Differential Motor Neuron Impairment and Axonal Regeneration in Sporadic and Familiar Amyotrophic Lateral Sclerosis with SOD-1 Mutations: Lessons from Neurophysiology

Abstract
:1. Introduction
2. Results and Discussion
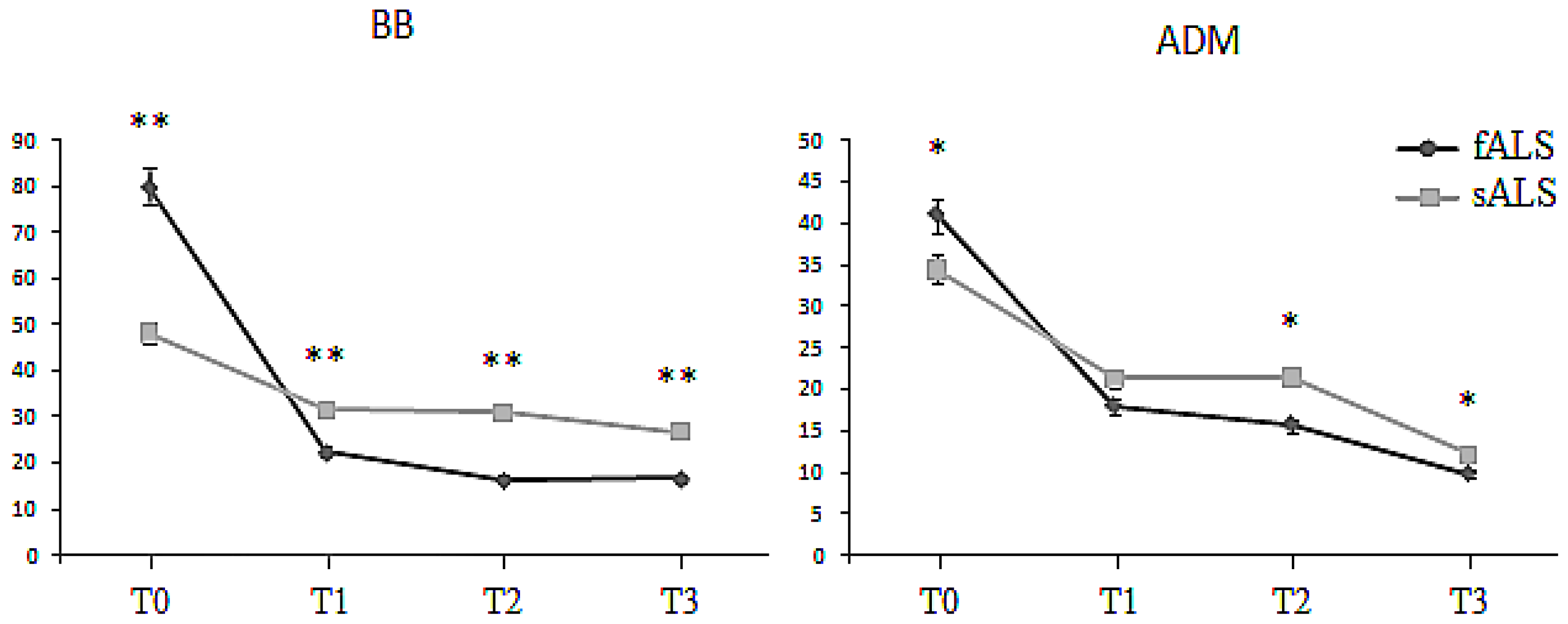
2.1. Motor Unit Number Estimation (MUNE)
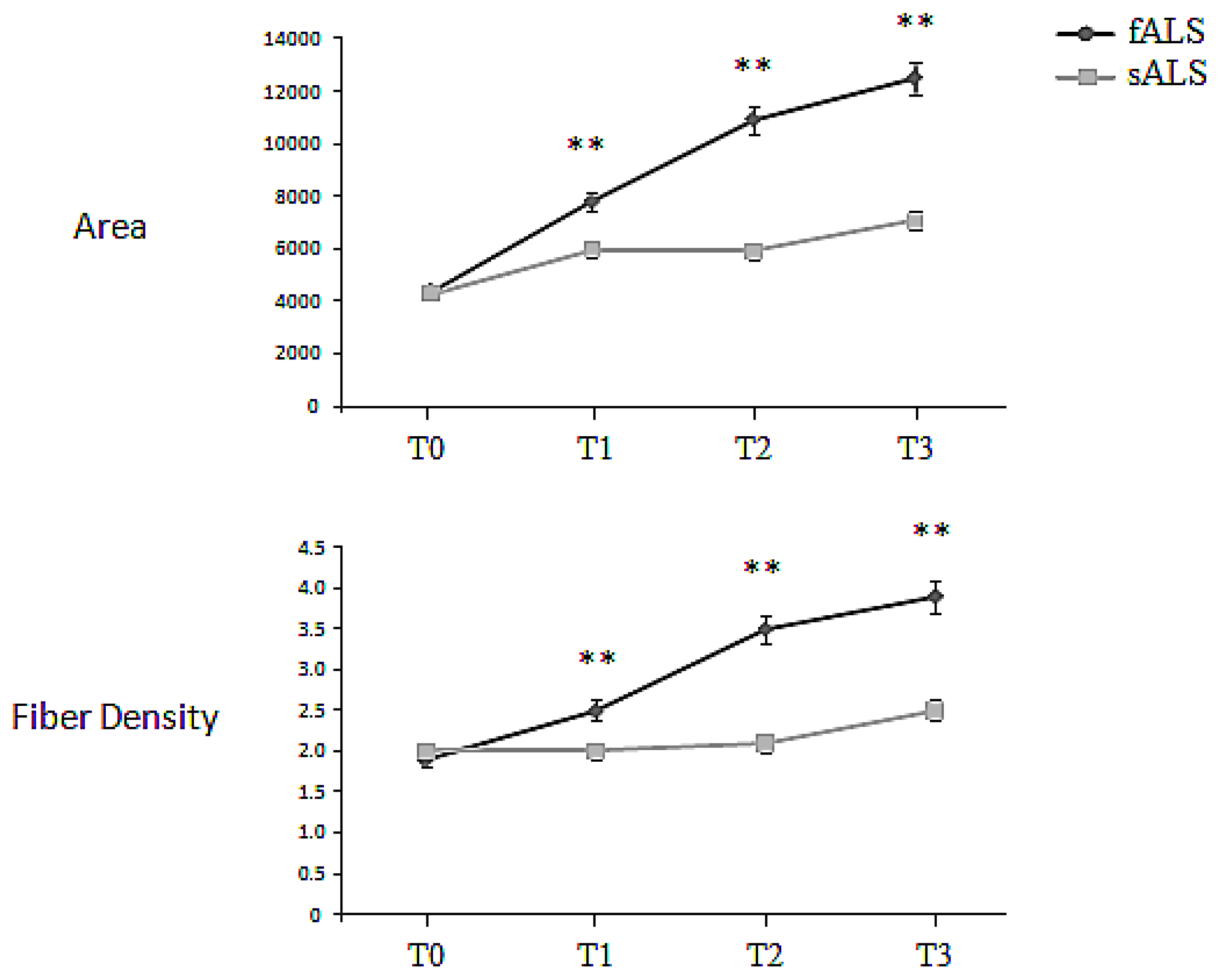
2.2. Macro-EMG
3. Experimental Section
3.1. Patients and Methods
3.2. Macro-EMG
3.3. Motor Unit Number Estimation (MUNE)
3.4. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Juergens, S.M.; Kurland, L.T.; Okazaki, H.; Mulder, D.W. ALS in rochester, minnesota, 1925–1977. Neurology 1980, 30, 463–470. [Google Scholar]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar]
- Amyotrophic Lateral Sclerosis Online Genetics Database. http://alsod.iop.kcl.ac.uk/ accessed on 8 October 2011.
- de Belleroche, J.; Orrell, R.; King, A. Familial amyotrophic lateral sclerosis/motor neuron disease (FALS): A review of current developments. J. Med. Genet 1995, 32, 841–847. [Google Scholar]
- Aggarwal, A.; Nicholson, G. Detection of preclinical motor neuron loss in sod1 mutation carriers using motor unit number estimation. J. Neurol. Neurosurg. Psychiatry 2002, 73, 199–201. [Google Scholar]
- Clarke, G.; Lumsden, C.J.; McInnes, R.R. Inherited neurodegenerative diseases: The one-hit model of neurodegeneration. Hum. Mol. Genet 2001, 10, 2269–2275. [Google Scholar]
- Johnston, J.A.; Dalton, M.J.; Gurney, M.E.; Kopito, R.R. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 12571–12576. [Google Scholar]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar]
- Deng, H.X.; Shi, Y.; Furukawa, Y.; Zhai, H.; Fu, R.; Liu, E.; Gorrie, G.H.; Khan, M.S.; Hung, W.Y.; Bigio, E.H.; et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7142–7147. [Google Scholar]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci 1998, 18, 3241–3250. [Google Scholar]
- Karch, C.M.; Prudencio, M.; Winkler, D.D.; Hart, P.J.; Borchelt, D.R. Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc. Natl. Acad. Sci. USA 2009, 106, 7774–7779. [Google Scholar]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol 2004, 185, 232–240. [Google Scholar]
- Jaarsma, D.; Teuling, E.; Haasdijk, E.D.; de Zeeuw, C.I.; Hoogenraad, C.C. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci 2008, 28, 2075–2088. [Google Scholar]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar]
- Siddique, T.; Deng, H.X. Genetics of amyotrophic lateral sclerosis. Hum. Mol. Genet 1996, 5, 1465–1470. [Google Scholar]
- Andersen, P.M.; Nilsson, P.; Keranen, M.L.; Forsgren, L.; Hagglund, J.; Karlsborg, M.; Ronnevi, L.O.; Gredal, O.; Marklund, S.L. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain 1997, 120, 1723–1737. [Google Scholar]
- Swash, M.; Ingram, D. Preclinical and subclinical events in motor neuron disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 165–168. [Google Scholar]
- Beghi, E.; Balzarini, C.; Bogliun, G.; Logroscino, G.; Manfredi, L.; Mazzini, L.; Micheli, A.; Millul, A.; Poloni, M.; Riva, R.; et al. Reliability of the El Escorial diagnostic criteria for amyotrophic lateral sclerosis. Neuroepidemiology 2002, 21, 265–270. [Google Scholar]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord 2000, 1, 293–299. [Google Scholar]
- Olney, R.K.; Lomen-Hoerth, C. Motor unit number estimation (MUNE): How may it contribute to the diagnosis of ALS? Amyotroph. Lateral Scler. Other Motor Neuron Disord 2000, 1, S41–S44. [Google Scholar]
- Bromberg, M.B.; Forshew, D.A.; Nau, K.L.; Bromberg, J.; Simmons, Z.; Fries, T.J. Motor unit number estimation, isometric strength, and electromyographic measures in amyotrophic lateral sclerosis. Muscle Nerve 1993, 16, 1213–1219. [Google Scholar]
- de Carvalho, M.; Costa, J.; Swash, M. Clinical trials in ALS: A review of the role of clinical and neurophysiological measurements. Amyotroph. Lateral Scler. Other Motor Neuron Disord 2005, 6, 202–212. [Google Scholar]
- Eisen, A. Clinical electrophysiology of the upper and lower motor neuron in amyotrophic lateral sclerosis. Semin. Neurol 2001, 21, 141–154. [Google Scholar]
- Finsterer, J.; Fuglsang-Frederiksen, A. Concentric-needle versus macro EMG. II. Detection of neuromuscular disorders. Clin. Neurophysiol 2001, 112, 853–860. [Google Scholar]
- Stålberg, E. Macro EMG, a new recording technique. J. Neurol. Neurosurg. Psychiatry 1980, 43, 475–482. [Google Scholar]
- Dengler, R.; Konstanzer, A.; Kuther, G.; Hesse, S.; Wolf, W.; Struppler, A. Amyotrophic lateral sclerosis: Macro-EMG and twitch forces of single motor units. Muscle Nerve 1990, 13, 545–550. [Google Scholar]
- Guiloff, R.J.; Modarres-Sadeghi, H.; Stålberg, E.; Rogers, H. Short-term stability of single motor unit recordings in motor neuron disease: A macro EMG study. J. Neurol. Neurosurg. Psychiatry 1988, 51, 671–676. [Google Scholar]
- Stålberg, E. Macro EMG. Muscle Nerve 1983, 6, 619–630. [Google Scholar]
- Daube, J.R. Motor unit number estimates—From A to Z. J. Neurol. Sci 2006, 242, 23–35. [Google Scholar]
- Gooch, C.L.; Shefner, J.M. ALS surrogate markers. MUNE. Amyotroph. Lateral Scler. Other Motor Neuron Disord 2004, 5, 104–107. [Google Scholar]
- McComas, A.J.; Fawcett, P.R.; Campbell, M.J.; Sica, R.E. Electrophysiological estimation of the number of motor units within a human muscle. J. Neurol. Neurosurg. Psychiatry 1971, 34, 121–131. [Google Scholar]
- Sartucci, F.; Maritato, P.; Moscato, G.; Orlandi, G.; Calabrese, R.; Domenici, G.L.; Murri, L. Motor unit number estimation (MUNE) as a quantitative measure of disease progression and motor unit reorganization in amyotrophic lateral sclerosis. Int. J. Neurosci 2007, 117, 1229–1236. [Google Scholar]
- Sartucci, F.; Moscato, G.; Rossi, C.; Caleo, M.; Bocci, T.; Murri, L.; Giannini, F.; Rossi, A. Macro-EMG and MUNE changes in patients with amyotrophic lateral sclerosis: One-year follow up. Int. J. Neurosci 2011, 121, 257–266. [Google Scholar]
- Cuturic, M.; Shamsnia, M.; Palliyath, S. Lateral asymmetry of motor unit number estimate (MUNE). Electromyogr. Clin. Neurophysiol 2005, 45, 233–239. [Google Scholar]
- Aggarwal, A.; Nicholson, G. Normal complement of motor units in asymptomatic familial (SOD1 mutation) amyotrophic lateral sclerosis carriers. J. Neurol. Neurosurg. Psychiatry 2001, 71, 478–481. [Google Scholar]
- Bauermeister, W.; Jabre, J.F. The spectrum of concentric macro EMG correlations. Part I. Normal subjects. Muscle Nerve 1992, 15, 1081–1084. [Google Scholar]
- Sanders, D.B.; Stalberg, E.V. Aaem minimonograph #25: Single-fiber electromyography. Muscle Nerve 1996, 19, 1069–1083. [Google Scholar]
- Keypoint Service Manual; DANTEC Keypoint: Copenhagen, Denmark, 1996.
- McComas, A.J. Motor unit estimation: Anxieties and achievements. Muscle Nerve 1995, 18, 369–379. [Google Scholar]
- Hong, Y.H.; Sung, J.J.; Park, K.S.; Kwon, O.; Min, J.H.; Lee, K.W. Statistical MUNE: A comparison of two methods of setting recording windows in healthy subjects and ALS patients. Clin. Neurophysiol 2007, 118, 2605–2611. [Google Scholar]
- Henderson, R.D.; Ridall, P.G.; Hutchinson, N.M.; Pettitt, A.N.; McCombe, P.A. Bayesian statistical MUNE method. Muscle Nerve 2007, 36, 206–213. [Google Scholar]
- STATISTICA, version 8; StatSoft, Inc: Tulsa, OK, USA, 2007.
- Strong, M.J.; Brown, W.F.; Hudson, A.J.; Snow, R. Motor unit estimates in the biceps-brachialis in amyotrophic lateral sclerosis. Muscle Nerve 1988, 11, 415–422. [Google Scholar]
- Gan, R.; Jabre, J.F. The spectrum of concentric macro EMG correlations. Part II. Patients with diseases of muscle and nerve. Muscle Nerve 1992, 15, 1085–1088. [Google Scholar]
- Ilieva, H.S.; Yamanaka, K.; Malkmus, S.; Kakinohana, O.; Yaksh, T.; Marsala, M.; Cleveland, D.W. Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proc. Natl. Acad. Sci. USA 2008, 105, 12599–12604. [Google Scholar]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar]
- Blain, C.R.; Brunton, S.; Williams, V.C.; Leemans, A.; Turner, M.R.; Andersen, P.M.; Catani, M.; Stanton, B.R.; Ganesalingham, J.; Jones, D.K.; et al. Differential corticospinal tract degeneration in homozygous “D90A” SOD-1 ALS and sporadic ALS. J. Neurol. Neurosurg. Psychiatry 2011, 82, 843–849. [Google Scholar]
- Agosta, F.; Pagani, E.; Petrolini, M.; Caputo, D.; Perini, M.; Prelle, A.; Salvi, F.; Filippi, M. Assessment of white matter tract damage in patients with amyotrophic lateral sclerosis: A diffusion tensor MR imaging tractography study. AJNR Am. J. Neuroradiol 2010, 31, 1457–1461. [Google Scholar]
- Agosta, F.; Chio, A.; Cosottini, M.; de Stefano, N.; Falini, A.; Mascalchi, M.; Rocca, M.A.; Silani, V.; Tedeschi, G.; Filippi, M. The present and the future of neuroimaging in amyotrophic lateral sclerosis. AJNR Am. J. Neuroradiol 2010, 31, 1769–1777. [Google Scholar]
- de Carvalho, M.; Chio, A.; Dengler, R.; Hecht, M.; Weber, M.; Swash, M. Neurophysiological measures in amyotrophic lateral sclerosis: Markers of progression in clinical trials. Amyotroph. Lateral Scler. Other Motor Neuron Disord 2005, 6, 17–28. [Google Scholar]
- Zhai, J.; Ström, A.L.; Kilty, R.; Venkatakrishnan, P.; White, J.; Everson, W.V.; Smart, E.J.; Zhu, H. Proteomic characterization of lipid raft proteins in amyotrophic lateral sclerosis mouse spinal cord. FEBS J 2009, 276, 3308–3323. [Google Scholar]
- Basso, M.; Samengo, G.; Nardo, G.; Massignan, T.; D’Alessandro, G.; Tartari, S.; Cantoni, L.; Marino, M.; Cheroni, C.; de Biasi, S.; et al. Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Migliore, L.; Coppedè, F. Genetics, enviromental factors and the emergin role of epigenetics in neurodegenerative diseases. Mutat. Res 2009, 667, 82–97. [Google Scholar]
- Hegedus, J.; Putman, C.T.; Gordon, T. Progressive motor unit loss in the G93A mouse model of amyotrophic lateral sclerosis is unaffected by gender. Muscle Nerve 2009, 39, 318–327. [Google Scholar]
- Nakamizo, T.; Urushitani, M.; Inoue, R.; Shinohara, A.; Sawada, H.; Honda, K.; Kihara, T.; Akaike, A.; Shimohama, S. Protection of cultured spinal motor neurons by estradiol. Neuroreport 2000, 11, 3493–3497. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T0 | T1 | T2 | T3 | ||
|---|---|---|---|---|---|
| (mean ± 1 SD) | (mean ± 1 SD) | (mean ± 1 SD) | (mean ± 1 SD) | ||
| BB Macro EMG | sALS | 4397.6 ± 255.9 | 6389.2 ± 586.3 | 6553 ± 498.7 | 8072.4 ± 1023.4 |
| Area (μVms) | fALS | 4378.9 ± 319.6 | 7791.0 ± 953.4 | 10922.8 ± 1123.7 | 12499.3 ± 1874.4 |
| BB Macro-EMG | sALS | 2.0 ± 0.3 | 2.1 ± 0.1 | 2.3 ± 0.3 | 2.5 ± 0.2 |
| Fiber Density | fALS | 1.9 ± 0.3 | 2.5 ± 0.3 | 3.5 ± 0.6 | 3.9 ± 0.5 |
| BB MUNE | sALS | 48.3 ± 9.3 | 34.1 ± 1.9 | 35.4 ± 1.4 | 30.8 ± 2.2 |
| fALS | 80.2 ± 7.8 | 21.8 ± 2.2 | 16.8 ± 1.0 | 16.5 ± 2.0 | |
| ADM MUNE | sALS | 35.6 ± 3.4 | 20.1 ± 1.8 | 22.8 ± 5.2 | 12.1 ± 5.8 |
| fALS | 42.8 ± 6.6 | 18.4 ± 3.1 | 15.3 ± 2.1 | 9.0 ± 2.1 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bocci, T.; Pecori, C.; Giorli, E.; Briscese, L.; Tognazzi, S.; Caleo, M.; Sartucci, F. Differential Motor Neuron Impairment and Axonal Regeneration in Sporadic and Familiar Amyotrophic Lateral Sclerosis with SOD-1 Mutations: Lessons from Neurophysiology. Int. J. Mol. Sci. 2011, 12, 9203-9215. https://doi.org/10.3390/ijms12129203
Bocci T, Pecori C, Giorli E, Briscese L, Tognazzi S, Caleo M, Sartucci F. Differential Motor Neuron Impairment and Axonal Regeneration in Sporadic and Familiar Amyotrophic Lateral Sclerosis with SOD-1 Mutations: Lessons from Neurophysiology. International Journal of Molecular Sciences. 2011; 12(12):9203-9215. https://doi.org/10.3390/ijms12129203
Chicago/Turabian StyleBocci, Tommaso, Chiara Pecori, Elisa Giorli, Lucia Briscese, Silvia Tognazzi, Matteo Caleo, and Ferdinando Sartucci. 2011. "Differential Motor Neuron Impairment and Axonal Regeneration in Sporadic and Familiar Amyotrophic Lateral Sclerosis with SOD-1 Mutations: Lessons from Neurophysiology" International Journal of Molecular Sciences 12, no. 12: 9203-9215. https://doi.org/10.3390/ijms12129203