Genetic and Epigenetic Traits as Biomarkers in Colorectal Cancer

Abstract
:
1. Introduction
2. Phenotypic Subgroups of CRC
3. Biomolecules
4. Adenoma Carcinoma Sequence
5. Biomarkers
5.1. Markers Aiding in Prediction of Risk
5.2. Diagnostic Markers
5.3. Prognostic Markers
5.4. Predictive Markers
6. Conclusions and the Way Forward
Supplementary Information
ijms-12-009426-s001.pdfAcknowledgments
References
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med 2009, 361, 2449–2460. [Google Scholar]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar]
- Rajagopalan, H.; Nowak, M.A.; Vogelstein, B.; Lengauer, C. The significance of unstable chromosomes in colorectal cancer. Nat. Rev. Cancer 2003, 3, 695–701. [Google Scholar]
- Barber, T.D.; McManus, K.; Yuen, K.W.; Reis, M.; Parmigiani, G.; Shen, D.; Barrett, I.; Nouhi, Y.; Spencer, F.; Markowitz, S.; et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 3443–3448. [Google Scholar]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of chromosomal instability. Curr. Biol 2010, 20, R285–R295. [Google Scholar]
- Soreide, K.; Janssen, E.A.; Soiland, H.; Korner, H.; Baak, J.P. Microsatellite instability in colorectal cancer. Br. J. Surg 2006, 93, 395–406. [Google Scholar]
- Baylin, S.B.; Hoppener, J.W.; de Bustros, A.; Steenbergh, P.H.; Lips, C.J.; Nelkin, B.D. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res 1986, 46, 2917–2922. [Google Scholar]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. Cpg island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar]
- Walther, A.; Johnstone, E.; Swanton, C.; Midgley, R.; Tomlinson, I.; Kerr, D. Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer 2009, 9, 489–499. [Google Scholar]
- Sastre, L. New DNA sequencing technologies open a promising era for cancer research and treatment. Clin. Transl. Oncol 2011, 13, 301–306. [Google Scholar]
- Chang, H.; Jackson, D.G.; Kayne, P.S.; Ross-Macdonald, P.B.; Ryseck, R.P.; Siemers, N.O. Exome sequencing reveals comprehensive genomic alterations across eight cancer cell lines. PLoS One 2011, 6, e21097. [Google Scholar]
- Timmermann, B.; Kerick, M.; Roehr, C.; Fischer, A.; Isau, M.; Boerno, S.T.; Wunderlich, A.; Barmeyer, C.; Seemann, P.; Koenig, J.; et al. Somatic mutation profiles of msi and mss colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One 2010, 5, e15661. [Google Scholar]
- Curtin, K.; Slattery, M.L.; Samowitz, W.S. Cpg island methylation in colorectal cancer: Past, present and future. Patholog. Res. Int 2011, 2011, 902674:1–902674:8. [Google Scholar]
- Schee, K.; Fodstad, O.; Flatmark, K. Micrornas as biomarkers in colorectal cancer. Am. J. Pathol 2010, 177, 1592–1599. [Google Scholar]
- van Kouwenhove, M.; Kedde, M.; Agami, R. Microrna regulation by rna-binding proteins and its implications for cancer. Nat. Rev. Cancer 2011, 11, 644–656. [Google Scholar]
- de Krijger, I.; Mekenkamp, L.J.; Punt, C.J.; Nagtegaal, I.D. Micrornas in colorectal cancer metastasis. J. Pathol 2011, 224, 438–447. [Google Scholar]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar]
- Jones, S.J.; Laskin, J.; Li, Y.Y.; Griffith, O.L.; An, J.; Bilenky, M.; Butterfield, Y.S.; Cezard, T.; Chuah, E.; Corbett, R.; et al. Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol 2010, 11, R82. [Google Scholar]
- Torlakovic, E.; Snover, D.C. Serrated adenomatous polyposis in humans. Gastroenterology 1996, 110, 748–755. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar]
- Laurent-Puig, P.; Blons, H.; Cugnenc, P.H. Sequence of molecular genetic events in colorectal tumorigenesis. Eur. J. Cancer Prev 1999, 8, S39–S47. [Google Scholar]
- Snover, D.C.; Jass, J.R.; Fenoglio-Preiser, C.; Batts, K.P. Serrated polyps of the large intestine: A morphologic and molecular review of an evolving concept. Am. J. Clin. Pathol 2005, 124, 380–391. [Google Scholar]
- O’Brien, M.J.; Yang, S.; Mack, C.; Xu, H.; Huang, C.S.; Mulcahy, E.; Amorosino, M.; Farraye, F.A. Comparison of microsatellite instability, cpg island methylation phenotype, braf and kras status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am. J. Surg. Pathol 2006, 30, 1491–1501. [Google Scholar]
- Kim, K.M.; Lee, E.J.; Ha, S.; Kang, S.Y.; Jang, K.T.; Park, C.K.; Kim, J.Y.; Kim, Y.H.; Chang, D.K.; Odze, R.D. Molecular features of colorectal hyperplastic polyps and sessile serrated adenoma/polyps from korea. Am. J. Surg. Pathol 2011, 35, 1274–1286. [Google Scholar]
- Berg, M. Genomics of Colorectal Carcinomas from Young and Elderly Patients. Ph.D. Thesis, University of Oslo, Oslo, Norway, 2010. [Google Scholar]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American society of clinical oncology provisional clinical opinion: Testing for kras gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol 2009, 27, 2091–2096. [Google Scholar]
- Jaeger, E.; Webb, E.; Howarth, K.; Carvajal-Carmona, L.; Rowan, A.; Broderick, P.; Walther, A.; Spain, S.; Pittman, A.; Kemp, Z.; et al. Common genetic variants at the crac1 (hmps) locus on chromosome 15q13.3 influence colorectal cancer risk. Nat. Genet 2008, 40, 26–28. [Google Scholar]
- Migliore, L.; Migheli, F.; Spisni, R.; Coppede, F. Genetics, cytogenetics, and epigenetics of colorectal cancer. J. Biomed. Biotechnol 2011, 2011, 792362. [Google Scholar]
- Bacolod, M.D.; Barany, F. Molecular profiling of colon tumors: The search for clinically relevant biomarkers of progression, prognosis, therapeutics, and predisposition. Ann. Surg. Oncol 2011, 18, 3694–3700. [Google Scholar]
- Broderick, P.; Carvajal-Carmona, L.; Pittman, A.M.; Webb, E.; Howarth, K.; Rowan, A.; Lubbe, S.; Spain, S.; Sullivan, K.; Fielding, S.; et al. A genome-wide association study shows that common alleles of smad7 influence colorectal cancer risk. Nat. Genet 2007, 39, 1315–1317. [Google Scholar]
- Pittman, A.M.; Webb, E.; Carvajal-Carmona, L.; Howarth, K.; Di Bernardo, M.C.; Broderick, P.; Spain, S.; Walther, A.; Price, A.; Sullivan, K.; et al. Refinement of the basis and impact of common 11q23.1 variation to the risk of developing colorectal cancer. Hum. Mol. Genet 2008, 17, 3720–3727. [Google Scholar]
- Zanke, B.W.; Greenwood, C.M.; Rangrej, J.; Kustra, R.; Tenesa, A.; Farrington, S.M.; Prendergast, J.; Olschwang, S.; Chiang, T.; Crowdy, E.; et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat. Genet 2007, 39, 989–994. [Google Scholar]
- Haiman, C.A.; Le, M.L.; Yamamato, J.; Stram, D.O.; Sheng, X.; Kolonel, L.N.; Wu, A.H.; Reich, D.; Henderson, B.E. A common genetic risk factor for colorectal and prostate cancer. Nat. Genet 2007, 39, 954–956. [Google Scholar]
- Tomlinson, I.; Webb, E.; Carvajal-Carmona, L.; Broderick, P.; Kemp, Z.; Spain, S.; Penegar, S.; Chandler, I.; Gorman, M.; Wood, W.; et al. A genome-wide association scan of tag snps identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat. Genet 2007, 39, 984–988. [Google Scholar]
- He, J.; Wilkens, L.R.; Stram, D.O.; Kolonel, L.N.; Henderson, B.E.; Wu, A.H.; Le, M.L.; Haiman, C.A. Generalizability and epidemiologic characterization of eleven colorectal cancer gwas hits in multiple populations. Cancer Epidemiol. Biomark. Prev 2011, 20, 70–81. [Google Scholar]
- Houlston, R.S.; Webb, E.; Broderick, P.; Pittman, A.M.; Di Bernardo, M.C.; Lubbe, S.; Chandler, I.; Vijayakrishnan, J.; Sullivan, K.; Penegar, S.; et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat. Genet 2008, 40, 1426–1435. [Google Scholar]
- Tenesa, A.; Farrington, S.M.; Prendergast, J.G.; Porteous, M.E.; Walker, M.; Haq, N.; Barnetson, R.A.; Theodoratou, E.; Cetnarskyj, R.; Cartwright, N.; et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat. Genet 2008, 40, 631–637. [Google Scholar]
- Tomlinson, I.P.; Webb, E.; Carvajal-Carmona, L.; Broderick, P.; Howarth, K.; Pittman, A.M.; Spain, S.; Lubbe, S.; Walther, A.; Sullivan, K.; et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat. Genet 2008, 40, 623–630. [Google Scholar]
- Bosch, L.J.; Carvalho, B.; Fijneman, R.J.; Jimenez, C.R.; Pinedo, H.M.; van, E.M.; Meijer, G.A. Molecular tests for colorectal cancer screening. Clin. Colorectal Cancer 2011, 10, 8–23. [Google Scholar]
- Ned, R.M.; Melillo, S.; Marrone, M. Fecal DNA testing for colorectal cancer screening: The colosure test. PLoS Curr 2011, 3. [Google Scholar] [CrossRef]
- Lofton-Day, C.; Model, F.; deVos, T.; Tetzner, R.; Distler, J.; Schuster, M.; Song, X.; Lesche, R.; Liebenberg, V.; Ebert, M.; et al. DNA methylation biomarkers for blood-based colorectal cancer screening. Clin. Chem 2008, 54, 414–423. [Google Scholar]
- Ng, E.K.; Chong, W.W.; Jin, H.; Lam, E.K.; Shin, V.Y.; Yu, J.; Poon, T.C.; Ng, S.S.; Sung, J.J. Differential expression of micrornas in plasma of patients with colorectal cancer: A potential marker for colorectal cancer screening. Gut 2009, 58, 1375–1381. [Google Scholar]
- Deschoolmeester, V.; Baay, M.; Specenier, P.; Lardon, F.; Vermorken, J.B. A review of the most promising biomarkers in colorectal cancer: One step closer to targeted therapy. Oncologist 2010, 15, 699–731. [Google Scholar]
- Tan, I.B.; Tan, P. Genetics: An 18-gene signature (coloprint(r)) for colon cancer prognosis. Nat. Rev. Clin. Oncol 2011, 8, 131–133. [Google Scholar]
- Salazar, R.; Roepman, P.; Capella, G.; Moreno, V.; Simon, I.; Dreezen, C.; Lopez-Doriga, A.; Santos, C.; Marijnen, C.; Westerga, J.; et al. Gene expression signature to improve prognosis prediction of stage ii and iii colorectal cancer. J. Clin. Oncol 2011, 29, 17–24. [Google Scholar]
- Clark-Langone, K.M.; Sangli, C.; Krishnakumar, J.; Watson, D. Translating tumor biology into personalized treatment planning: Analytical performance characteristics of the oncotype dx colon cancer assay. BMC Cancer 2010, 10, 691. [Google Scholar]
- Webber, E.M.; Lin, J.S.; Evelyn, P.W. Oncotype dx tumor gene expression profiling in stage ii colon cancer. Application: Prognostic, risk prediction. PLoS Curr 2010, 2. [Google Scholar] [CrossRef]
- Mejia, A.; Waldmana, S.A. Previstage gcc test for staging patients with colorectal cancer. Expert Rev. Mol. Diagn 2008, 8, 571–578. [Google Scholar]
- Carlson, M.R. Previstage gcc colorectal cancer staging test: A new molecular test to identify lymph node metastases and provide more accurate information about the stage of patients with colorectal cancer. Mol. Diagn. Ther 2009, 13, 11–14. [Google Scholar]
- Guo, C.; Sah, J.F.; Beard, L.; Willson, J.K.; Markowitz, S.D.; Guda, K. The noncoding rna, mir-126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3-kinase signaling and is frequently lost in colon cancers. Genes Chromosomes Cancer 2008, 47, 939–946. [Google Scholar]
- Schetter, A.J.; Leung, S.Y.; Sohn, J.J.; Zanetti, K.A.; Bowman, E.D.; Yanaihara, N.; Yuen, S.T.; Chan, T.L.; Kwong, D.L.; Au, G.K.; et al. Microrna expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 2008, 299, 425–436. [Google Scholar]
- Ross, J.S.; Torres-Mora, J.; Wagle, N.; Jennings, T.A.; Jones, D.M. Biomarker-based prediction of response to therapy for colorectal cancer: Current perspective. Am. J. Clin. Pathol 2010, 134, 478–490. [Google Scholar]
- Garcia-Aguilar, J.; Chen, Z.; Smith, D.D.; Li, W.; Madoff, R.D.; Cataldo, P.; Marcet, J.; Pastor, C. Identification of a biomarker profile associated with resistance to neoadjuvant chemoradiation therapy in rectal cancer. Ann. Surg 2011, 254, 486–493. [Google Scholar]
- Song, B.; Wang, Y.; Xi, Y.; Kudo, K.; Bruheim, S.; Botchkina, G.I.; Gavin, E.; Wan, Y.; Formentini, A.; Kornmann, M.; et al. Mechanism of chemoresistance mediated by mir-140 in human osteosarcoma and colon cancer cells. Oncogene 2009, 28, 4065–4074. [Google Scholar]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med 2004, 10, 789–799. [Google Scholar]
- Gruber, S.B.; Ellis, N.A.; Scott, K.K.; Almog, R.; Kolachana, P.; Bonner, J.D.; Kirchhoff, T.; Tomsho, L.P.; Nafa, K.; Pierce, H.; et al. Blm heterozygosity and the risk of colorectal cancer. Science 2002, 297, 2013. [Google Scholar]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of msh2 in families with lynch syndrome due to deletion of the 3′ exons of tacstd1. Nat. Genet 2009, 41, 112–117. [Google Scholar]
- Ekelund, G.; Manjer, J.; Zackrisson, S. Population-based screening for colorectal cancer with faecal occult blood test--do we really have enough evidence? Int. J. Colorectal Dis 2010, 25, 1269–1275. [Google Scholar]
- Lenhard, K.; Bommer, G.T.; Asutay, S.; Schauer, R.; Brabletz, T.; Goke, B.; Lamerz, R.; Kolligs, F.T. Analysis of promoter methylation in stool: A novel method for the detection of colorectal cancer. Clin. Gastroenterol. Hepatol 2005, 3, 142–149. [Google Scholar]
- Itzkowitz, S.; Brand, R.; Jandorf, L.; Durkee, K.; Millholland, J.; Rabeneck, L.; Schroy, P.C., III; Sontag, S.; Johnson, D.; Markowitz, S.; et al. A simplified, noninvasive stool DNA test for colorectal cancer detection. Am. J. Gastroenterol 2008, 103, 2862–2870. [Google Scholar]
- Itzkowitz, S.H.; Jandorf, L.; Brand, R.; Rabeneck, L.; Schroy, P.C., III; Sontag, S.; Johnson, D.; Skoletsky, J.; Durkee, K.; Markowitz, S.; et al. Improved fecal DNA test for colorectal cancer screening. Clin. Gastroenterol. Hepatol 2007, 5, 111–117. [Google Scholar]
- Azuara, D.; Rodriguez-Moranta, F.; de, O.J.; Soriano-Izquierdo, A.; Mora, J.; Guardiola, J.; Biondo, S.; Blanco, I.; Peinado, M.A.; Moreno, V.; et al. Novel methylation panel for the early detection of colorectal tumors in stool DNA. Clin. Colorectal Cancer 2010, 9, 168–176. [Google Scholar]
- Lind, G.E.; Danielsen, S.A.; Ahlquist, T.; Merok, M.A.; Andresen, K.; Skotheim, R.I.; Hektoen, M.; Rognum, T.O.; Meling, G.I.; Hoff, G.; et al. Identification of an epigenetic biomarker panel with high sensitivity and specificity for colorectal cancer and adenomas. Mol. Cancer 2011, 10, 85. [Google Scholar]
- Model, F.; Osborn, N.; Ahlquist, D.; Gruetzmann, R.; Molnar, B.; Sipos, F.; Galamb, O.; Pilarsky, C.; Saeger, H.D.; Tulassay, Z.; et al. Identification and validation of colorectal neoplasia-specific methylation markers for accurate classification of disease. Mol. Cancer Res 2007, 5, 153–163. [Google Scholar]
- Guastadisegni, C.; Colafranceschi, M.; Ottini, L.; Dogliotti, E. Microsatellite instability as a marker of prognosis and response to therapy: A meta-analysis of colorectal cancer survival data. Eur. J. Cancer 2010, 46, 2788–2798. [Google Scholar]
- Bardelli, A.; Siena, S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J. Clin. Oncol 2010, 28, 1254–1261. [Google Scholar]
- Samowitz, W.S.; Sweeney, C.; Herrick, J.; Albertsen, H.; Levin, T.R.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Poor survival associated with the braf v600e mutation in microsatellite-stable colon cancers. Cancer Res 2005, 65, 6063–6069. [Google Scholar]
- Sartore-Bianchi, A.; Di, N.F.; Nichelatti, M.; Molinari, F.; De, D.S.; Saletti, P.; Martini, M.; Cipani, T.; Marrapese, G.; Mazzucchelli, L.; et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to egfr-targeted monoclonal antibodies in colorectal cancer. PLoS One 2009, 4, e7287. [Google Scholar]
- Li, Z.; Jin, K.; Lan, H.; Teng, L. Heterogeneity in primary colorectal cancer and its corresponding metastases: A potential reason of egfr-targeted therapy failure? Hepatogastroenterology 2011, 58, 411–416. [Google Scholar]
- Oltedal, S.; Aasprong, O.G.; Moller, J.H.; Korner, H.; Gilje, B.; Tjensvoll, K.; Birkemeyer, E.M.; Heikkila, R.; Smaaland, R.; Nordgard, O. Heterogeneous distribution of k-ras mutations in primary colon carcinomas: Implications for egfr-directed therapy. Int. J. Colorectal Dis 2011, 26, 1271–1277. [Google Scholar]
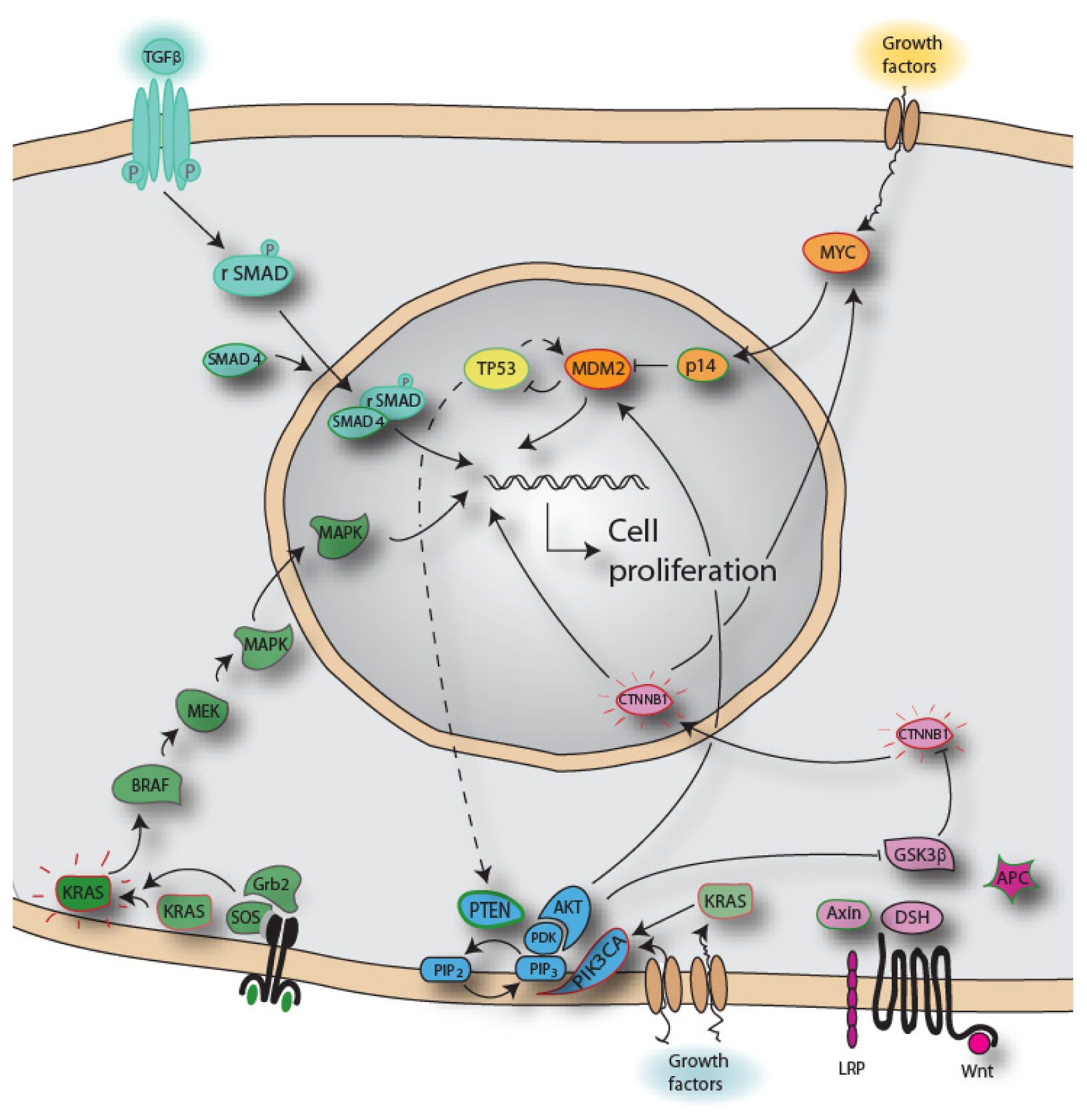

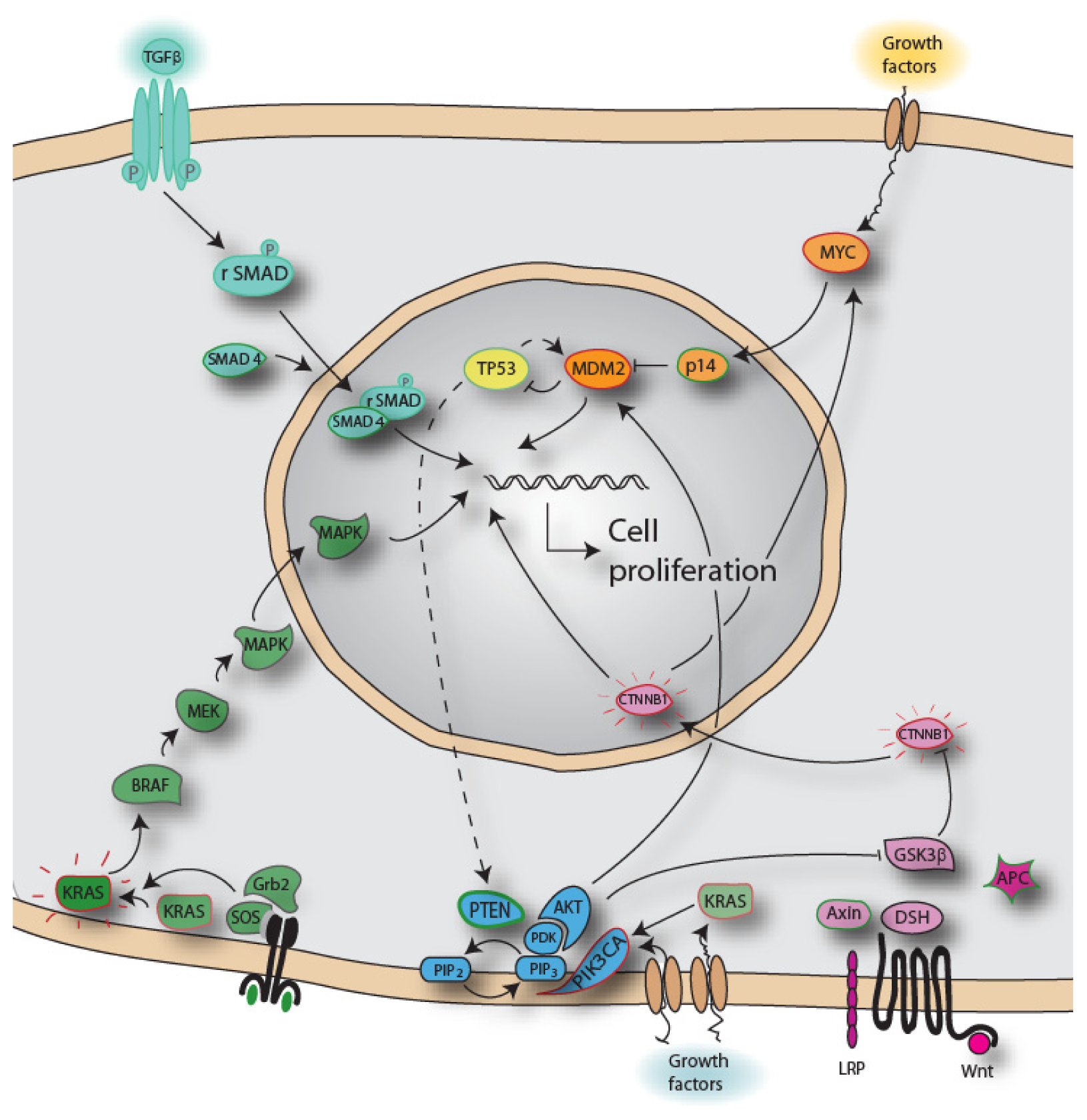
{kind=link}
{kind=link}
| Type of Biomarker | Objective for use | Biological marker | References |
|---|---|---|---|
| Risk stratification | Assess the likelihood that cancers will develop | APC, AXIN2, BMPR1A, SMAD4, MUTYH, MSH2, MLH1, MSH6, PMS2, STK11, PTEN, EPCAM, 8q24, 15q13.3, SMAD7, LOC120376 | [29–40] |
| Screening | Detect cancers in the asymptomatic population | Stool tests, blood based tests | [41] |
| Diagnosis | Definitively establish the presence of cancer | Vimentin (ColoSure *), SEPT9 (ColoVantage *), miR-17-3p, miR-92 | [42–44] |
| Classification | Classify patients by disease subset | MSI, CIN, CIMP | |
| Prognosis | Predict the probable outcome of cancer regardless of therapy | MSI, 18-gene signature (ColoPrint *), 12-gene signature (OncoType DX *), GCC expression (Previstage *), miR-21 | [45–53] |
| Prediction/treatment stratification | Predict response to particular therapies and choose the drug that is mostly likely to yield a favorable response in a given patient | EGFR, KRAS, BRAF, PIK3CA, PTEN, TP53, miR-140 Panel (TP53, KRAS, CCDN1, MTHFR) | [54–56] |
| Test name | Biological material | Biomarker(s) |
|---|---|---|
| ColoSureTM | Methylated DNA in feces | Vimentin |
| ColoVantage® | Methylated DNA in plasma | SEPT9 |
| ColoPrint® | mRNA expression in tumor tissue | MCTP1, LAMA3, CTSC, PYROX D1, EDEM1, IL2RB, ZNF697, SLC6A11, IL2RA, CYFIP2, PIM3, LIF, PLIN3, HSD3B1, ZBED4, PPARA, THNSL2, CA4388O2 |
| OncoType DX® | mRNA expression in tumor tissue | Ki-67, C-MYC, MYBL2, FAP, BGN, INHBA, GADD45B, ATP5E, PGK1, GPX1, UBB, VDAC2 |
| PrevistageTM | mRNA expression in lymph node tissue | GCC (GUCY2C) |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Berg, M.; Søreide, K. Genetic and Epigenetic Traits as Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2011, 12, 9426-9439. https://doi.org/10.3390/ijms12129426
Berg M, Søreide K. Genetic and Epigenetic Traits as Biomarkers in Colorectal Cancer. International Journal of Molecular Sciences. 2011; 12(12):9426-9439. https://doi.org/10.3390/ijms12129426
Chicago/Turabian StyleBerg, Marianne, and Kjetil Søreide. 2011. "Genetic and Epigenetic Traits as Biomarkers in Colorectal Cancer" International Journal of Molecular Sciences 12, no. 12: 9426-9439. https://doi.org/10.3390/ijms12129426
APA StyleBerg, M., & Søreide, K. (2011). Genetic and Epigenetic Traits as Biomarkers in Colorectal Cancer. International Journal of Molecular Sciences, 12(12), 9426-9439. https://doi.org/10.3390/ijms12129426