiro, L; Moerner, WE. Single Molecules of the Bacterial Actin MreB Undergo Directed Treadmilling Motion. Caulobacter crescentus. Proc. Natl. Acad. Sci. USA 2006, 103, 10929–10934. [Google Scholar]
- Biteen, JS; Thompson, MA; Tselentis, NK; Bowman, GR; Shapiro, L; Moerner, WE. Super-Resolution Imaging in Live Caulobacter crescentus Cells Using Photoswitchable EYFP. Nat. Meth 2008, 5, 947–949. [Google Scholar]
- Niu, L; Yu, J. Investigating Intracellular Dynamics of FtsZ Cytoskeleton with Photoactivation Single-Molecule Tracking. Biophys. J 2008, 95, 2009–2016. [Google Scholar]
- Fu, G; Huang, T; Buss, J; Coltharp, C; Hensel, Z; Xiao, J. In Vivo Structure of the E. coli FtsZ-Ring Revealed by Photoactivated Localization Microscopy (PALM). PLoS ONE 2010, 5, e12680. [Google Scholar]
- Jennings, PC; Cox, GC; Monahan, LG; Harry, EJ. Super-Resolution Imaging of the Bacterial Cytokinetic Protein FtsZ. Micron 2011, 42, 336–341. [Google Scholar]
- Howard, M; Gerdes, K. What Is the Mechanism of ParA-Mediated DNA Movement? Mol. Microbiol 2010, 78, 9–12. [Google Scholar]
- Ptacin, JL; Lee, SF; Garner, EC; Toro, E; Eckart, M; Comolli, LR; Moerner, WE; Shapiro, L. A Spindle-Like Apparatus Guides Bacterial Chromosome Segregation. Nat. Cell Biol 2010, 12, 791–798. [Google Scholar]
- Shebelut, CW; Guberman, JM; van Teeffelen, S; Yakhnina, AA; Gitai, Z. Caulobacter Chromosome Segregation Is an Ordered Multistep Process. Proc. Natl. Acad. Sci. USA 2010, 107, 14194–14198. [Google Scholar]
- Atmakuri, K; Cascales, E; Burton, OT; Banta, LM; Christie, PJ. Agrobacterium ParA/MinD-Like VirC1 Spatially Coordinates Early Conjugative DNA Transfer Reactions. EMBO J 2007, 26, 2540–2551. [Google Scholar]
- Savage, DF; Afonso, B; Chen, AH; Silver, PA. Spatially Ordered Dynamics of the Bacterial Carbon Fixation Machinery. Science 2010, 327, 1258–1261. [Google Scholar]
- Thompson, SR; Wadhams, GH; Armitage, JP. The Positioning of Cytoplasmic Protein Clusters in Bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 8209–8214. [Google Scholar]
- Wallden, M; Elf, J. Studying Transcriptional Interactions in Single Cells at Sufficient Resolution. Curr. Opin. Biotechnol 2011, 22, 81–86. [Google Scholar]
- Megason, SG; Fraser, SE. Imaging in Systems Biology. Cell 2007, 130, 784–795. [Google Scholar]
- Pastrana, E. Optogenetics: Controlling Cell Function with Light. Nat. Meth 2011, 8, 24–25. [Google Scholar]
- Toettcher, JE; Voigt, CA; Weiner, OD; Lim, WA. The Promise of Optogenetics in Cell Biology: Interrogating Molecular Circuits in Space and Time. Nat. Meth 2011, 8, 35–38. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chiu, S.-W.; Leake, M.C. Functioning Nanomachines Seen in Real-Time in Living Bacteria Using Single-Molecule and Super-Resolution Fluorescence Imaging. Int. J. Mol. Sci. 2011, 12, 2518-2542. https://doi.org/10.3390/ijms12042518
Chiu S-W, Leake MC. Functioning Nanomachines Seen in Real-Time in Living Bacteria Using Single-Molecule and Super-Resolution Fluorescence Imaging. International Journal of Molecular Sciences. 2011; 12(4):2518-2542. https://doi.org/10.3390/ijms12042518
Chicago/Turabian StyleChiu, Sheng-Wen, and Mark C. Leake. 2011. "Functioning Nanomachines Seen in Real-Time in Living Bacteria Using Single-Molecule and Super-Resolution Fluorescence Imaging" International Journal of Molecular Sciences 12, no. 4: 2518-2542. https://doi.org/10.3390/ijms12042518