Dynamic Structure-Based Pharmacophore Model Development: A New and Effective Addition in the Histone Deacetylase 8 (HDAC8) Inhibitor Discovery

Abstract
:
1. Introduction
2. Results and Discussion
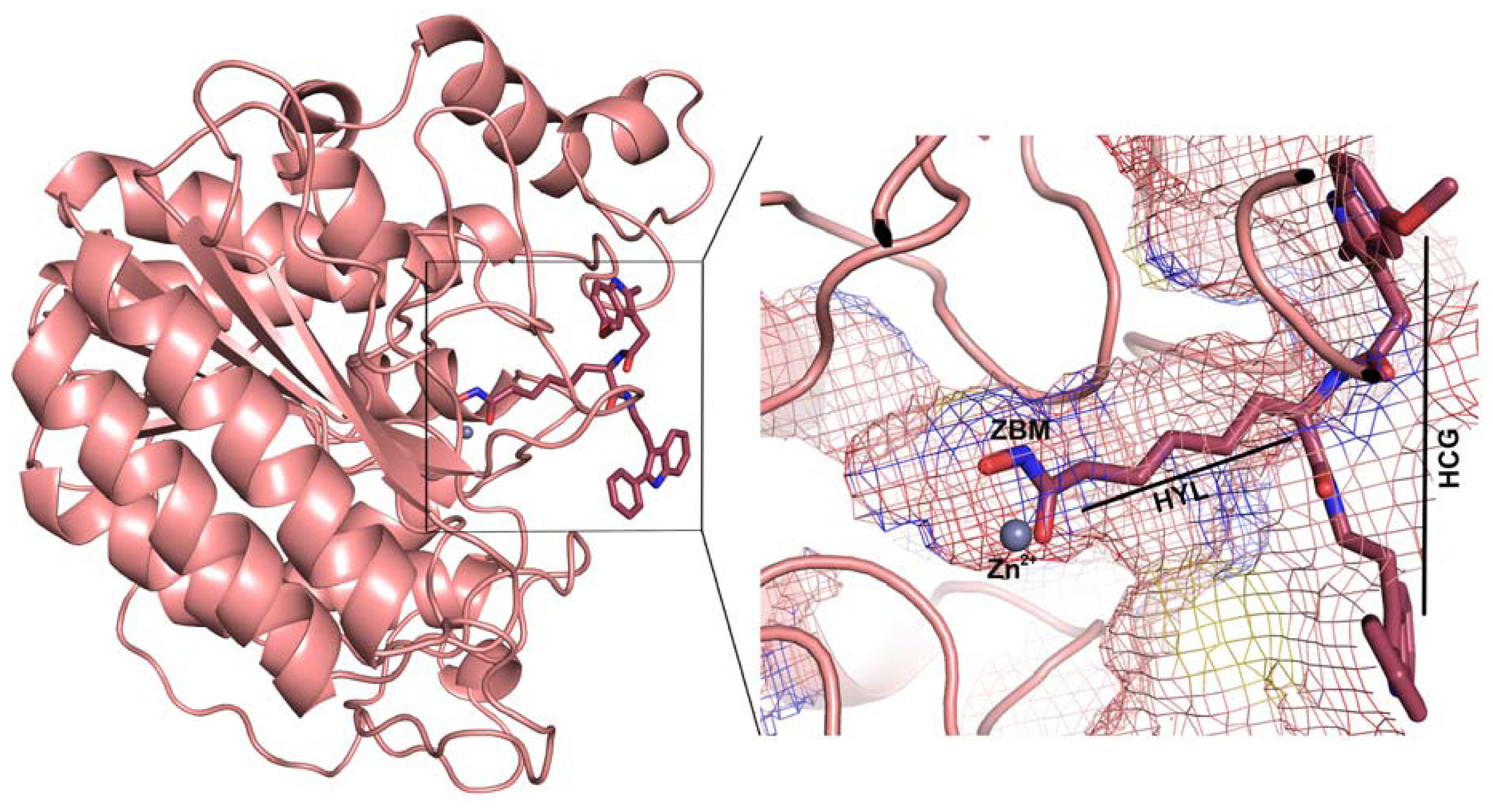
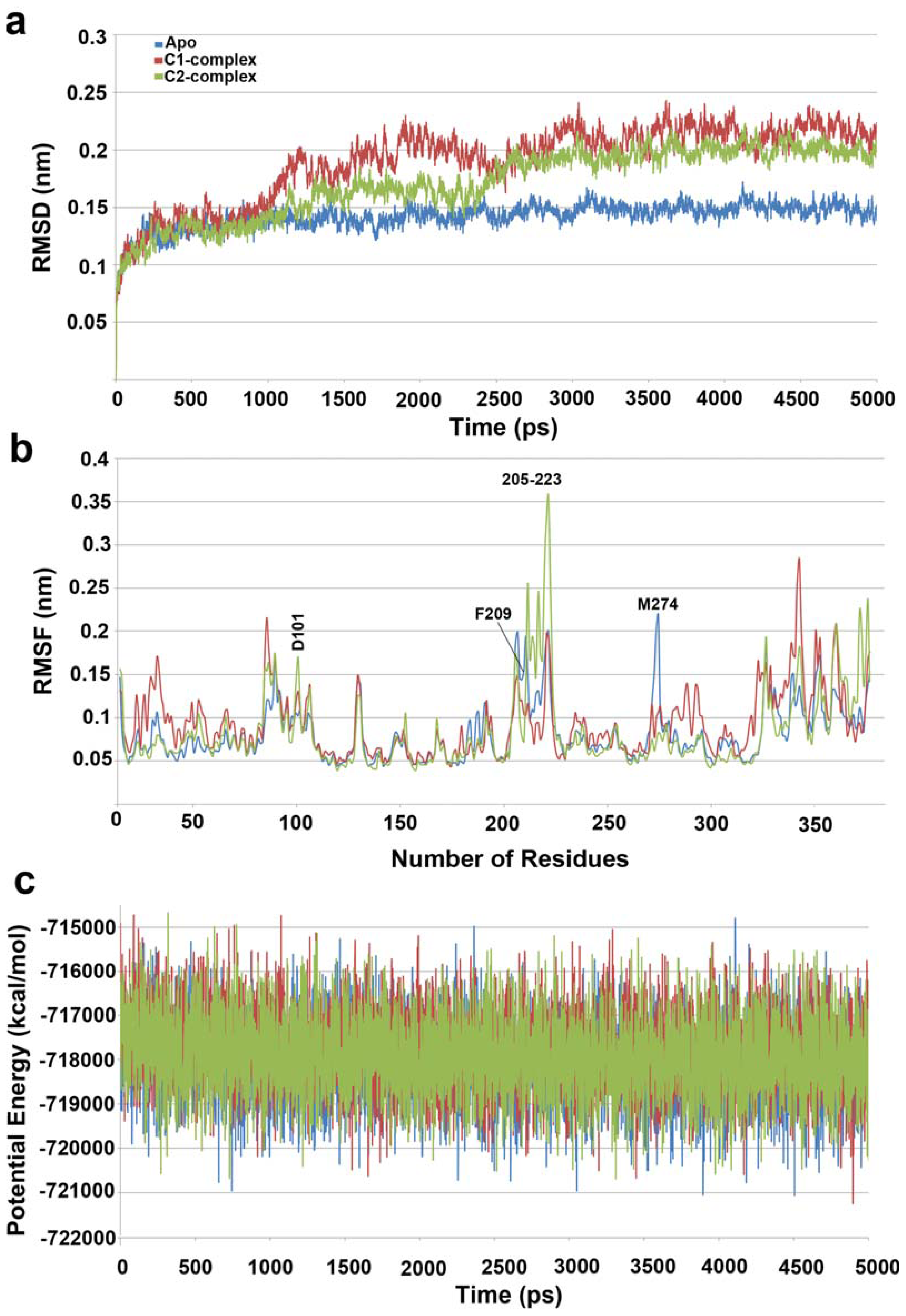
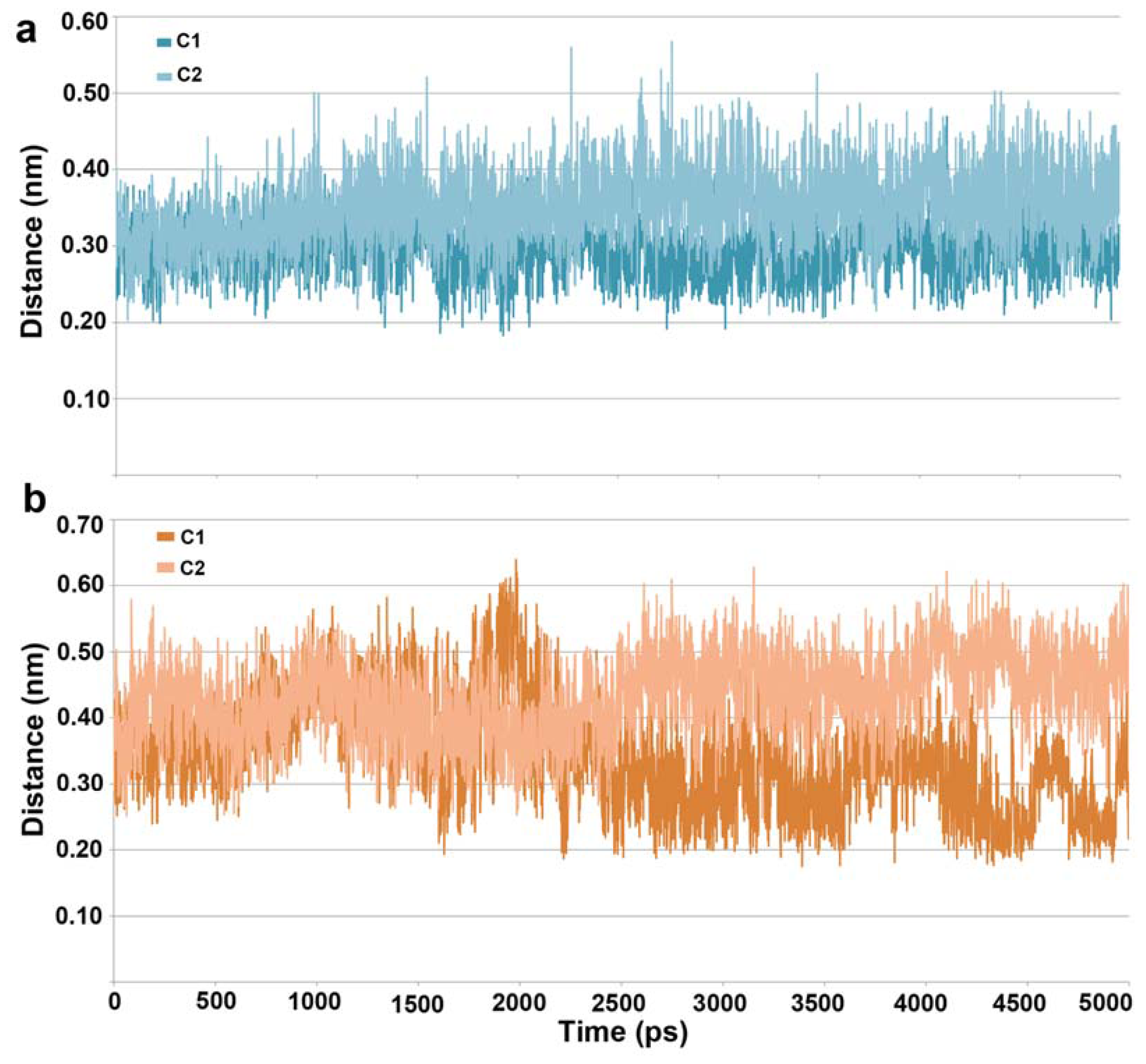
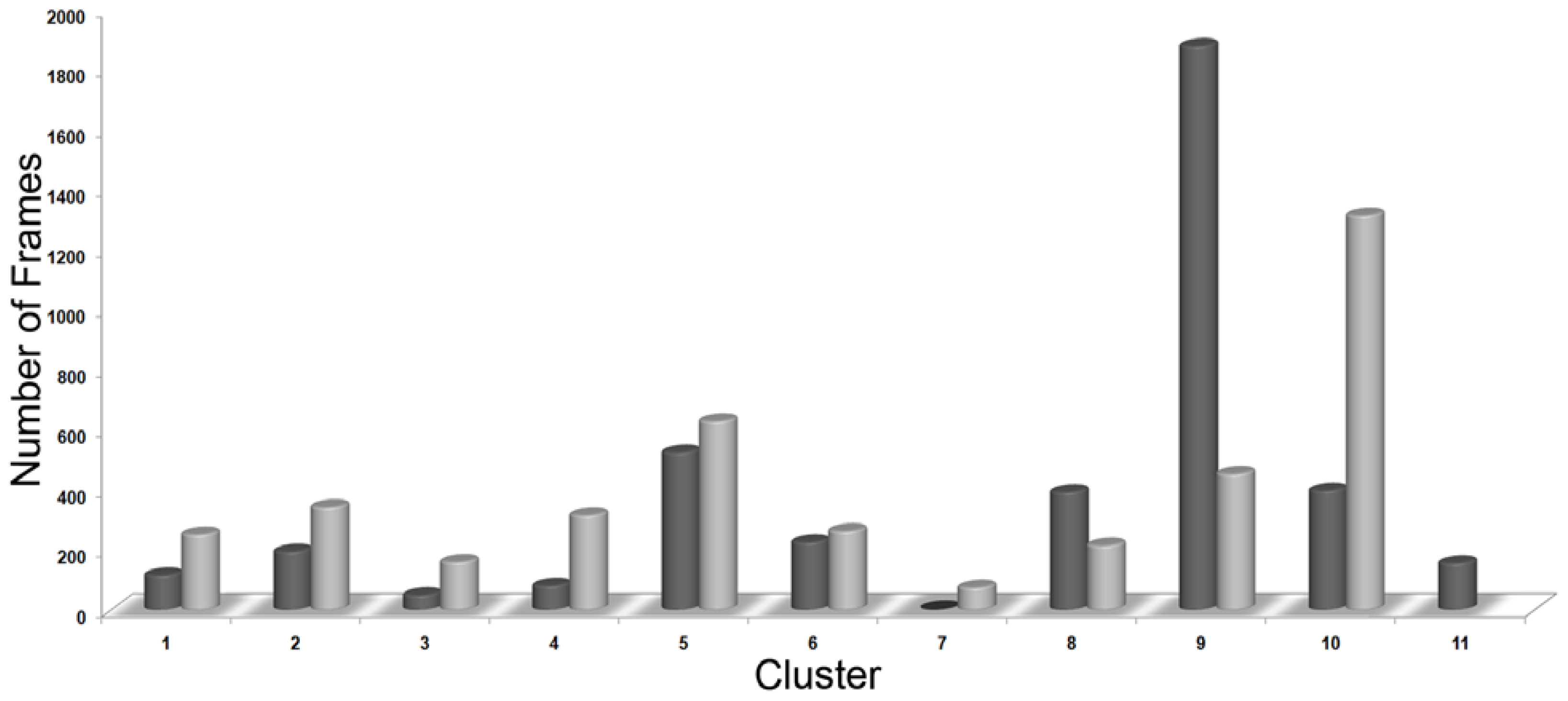
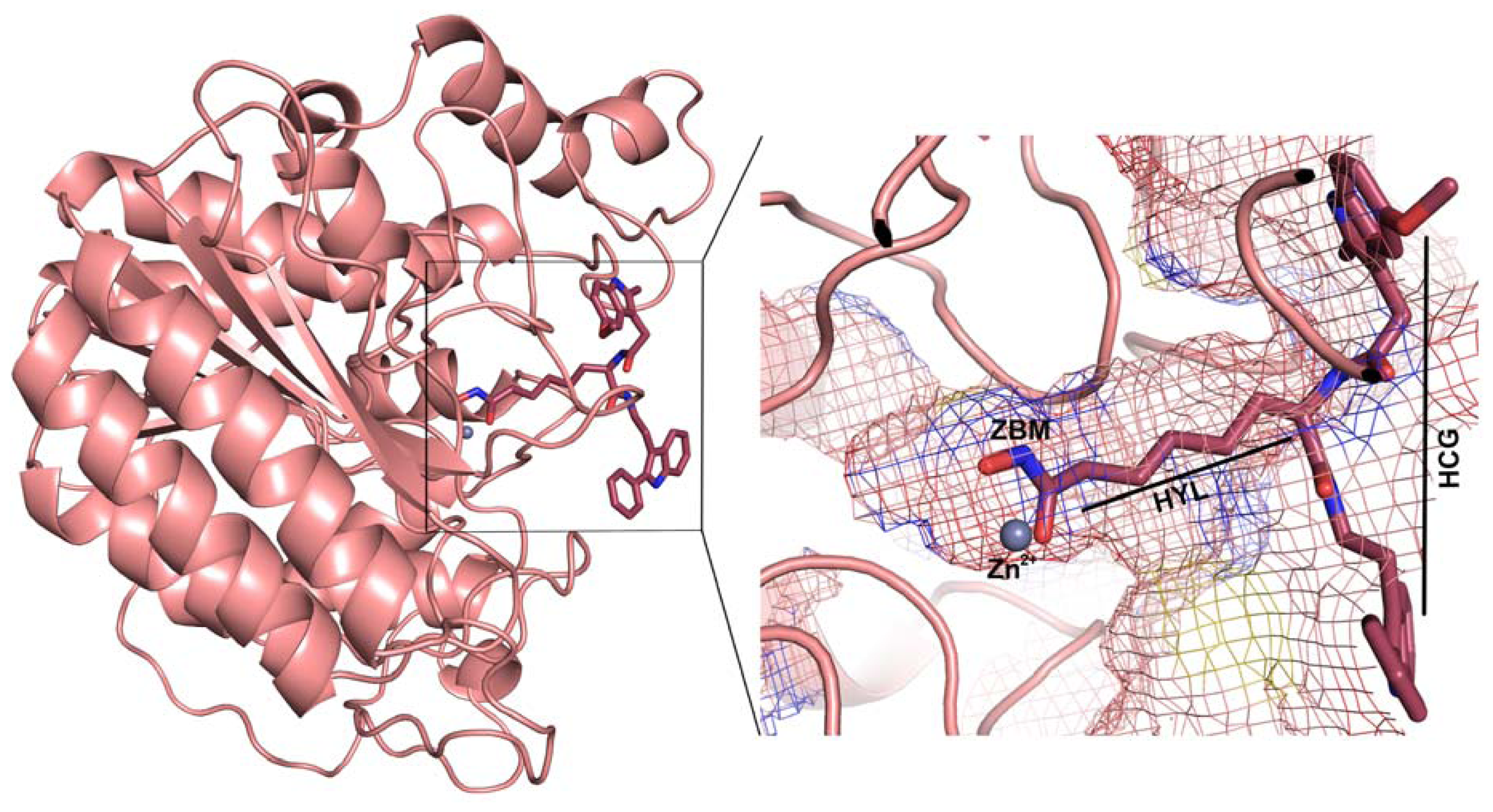
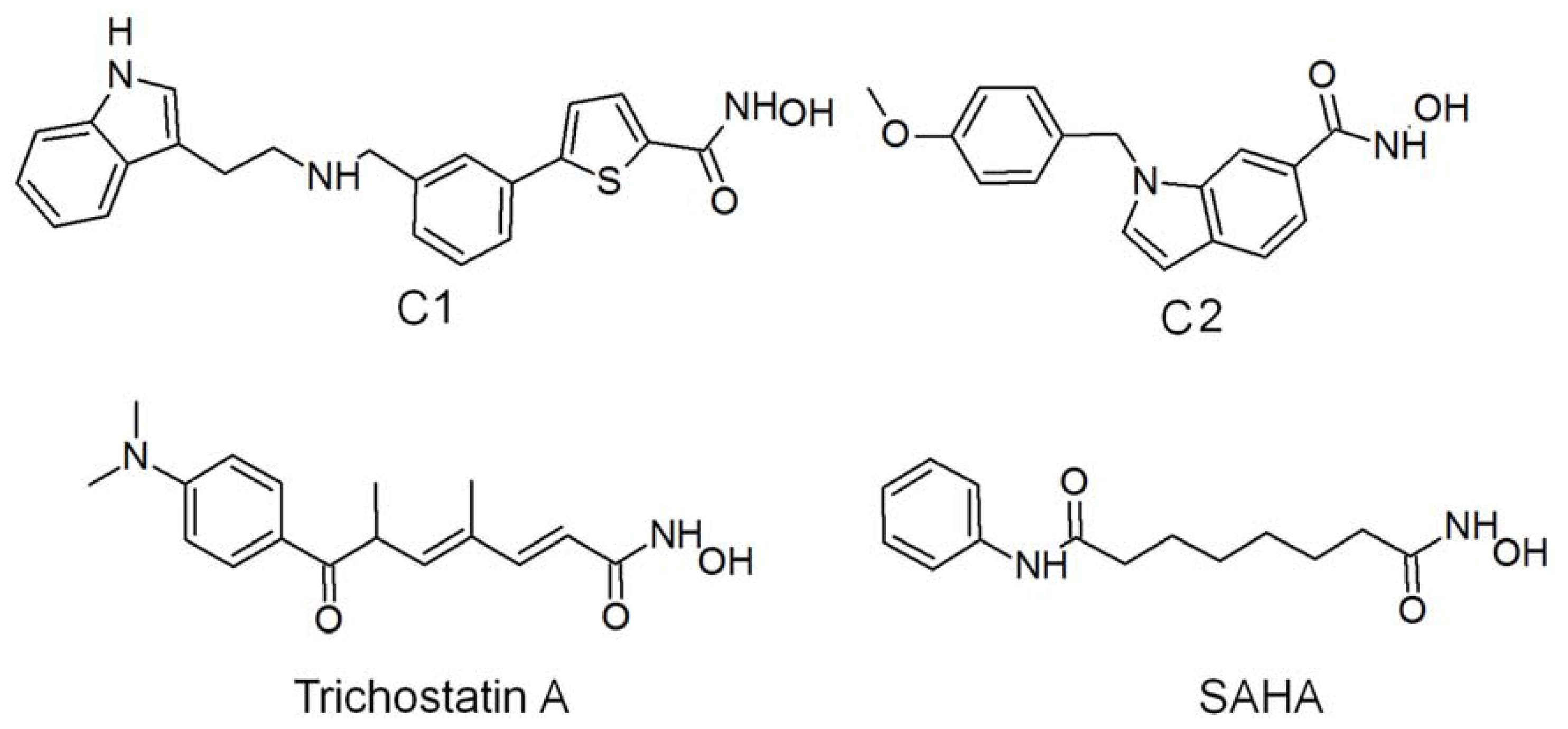
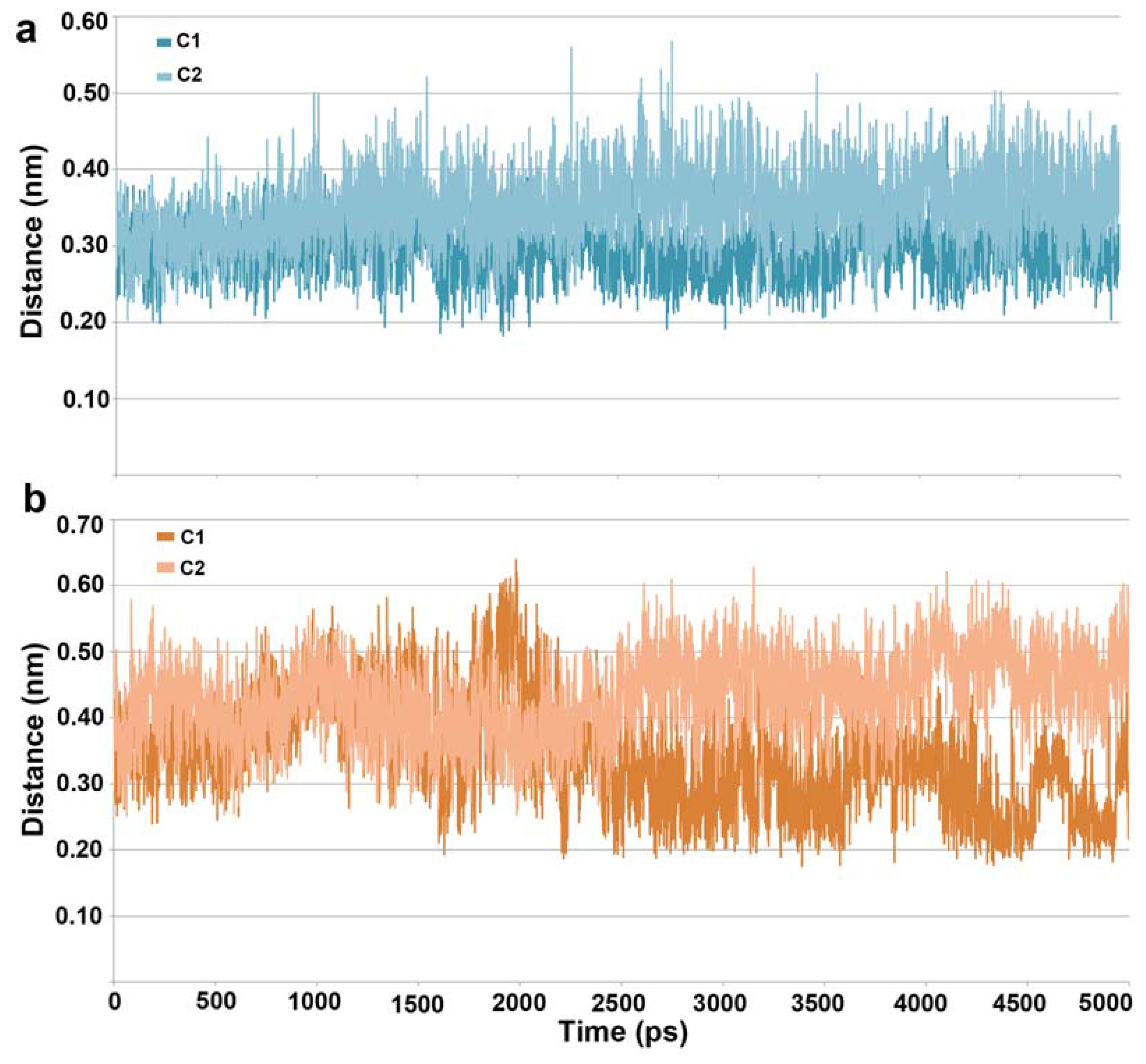
2.1. Molecular Dynamic Simulation Studies
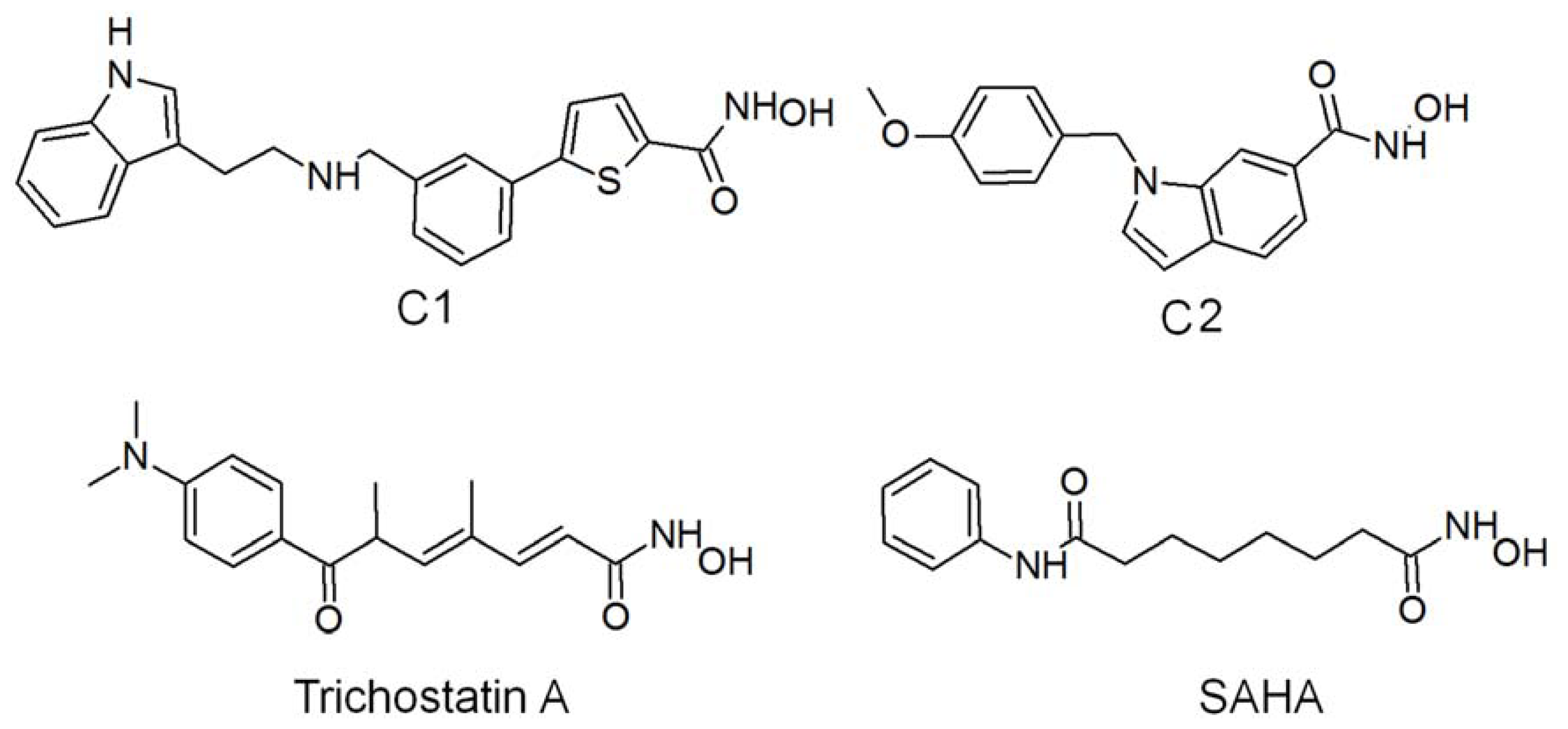
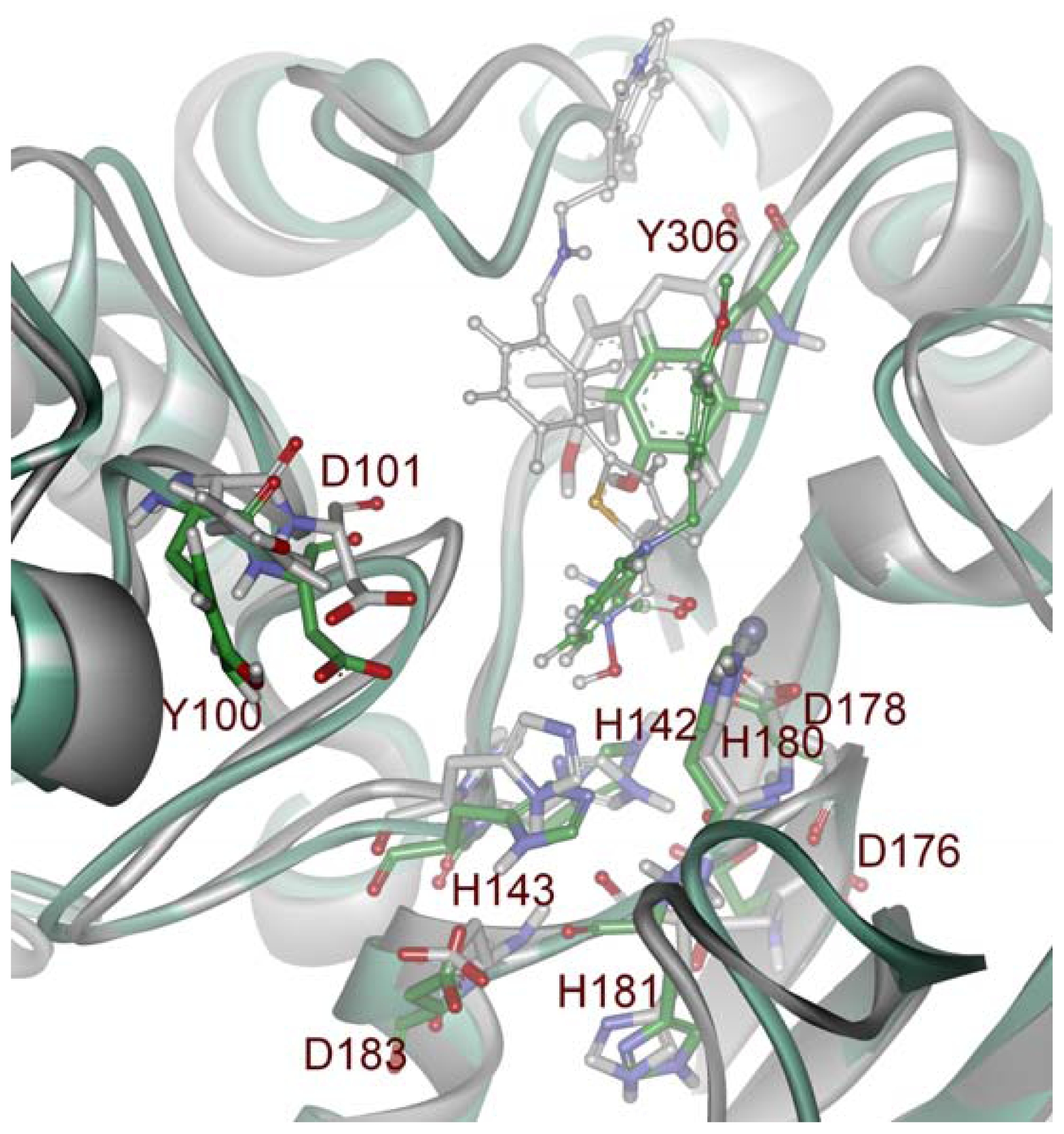
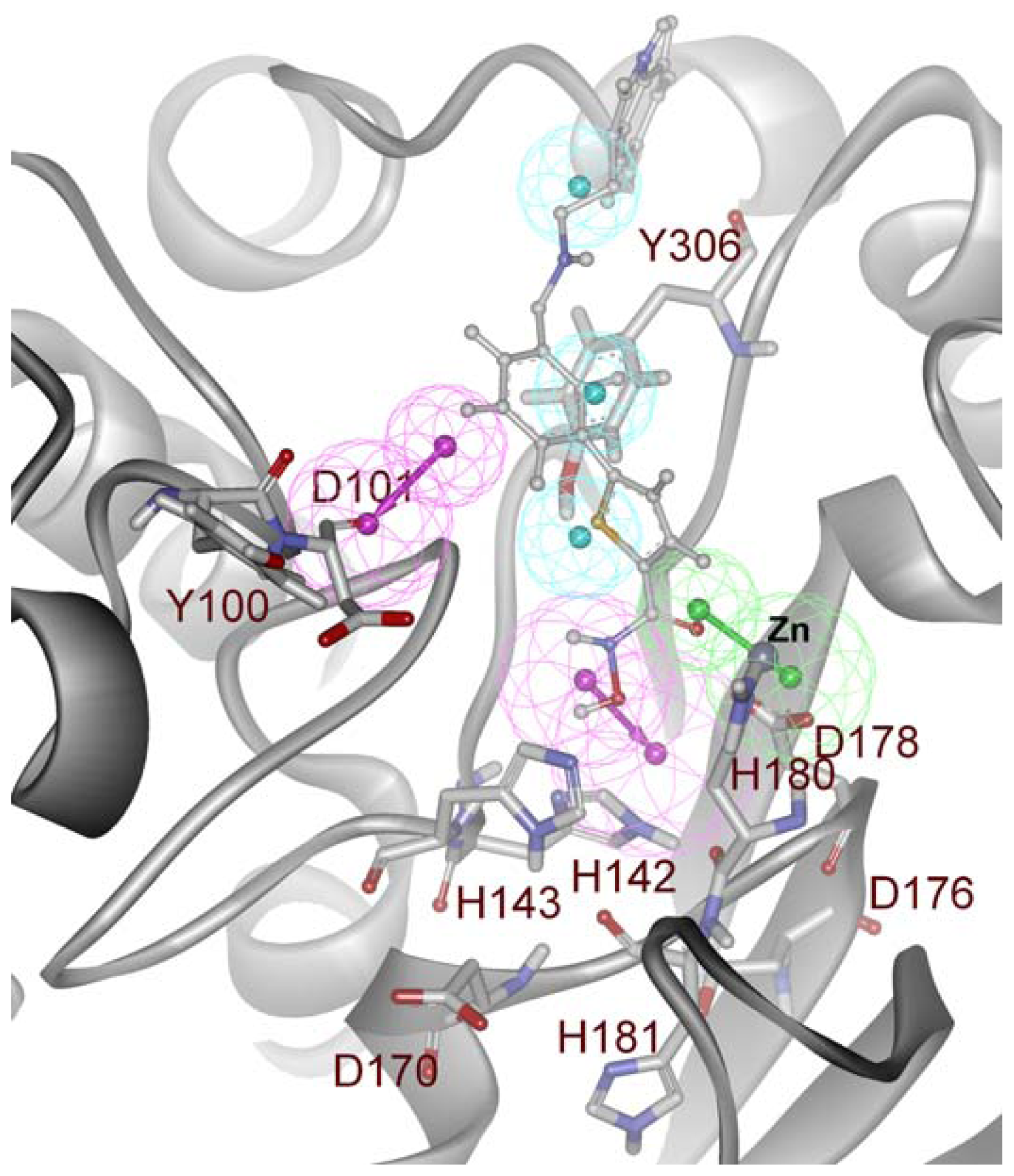
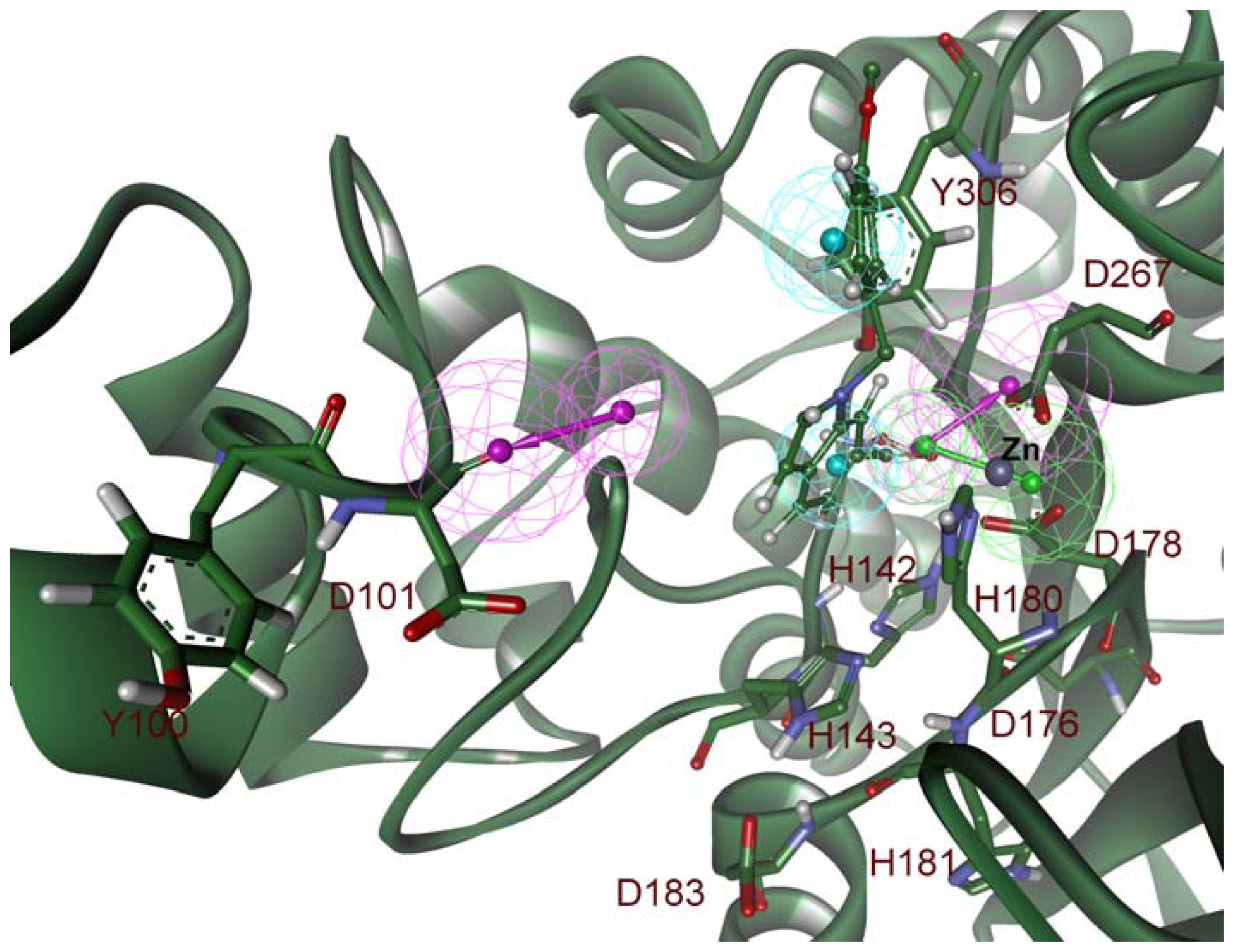
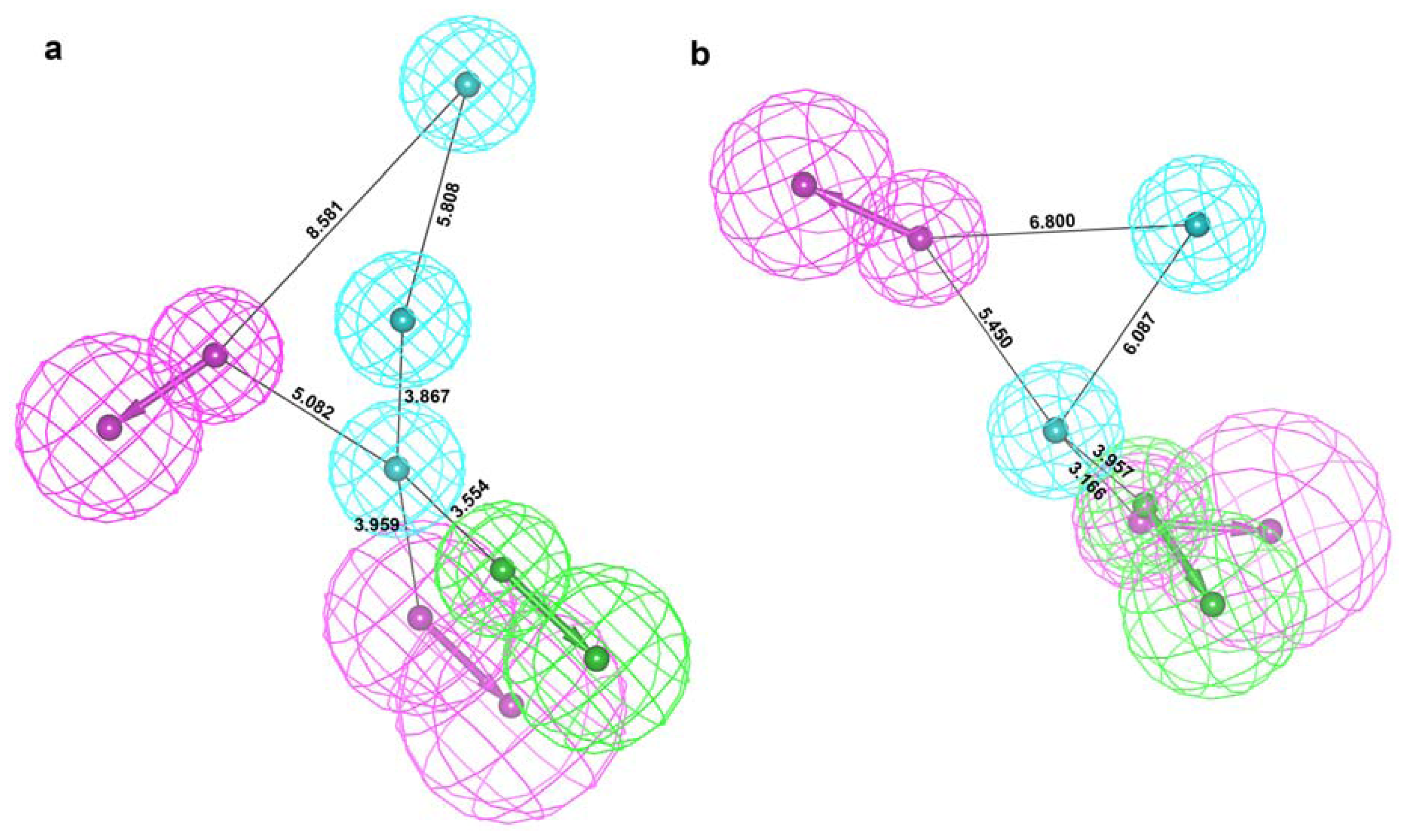
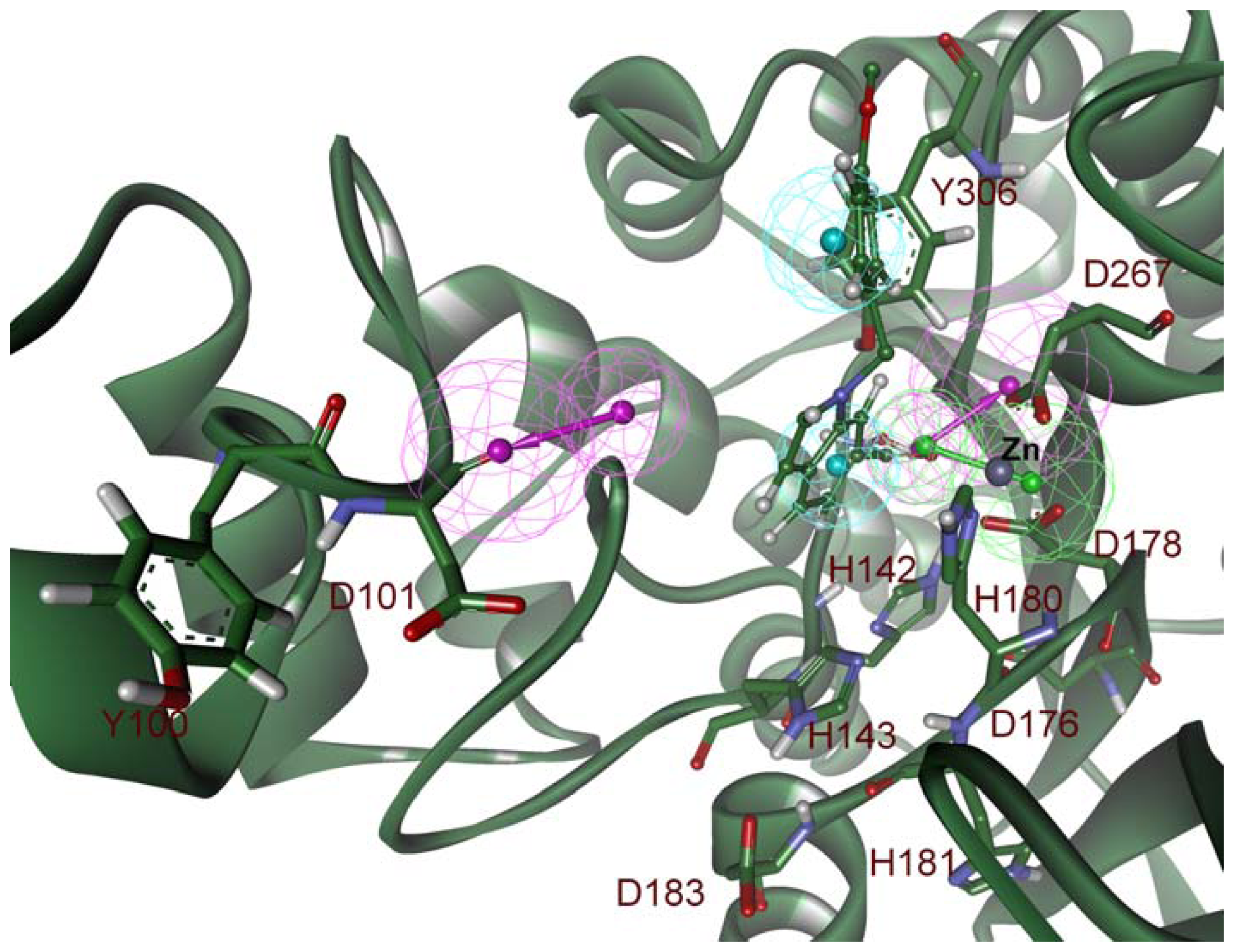
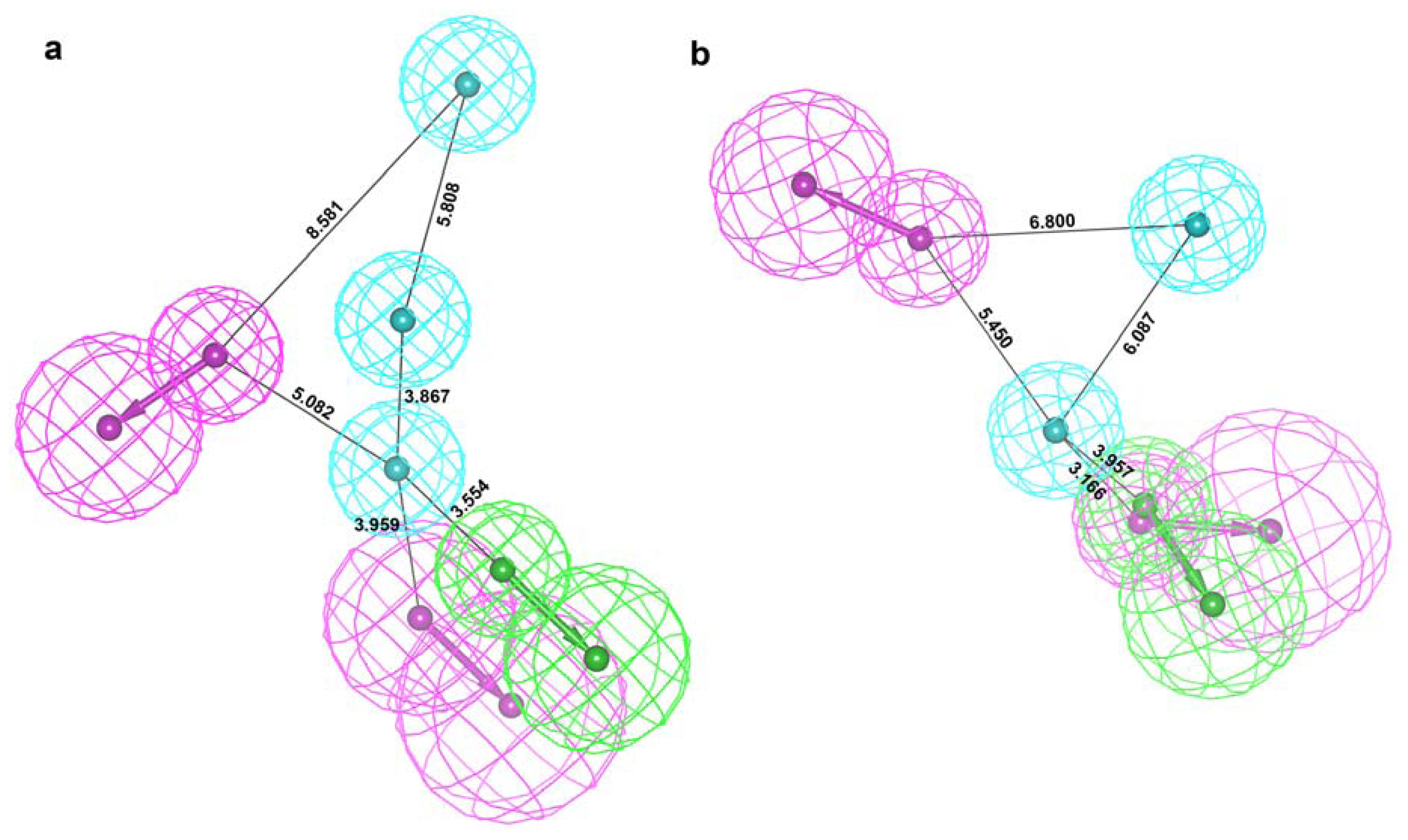
2.2. Structure-Based Pharmacophore Models
2.3. Validation of Generated Pharmacophore Models
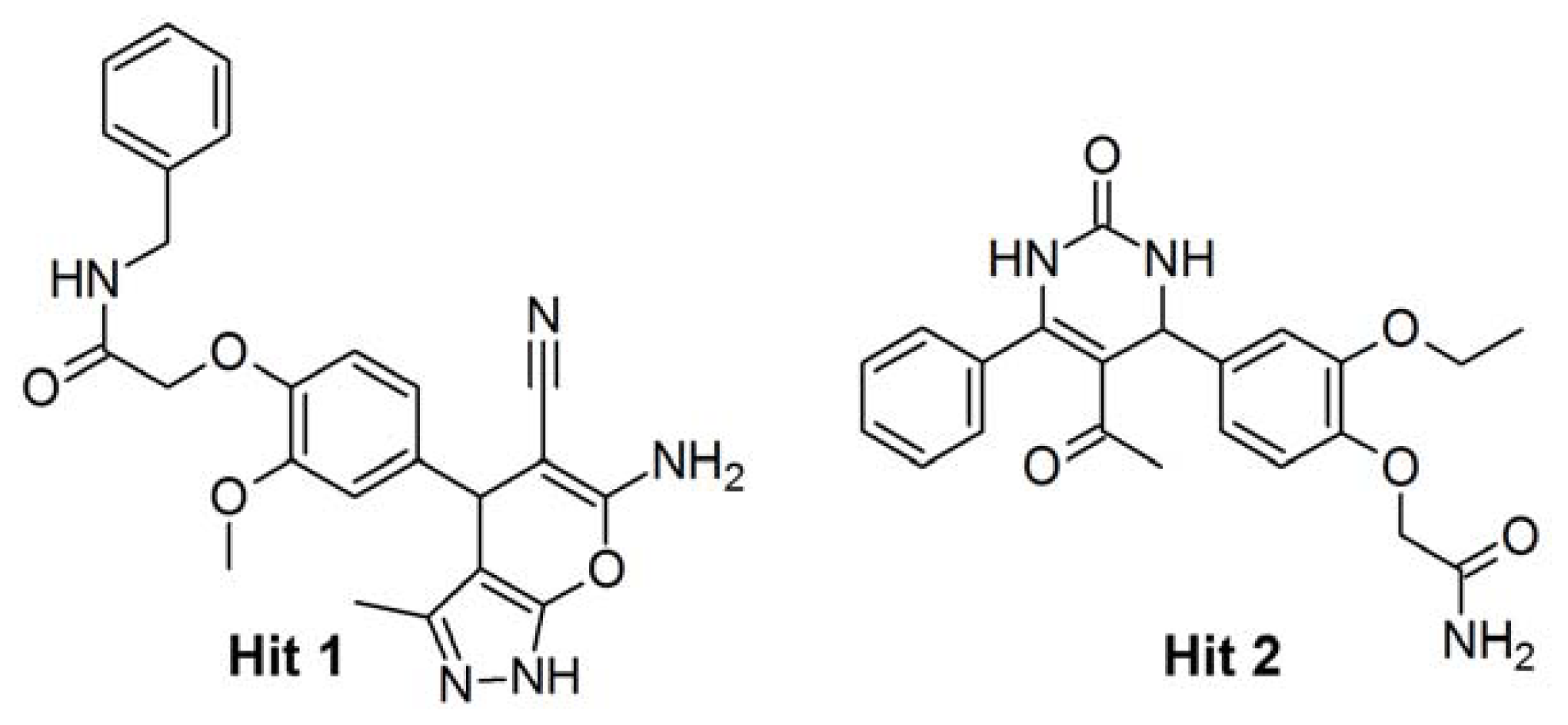
2.4. Database Screening
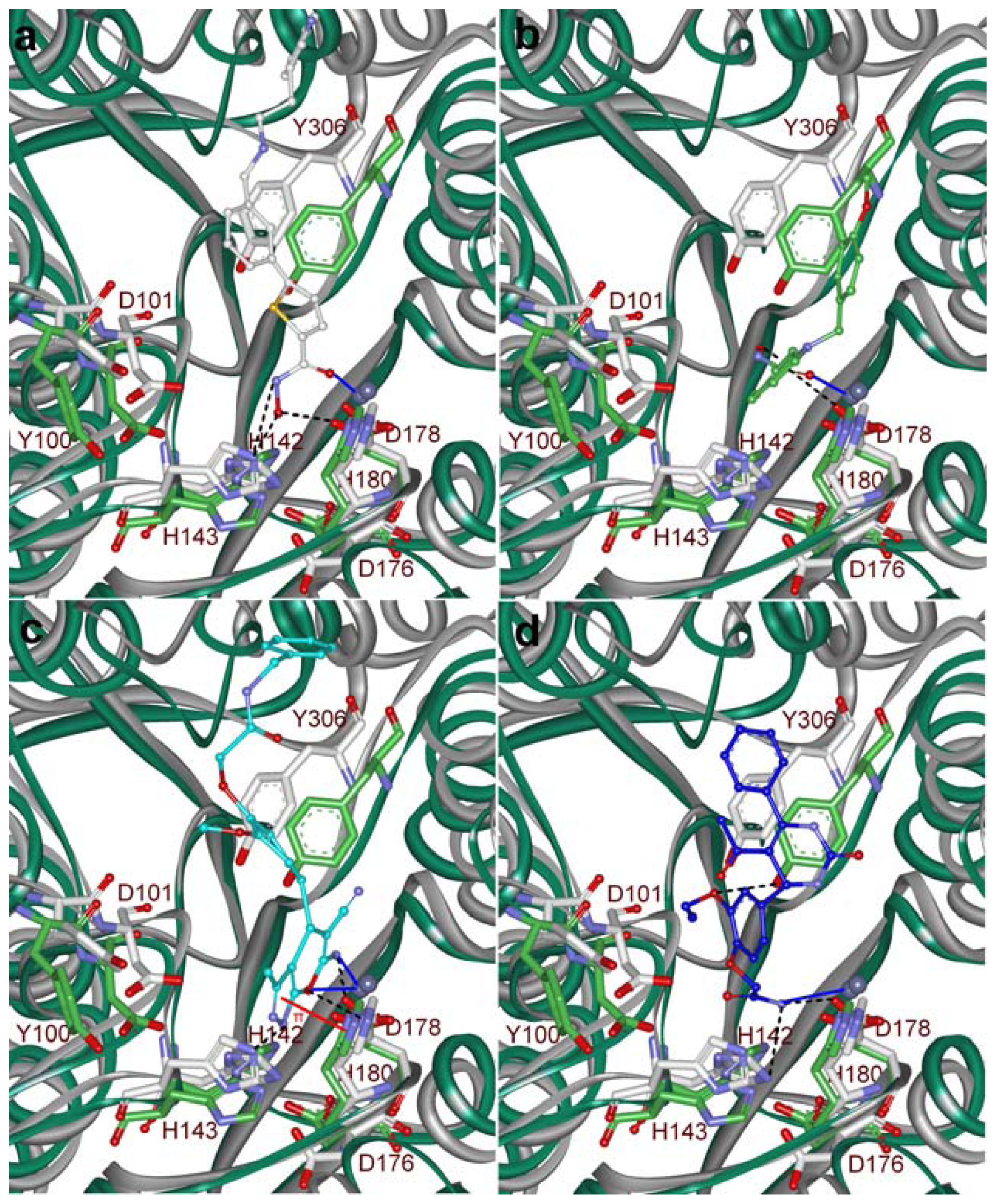
2.5. Molecular Docking
3. Experimental Section
3.1. Selection of Protein and Inhibitor Structures
3.2. Molecular Docking and Dynamic Simulations
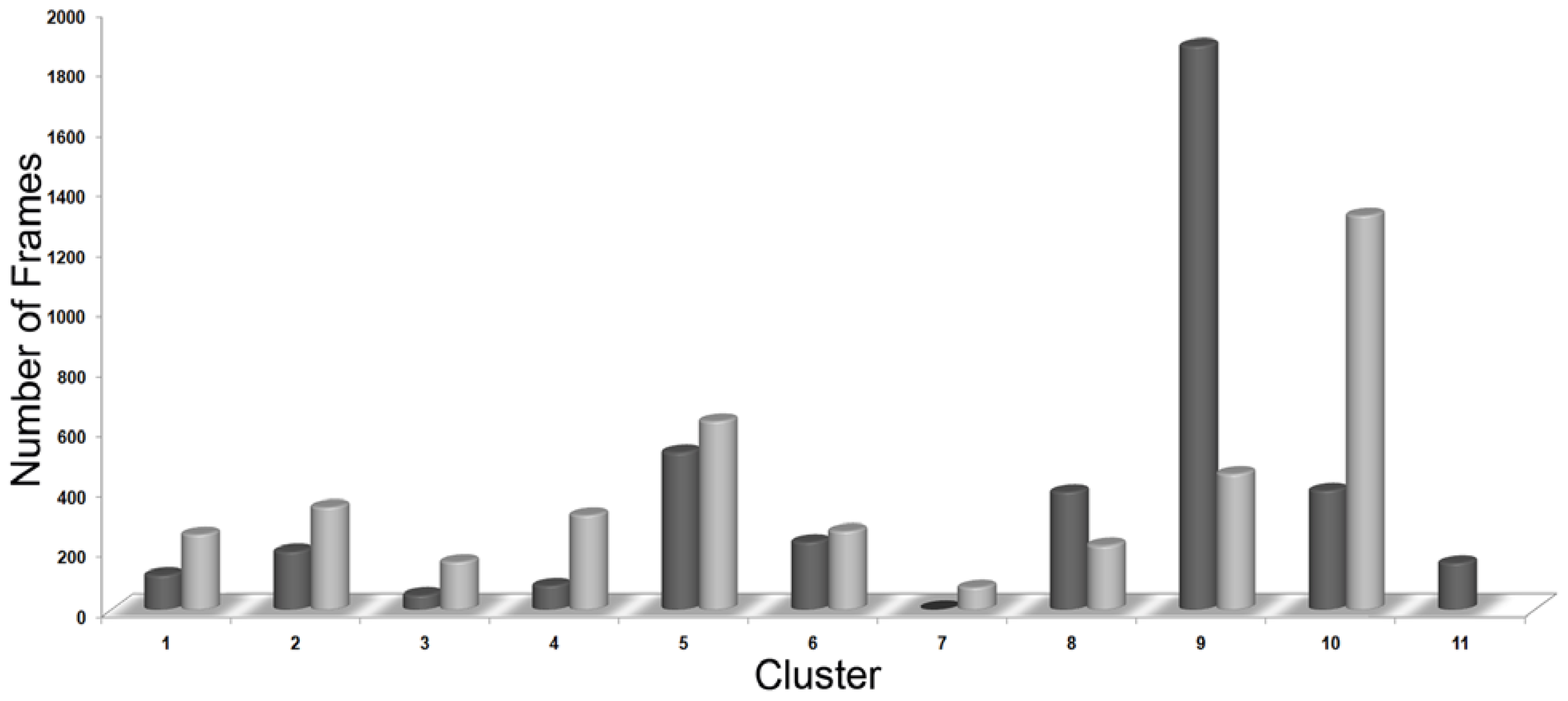
3.3. Clustering of Snapshots
3.4. Structure-Based Pharmacophore Generation and Validation
3.5. Database Screening
3.6. Molecular Docking and Lead Identification
4. Conclusions
Supplementary Material
ijms-12-09440-s001.pdfAcknowledgements
References
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar]
- Nielsen, T.K.; Hildmann, C.; Dickmanns, A.; Schwienhorst, A.; Ficner, R. Crystal structure of a bacterial class 2 histone deacetylase homologue. J. Mol. Biol 2005, 354, 107–120. [Google Scholar]
- Sternson, S.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Synthesis of 7200 small molecules based on a substructural analysis of the histone deacetylase inhibitors trichostatin and trapoxin. Org. Lett 2001, 3, 4239–4242. [Google Scholar]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar]
- Hassig, C.A.; Schreiber, S.L. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr. Opin. Chem. Biol 1997, 1, 300–308. [Google Scholar]
- Kurdistani, S.K.; Grunstein, M. Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol 2003, 4, 276–284. [Google Scholar]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Docking-enabled pharmacophore model for histone deacetylase 8 inhibitors and its application in anti-cancer drug discovery. J. Mol. Graph. Model 2010, 29, 382–395. [Google Scholar]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep 2002, 3, 224–229. [Google Scholar]
- Marks, P.A.; Dokmanovic, M. Histone deacetylase inhibitors: Discovery and development as anticancer agents. Expert Opin. Investig. Drugs 2005, 14, 1497–1511. [Google Scholar]
- Bi, G.; Jiang, G. The Molecular mechanism of HDAC inhibitors in anticancer effects. Cell. Mol. Immunol 2006, 3, 285–290. [Google Scholar]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell. Biochem 2005, 96, 293–304. [Google Scholar]
- Hahnen, E.; Hauke, J.; Tränkle, C.; Eyupoglu, I.Y.; Wirth, B.; Blümcke, I. Histone deacetylase inhibitors: Possible implications for neurodegenerative disorders. Expert Opin. Investig. Drugs 2008, 17, 169–184. [Google Scholar]
- Morrison, B.E.; Majdzadeh, N.; D’Mello, S.R. Histone deacetylases: Focus on the nervous system. Cell. Mol. Life Sci 2007, 64, 2258–2269. [Google Scholar]
- Brichta, L.; Hofmann, Y.; Hahnen, E.; Siebzehnrubl, F.A.; Raschke, H.; Blumcke, I.; Eyupoglu, I.Y.; Wirth, B. Valproic acid increases the SMN2 protein level: A well-known drug as a potential therapy for spinal muscular atrophy. Hum. Mol. Genet 2003, 12, 2481–2489. [Google Scholar]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar]
- Adcock, I.M. HDAC inhibitors as anti-inflammatory agents. Br. J. Pharmacol 2009, 150, 829–831. [Google Scholar]
- Mottet, D.; Castronova, V. Histone deacetylases: Target enzymes for cancer therapy. Clin. Exp. Metastasis 2008, 25, 183–189. [Google Scholar]
- Pan, L.; Lu, J.; Huang, B. HDAC Inhibitors: A potential new category of anti-tumor agents. Cell. Mol. Immunol 2007, 4, 337–343. [Google Scholar]
- Krennhrubec, K.; Marshall, B.L.; Hedglin, M.; Verdin, E.; Ulrich, S.M. Design and evaluation of linkerless hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 2874–2878. [Google Scholar]
- Estiu, G.; Greenberg, E.; Harrison, C.B.; Kwiatkowski, N.P.; Mazitschek, R.; Bradner, J.E.; Wiest, O. Structural origin of selectivity in class II-selective histone deacetylase inhibitors. J. Med. Chem 2008, 51, 2898–2906. [Google Scholar]
- Park, J.H.; Jung, Y.; Kim, T.; Kim, S.; Jong, H.S.; Lee, J.W.; Kim, D.K.; Lee, J.S.; Kim, N.K.; Kim, T.Y.; et al. Class 1 histone deacetylase-selective novel synthetic inhibitors potently inhibit human tumor proliferation. Clin. Cancer Res 2004, 10, 5271–5281. [Google Scholar]
- Bhalla, K.N. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J. Clin. Oncol 2005, 23, 3971–3993. [Google Scholar]
- Vannini, A.; Volpari, C.; Gallinari, P.; Jones, P.; Mattu, M.; Carfí, A.; de Francesco, R.; Steinkühler, C.; Di Marco, S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep 2007, 9, 879–884. [Google Scholar]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res 2007, 17, 195–211. [Google Scholar]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisiawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas HDAC8-specific inhibitor PCI-34051 in T-cell lymphoma. Leukemia 2008, 22, 1026–1034. [Google Scholar]
- Rasheed, W.K.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors in cancer therapy. Expert Opin. Investig. Drugs 2007, 16, 659–677. [Google Scholar]
- Gabriella, O.; Francesco, D.C.; Adriano, M. Docking of hydroxamic acids into HDAC1 and HDAC8: A rationalization of activity trends and selectivities. J. Chem. Inf. Model 2009, 49, 2774–2785. [Google Scholar]
- McKinsey, T.A. Isoform-selective HDAC inhibitors: Closing in on translational medicine for the heart. J. Mol. Cell. Cardiol 2011, 51, 491–496. [Google Scholar]
- Estiu, G.; West, N.; Mazitschek, R.; Greenberg, E.; Bradner, J.E.; Wiest, O. On the inhibition of histone deacetylase 8. Bioorg. Med. Chem 2010, 18, 4103–4110. [Google Scholar]
- Bharatham, K.; Bharatham, N.; Kwon, Y.J.; Lee, K.W. Molecular dynamics simulation study of PTP1B with allosteric inhibitor and its application in receptor based pharmacophore modeling. J. Comput. Aided Mol. Des 2008, 22, 925–933. [Google Scholar]
- Deng, J.; Lee, K.W.; Sanchez, T.; Cui, M.; Neamati, N.; Brigg, J.M. Dynamic receptor-based pharmacophore model development and its application in designing novel HIV-1 integrase inhibitors. J. Med. Chem 2005, 48, 1496–1505. [Google Scholar]
- GOLD, version 4.1 UserGuide; CCDC Software Ltd: Cambridge, UK, 2010.
- Vanommeslaeghe, K.; de Proft, F.; Loverix, S.; Tourwe, D.; Geerlings, P. Theoretical study revealing the functioning of a novel combination of catalytic motifs in histone deacetylase. Bioorg. Med. Chem 2005, 13, 3987–3992. [Google Scholar]
- Wu, R.; Wang, S.; Zhou, N.; Cao, Z.; Zhang, Y. A proton-shuttle reaction mechanism for histone deacetylase 8 and the catalytic role of metal ions. J. Am. Chem. Soc 2010, 132, 9471–9479. [Google Scholar]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Ligand and structure based pharmacophore modeling to facilitate novel histone deacetylase 8 inhibitor design. Eur. J. Med. Chem 2010, 45, 4409–4417. [Google Scholar]
- Buggy, J.J.; Sideris, M.L.; Mak, P.; Lorimer, D.D.; McIntosh, B.; Clark, J.M. Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem. J 2000, 350, 199–205. [Google Scholar]
- Wagner, A.B. SciFinder scholar 2006: An empirical analysis of research topic query processing. J. Chem. Inf. Model 2006, 46, 767–774. [Google Scholar]
- Wang, Y.; Bolton, E.; Dracheva, S.; Karapetyan, K.; Shoemaker, B.A.; Suzek, T.O.; Wang, J.; Xiao, J.; Zhang, J.; Bryant, S.H. An overview of the PubChem BioAssay resource. Nucleic Acids Res 2010, 38, D255–D266. [Google Scholar]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; Francesco, R.D.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar]
- Somoza, L.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar]
- Dowling, D.P.; Gantt, S.L.; Gattis, S.G.; Fierke, C.A.; Christianson, D.W. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry 2008, 47, 13554–13563. [Google Scholar]
- Dowling, D.P.; Gattis, S.G.; Fierke, C.A.; Christianson, D.W. Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry 2010, 49, 5048–5056. [Google Scholar]
- Stunkel, W.; Wang, H.; Yin, Z. Biaryl Linked Hydroxamates: Preparation and Pharmaceutical Applications. U.S. Patent 2007/0167499 A1, 19 July 2007. [Google Scholar]
- Joseph, J.B.; Sriram, B. Uses of Selective Inhibitors of HDAC8 for Treatment of T-Cell Proliferative Disorders. U.S. Patent 2008/0112889 A1, 15 May 2008. [Google Scholar]
- Liu, J.; Wang, F.; Ma, Z.; Wang, X.; Wang, Y. Structural determination of three different series of compounds as Hsp90 inhibitors using 3D-QSAR modeling, molecular docking and molecular dynamics methods. Int. J. Mol. Sci 2011, 12, 946–970. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 2008, 4, 435–447. [Google Scholar]
- GROMACS, version 4; University of Groningen: Groningen, The Netherlands, 2005; Manual pages.
- GROMOS, version 96; Swiss Federal Institute of Technology: Zürich, Switzerland, 1996.
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr 2004, 60, 1355–1363. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys 2007, 126, 014101:1–014101:7. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- Discovery Studio, version 2.5; Accelrys, Inc: San Diego, CA, USA, 2009.
- Wenxin, G.; Inna, N.; Ronald, D.S.; Curt, M.H.; Richard, B.S. Carbonyl- and sulfur-containing analogs of suberoylanilide hydroxamic acid: Potent inhibition of histone deacetylases. Bioorg. Med. Chem 2006, 14, 3320–3329. [Google Scholar]
- Tom, Y.H.W.; Christian, H.; Yiqin, W.; Sheng, D.; Peter, G.S. Design, synthesis, and activity of HDAC inhibitors with a N-formyl hydroxylamine head group. Bioorg. Med. Chem. Lett 2004, 14, 449–453. [Google Scholar]
- Jeffrey, M.B.; Zuomei, L.; Daniel, D.; Claire, B. Methods for Specifically Inhibiting Histone-7 and 8. U.S. Patent 2004/0072770 A1, 15 April 2004. [Google Scholar]
- Dizhong, C.; Weiping, D.; Kand, S.; Hong, Y.S.; Eric, T.S.; Niefang, Y.; Yong, Z. Benzimidazole Derivatives: Preparation and Pharmaceutical Applications. U.S. Patent 2007/0043043 A1, 22 February 2007. [Google Scholar]
- Walter, S.; Haishan, W.; Zheng, Y. Biaryl Linked Hydroxamates: Preparation and Pharmaceutical Applications. U.S. Patent 2007/0167499 A1, 19 July 2007. [Google Scholar]
- Ze-Yi, L.; Haishan, W.; Yan, Z. Aclyurea Connected and Sulfonamide Connected Hydroxamates. U.S. Patent 2008/0070954 A1, 20 March 2008. [Google Scholar]
- Joseph, J.B.; Sriram, B. Uses of Selective Inhibitors of HDAC8 for Treatment of T-Cell Proliferative Disorders. U.S. Patent 2008/0112889 A1, 15 May 2008. [Google Scholar]
- Langer, L.; Wolber, G. Pharmacophore definition and 3D searches. Drug Discov. Today Technol 2004, 1, 203–207. [Google Scholar]
- Zhu, R.; Hu, L.; Li, H.; Su, J.; Cao, Z.; Zhang, W. Novel natural inhibitors of CYP1A2 identified by in silico and in vitro screening. Int. J. Mol. Sci 2011, 12, 3250–3262. [Google Scholar]
- Lee, C.-H.; Huang, H.-C.; Juan, H.-F. Reviewing ligand-based rational drug design: The search for an ATP synthase inhibitor. Int. J. Mol. Sci 2011, 12, 5304–5318. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 2001, 46, 3–26. [Google Scholar]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar]
- Li, X.; Li, Y.; Cheng, T.; Liu, Z.; Wang, R. Evaluation of the performance of four molecular docking programs on a diverse set of protein-ligand complexes. J. Comput. Chem 2010, 31, 2109–2125. [Google Scholar]
- Plewczynski, D.; Lazniewski, M.; Augustyniak, R.; Ginalski, K. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J. Comput. Chem 2011, 32, 742–755. [Google Scholar]
- Cheng, F.; Wang, Q.; Chen, M.; Quiocho, F.A.; Ma, J. Molecular docking study of the interactions between the thioesterase domain of human fatty acid synthase and its ligands. Proteins 2008, 70, 1228–1234. [Google Scholar]
- Molegro Molecular Viewer, version 2.2.0; Molegro ApS: Aarhus, Denmark, 2011.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDAC8 Dataset | Total | Pharm-A | Pharm-B | Pharm-X | |||
|---|---|---|---|---|---|---|---|
| Screened | Percentage (%) | Screened | Percentage (%) | Screened | Percentage (%) | ||
| Total | 100 | 52 | 52 | 57 | 57 | 18 | 18 |
| Active <0.1 μM | 17 | 15 | 88.24 | 13 | 76.47 | 9 | 52.94 |
| Moderately active ≥0.1 <1 μM | 68 | 35 | 51.47 | 40 | 58.82 | 8 | 11.76 |
| Inactive >1 μM | 15 | 2 | 13.33 | 4 | 26.66 | 1 | 6.66 |
| PDB ID | Resolution (Å) | Bound Ligand | Mutation | Metal Ion |
|---|---|---|---|---|
| 1T64 | 1.90 | Trichostatin A | - | Zn2+ |
| 1T67 | 2.31 | M344 | - | Zn2+ |
| 1T69 | 2.91 | SAHA | - | Zn2+ |
| 1VKG | 2.20 | CRA-19156 | - | Zn2+ |
| 1W22 | 2.50 | NHB | - | Zn2+ |
| 2V5W | 2.00 | Substrate | - | Zn2+ |
| 2V5X | 2.25 | V5X | - | Zn2+ |
| 3EW8 | 1.80 | M344 | D101L | Zn2+ |
| 3EWF | 2.50 | Substrate | H143A | Zn2+ |
| 3EZP | 2.65 | M344 | D101N | Zn2+ |
| 3EZT | 2.85 | M344 | D101E | Zn2+ |
| 3F06 | 2.55 | M344 | D101A | Zn2+ |
| 3F07 | 3.30 | APHA | - | Zn2+ |
| 3F0R | 2.54 | Trichostatin A | - | Zn2+ |
| 3MZ3 | 3.20 | M344 | - | Co2+ |
| 3MZ4 | 1.85 | M344 | D101L | Mn2+ |
| 3MZ6 | 2.00 | M344 | D101L | Fe2+ |
| 3MZ7 | 1.90 | M344 | D101L | Co2+ |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Thangapandian, S.; John, S.; Lee, Y.; Kim, S.; Lee, K.W. Dynamic Structure-Based Pharmacophore Model Development: A New and Effective Addition in the Histone Deacetylase 8 (HDAC8) Inhibitor Discovery. Int. J. Mol. Sci. 2011, 12, 9440-9462. https://doi.org/10.3390/ijms12129440
Thangapandian S, John S, Lee Y, Kim S, Lee KW. Dynamic Structure-Based Pharmacophore Model Development: A New and Effective Addition in the Histone Deacetylase 8 (HDAC8) Inhibitor Discovery. International Journal of Molecular Sciences. 2011; 12(12):9440-9462. https://doi.org/10.3390/ijms12129440
Chicago/Turabian StyleThangapandian, Sundarapandian, Shalini John, Yuno Lee, Songmi Kim, and Keun Woo Lee. 2011. "Dynamic Structure-Based Pharmacophore Model Development: A New and Effective Addition in the Histone Deacetylase 8 (HDAC8) Inhibitor Discovery" International Journal of Molecular Sciences 12, no. 12: 9440-9462. https://doi.org/10.3390/ijms12129440