Protein Misdirection Inside and Outside Motor Neurons in Amyotrophic Lateral Sclerosis (ALS): A Possible Clue for Therapeutic Strategies

Abstract
:1. Introduction
2. Superoxide Dismutase 1 (SOD1)
2.1. Molecular Basis
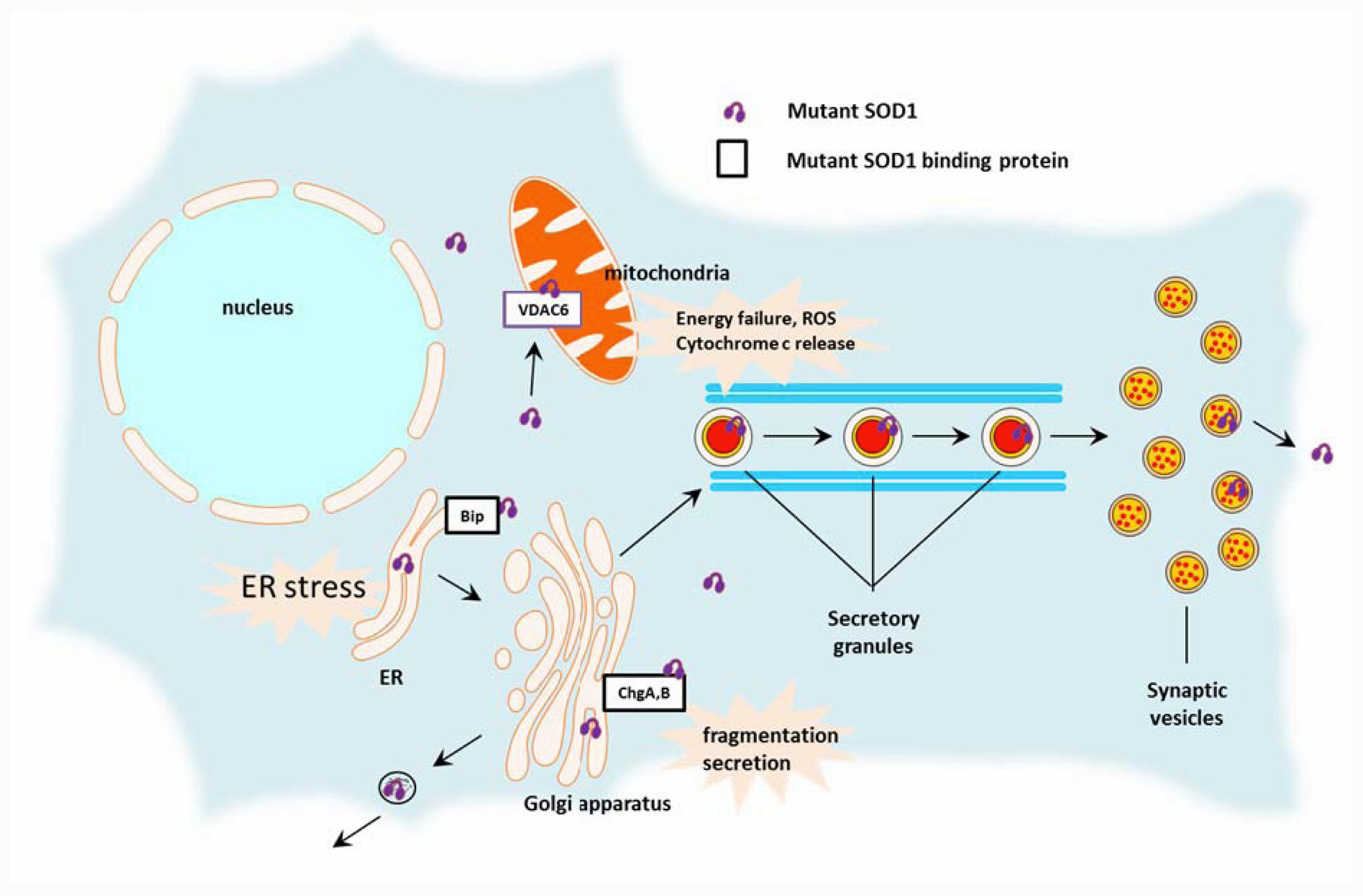
2.2. Toxic Cascades Caused by Mutant SOD1
2.2.1. Mitochondria
2.2.2. Endoplasmic Reticulum (ER)
2.2.3. Golgi Apparatus, Secretory Pathway
2.3. Cell-Autonomous and Non-Cell-Autonomous Motor Neuron Degeneration
2.4. The Role of Extracellular SOD1 Mutants in the Non-Cell-Autonomous Pathology of ALS
- The density of mutant SOD1-expressing cells in the anterior horns governs motor neuron survival;
- Mutant SOD1 in motor neurons or glial cells does indeed induce intracellular damage, and the motor neuron degeneration is accomplished by summation of these phenomena;
- Most of the chemical mediators identified so far are reactive oxygen species or proinflammatory molecules.
2.5. WT SOD1
3. TAR DNA Binding Protein-43kDa (TDP-43)
3.1. Cytosolic Redistribution of TDP-43 in ALS Pathogenesis
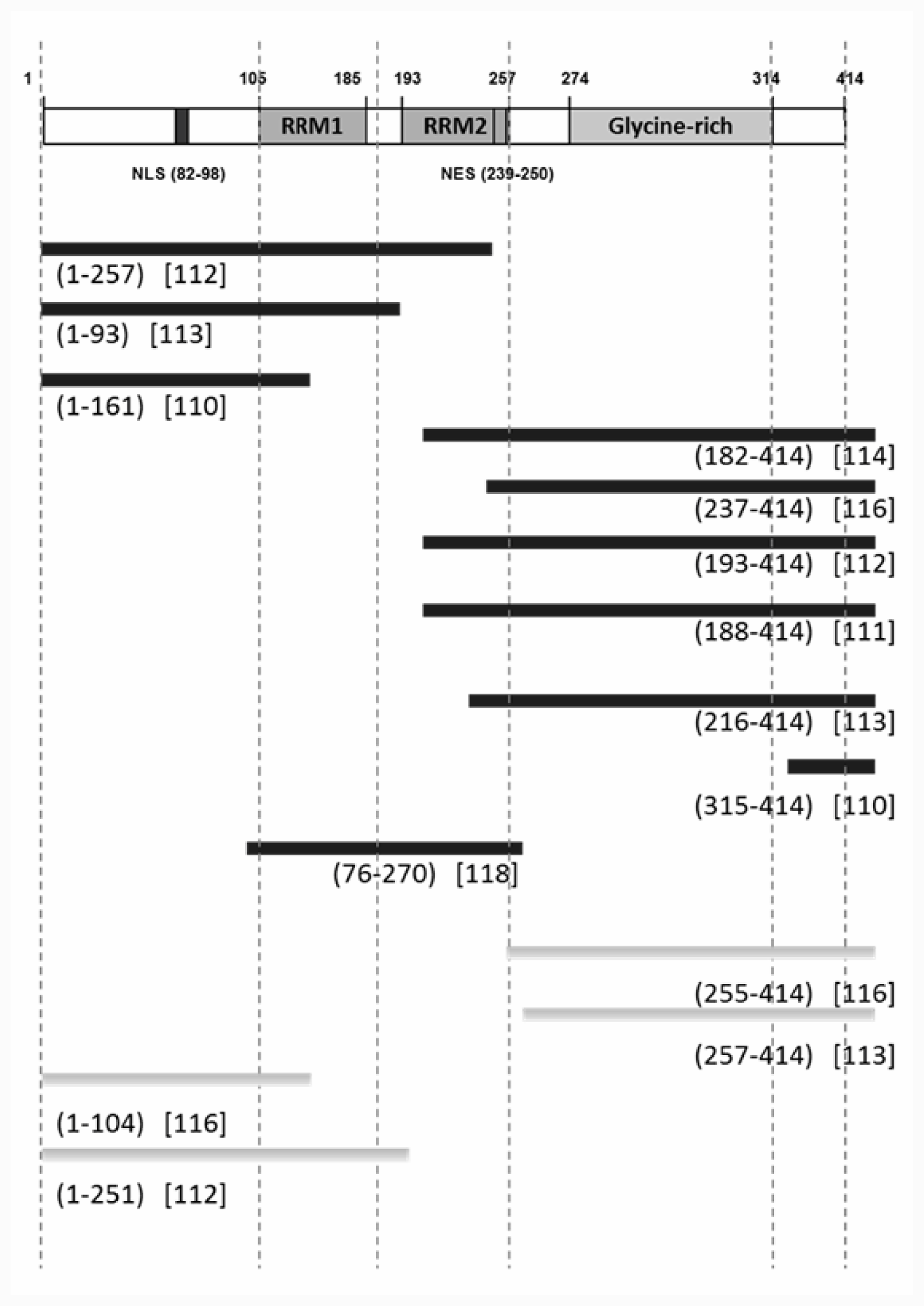
3.2. Nuclear Localizing Signal (NLS) and the Responsible Domains for Cytosolic Redistribution
3.3. Pathogenic Role of Mislocalized TDP-43 in the Cytosol
3.4. WT and Mutant TDP-43 in Motor Neuron Degeneration
4. Aberrant Localization and Therapeutic Hints
5. Conclusions
Acknowledgment
References
- Kabashi, E; Durham, HD. Failure of protein quality control in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1038–1050. [Google Scholar]
- Hart, PJ. Pathogenic superoxide dismutase structure, folding, aggregation and turnover. Curr. Opin. Chem. Biol 2006, 10, 131–138. [Google Scholar]
- Ticozzi, N; Ratti, A; Silani, V. Protein aggregation and defective RNA metabolism as mechanisms for motor neuron damage. CNS Neurol. Disord. Drug Targets 2010, 9, 285–296. [Google Scholar]
- Takahashi, K; Nakamura, H; Okada, E. Hereditary amyotrophic lateral sclerosis. Histochemical and electron microscopic study of hyaline inclusions in motor neurons. Arch. Neurol 1972, 27, 292–299. [Google Scholar]
- Sun, CN; Araoz, C; Lucas, G; Morgan, PN; White, HJ. Amyotrophic lateral sclerosis. Inclusion bodies in a case of the classic sporadic form. Ann. Clin. Lab. Sci 1975, 5, 38–44. [Google Scholar]
- Murayama, S; Ookawa, Y; Mori, H; Nakano, I; Ihara, Y; Kuzuhara, S; Tomonaga, M. Immunocytochemical and ultrastructural study of Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis. Acta Neuropathol. (Berl. ) 1989, 78, 143–152. [Google Scholar]
- Mizusawa, H; Nakamura, H; Wakayama, I; Yen, SH; Hirano, A. Skein-like inclusions in the anterior horn cells in motor neuron disease. J. Neurol. Sci 1991, 105, 14–21. [Google Scholar]
- Traub, R; Mitsumoto, H; Rowland, LP. Research advances in amyotrophic lateral sclerosis, 2009 to 2010. Curr. Neurol. Neurosci. Rep 2011, 11, 67–77. [Google Scholar]
- Maruyama, H; Morino, H; Ito, H; Izumi, Y; Kato, H; Watanabe, Y; Kinoshita, Y; Kamada, M; Nodera, H; Suzuki, H; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar]
- Deng, HX; Chen, W; Hong, ST; Boycott, KM; Gorrie, GH; Siddique, N; Yang, Y; Fecto, F; Shi, Y; Zhai, H; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar]
- Renton, AE; Majounie, E; Waite, A; Simón-Sánchez, J; Rollinson, S; Gibbs, JR; Schymick, JC; Laaksovirta, H; van Swieten, JC; Myllykangas, L; et al. A Hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron 2011, in press. [Google Scholar]
- Dejesus-Hernandez, M; Mackenzie, IR; Boeve, BF; Boxer, AL; Baker, M; Rutherford, NJ; Nicholson, AM; Finch, NA; Flynn, H; Adamson, J; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron 2011, in press. [Google Scholar]
- Ilieva, H; Polymenidou, M; Cleveland, DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol 2009, 187, 761–772. [Google Scholar]
- Bento-Abreu, A; Van Damme, P; Van Den Bosch, L; Robberecht, W. The neurobiology of amyotrophic lateral sclerosis. Eur. J. Neurosci 2010, 31, 2247–2265. [Google Scholar]
- Kato, S; Takikawa, M; Nakashima, K; Hirano, A; Cleveland, DW; Kusaka, H; Shibata, N; Kato, M; Nakano, I; Ohama, E. New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: Inclusions containing SOD1 in neurons and astrocytes. Amyotroph. Lateral Scler 2000, 1, 163–184. [Google Scholar]
- Yokoseki, A; Shiga, A; Tan, CF; Tagawa, A; Kaneko, H; Koyama, A; Eguchi, H; Tsujino, A; Ikeuchi, T; Kakita, A; et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol 2008, 63, 538–542. [Google Scholar]
- Tateishi, T; Hokonohara, T; Yamasaki, R; Miura, S; Kikuchi, H; Iwaki, A; Tashiro, H; Furuya, H; Nagara, Y; Ohyagi, Y; et al. Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol. (Berl. ) 2010, 119, 355–364. [Google Scholar]
- Rosen, DR; Siddique, T; Patterson, D; Figlewicz, DA; Sapp, P; Hentati, A; Donaldson, D; Goto, J; O’Regan, JP; Deng, HX; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar]
- University of Tokyo. ALS Mutation Database. 2011. Available online: https://reseq.lifesciencedb.jp/resequence/GeneDetail.do?targetId=1&geneId=EG6647 accessed on 12 October 2011.
- Reaume, AG; Elliott, JL; Hoffman, EK; Kowall, NW; Ferrante, RJ; Siwek, DF; Wilcox, HM; Flood, DG; Beal, MF; Brown, RH, Jr; et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet 1996, 13, 43–47. [Google Scholar]
- Furukawa, Y; Fu, R; Deng, HX; Siddique, T; O’halloran, TV. From the Cover: Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc. Natl. Acad. Sci. USA 2005, 280, 7148–7153. [Google Scholar]
- Wang, J; Xu, G; Borchelt, DR. Mapping superoxide dismutase 1 domains of non-native interaction: roles of intra- and intermolecular disulfide bonding in aggregation. J. Neurochem 2006, 96, 1277–1288. [Google Scholar]
- Watanabe, S; Nagano, S; Duce, J; Kiaei, M; Li, QX; Tucker, SM; Tiwari, A; Brown, RH, Jr; Beal, MF; Hayward, LJ; et al. Increased affinity for copper mediated by cysteine 111 in forms of mutant superoxide dismutase 1 linked to amyotrophic lateral sclerosis. Free Radic. Biol. Med 2007, 42, 1534–1542. [Google Scholar]
- Fujiwara, N; Nakano, M; Kato, S; Yoshihara, D; Ookawara, T; Eguchi, H; Taniguchi, N; Suzuki, K. Oxidative modification to cysteine sulfonic Acid of cys111 in human copper-zinc superoxide dismutase. J. Biol. Chem 2007, 282, 35933–35944. [Google Scholar]
- Tiwari, A; Hayward, LJ. Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J. Biol. Chem 2003, 278, 5984–5992. [Google Scholar]
- Rakhit, R; Crow, JP; Lepock, JR; Kondejewski, LH; Cashman, NR; Chakrabartty, A. Monomeric Cu, Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J. Biol. Chem 2002, 277, 15499–15504. [Google Scholar]
- Rakhit, R; Robertson, J; Vande Velde, C; Horne, P; Ruth, DM; Griffin, J; Cleveland, DW; Cashman, NR; Chakrabartty, A. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat. Med 2007, 13, 754–759. [Google Scholar]
- Urushitani, M; Kurisu, J; Tateno, M; Hatakeyama, S; Nakayama, K; Kato, S; Takahashi, R. CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J. Neurochem 2004, 90, 231–244. [Google Scholar]
- Urushitani, M; Kurisu, J; Tsukita, K; Takahashi, R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem 2002, 83, 1030–1042. [Google Scholar]
- Urushitani, M; Ezzi, SA; Matsuo, A; Tooyama, I; Julien, JP. The endoplasmic reticulum-Golgi pathway is a target for translocation and aggregation of mutant superoxide dismutase linked to ALS. FASEB J 2008, 22, 2476–2487. [Google Scholar]
- Okado-Matsumoto, A; Fridovich, I. Amyotrophic lateral sclerosis: a proposed mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 9010–9014. [Google Scholar]
- Shaw, BF; Valentine, JS. How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem. Sci 2007, 32, 78–85. [Google Scholar]
- Urushitani, M; Nakamizo, T; Inoue, R; Sawada, H; Kihara, T; Honda, K; Akaike, A; Shimohama, S. N-methyl-d-aspartate receptor-mediated mitochondrial Ca(2+) overload in acute excitotoxic motor neuron death: A mechanism distinct from chronic neurotoxicity after Ca(2+) influx. J. Neurosci. Res 2001, 63, 377–387. [Google Scholar]
- Wong, PC; Pardo, CA; Borchelt, DR; Lee, MK; Copeland, NG; Jenkins, NA; Sisodia, SS; Cleveland, DW; Price, DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar]
- Higgins, CM; Jung, C; Ding, H; Xu, Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci 2002, 22, RC215. [Google Scholar]
- Takeuchi, H; Kobayashi, Y; Ishigaki, S; Doyu, M; Sobue, G. Mitochondrial localization of mutant superoxide dismutase 1 triggers caspase-dependent cell death in a cellular model of familial amyotrophic lateral sclerosis. J. Biol. Chem 2002, 277, 50966–50972. [Google Scholar]
- Inoue, H; Tsukita, K; Iwasato, T; Suzuki, Y; Tomioka, M; Tateno, M; Nagao, M; Kawata, A; Saido, TC; Miura, M; et al. The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J 2003, 22, 6665–6674. [Google Scholar]
- Reyes, NA; Fisher, JK; Austgen, K; VandenBerg, S; Huang, EJ; Oakes, SA. Blocking the mitochondrial apoptotic pathway preserves motor neuron viability and function in a mouse model of amyotrophic lateral sclerosis. J. Clin. Invest 2010, 120, 3673–3679. [Google Scholar]
- Vande Velde, C; McDonald, KK; Boukhedimi, Y; McAlonis-Downes, M; Lobsiger, CS; Bel Hadj, S; Zandona, A; Julien, JP; Shah, SB; Cleveland, DW. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS One 2011, 6, e22031. [Google Scholar]
- Israelson, A; Arbel, N; Da Cruz, S; Ilieva, H; Yamanaka, K; Shoshan-Barmatz, V; Cleveland, DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar]
- Misawa, H; Nakata, K; Matsuura, J; Moriwaki, Y; Kawashima, K; Shimizu, T; Shirasawa, T; Takahashi, R. Conditional knockout of Mn superoxide dismutase in postnatal motor neurons reveals resistance to mitochondrial generated superoxide radicals. Neurobiol. Dis 2006, 23, 169–177. [Google Scholar]
- Zhu, YB; Sheng, ZH. Increased axonal mitochondrial mobility does not slow amyotrophic lateral sclerosis (ALS)-like disease in mutant SOD1 mice. J. Biol. Chem 2011, 286, 23432–23440. [Google Scholar]
- Chang, LY; Slot, JW; Geuze, HJ; Crapo, JD. Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J. Cell Biol 1988, 107, 2169–2179. [Google Scholar]
- Saito, T; Shinzawa, H; Togashi, H; Wakabayashi, H; Ukai, K; Takahashi, T; Ishikawa, M; Dobashi, M; Imai, Y. Ultrastructural localization of Cu, Zn-SOD in hepatocytes of patients with various liver diseases. Histol. Histopathol 1989, 4, 1–6. [Google Scholar]
- Tobisawa, S; Hozumi, Y; Arawaka, S; Koyama, S; Wada, M; Nagai, M; Aoki, M; Itoyama, Y; Goto, K; Kato, T. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem. Biophys. Res. Commun 2003, 303, 496–503. [Google Scholar]
- Urushitani, M; Sik, A; Sakurai, T; Nukina, N; Takahashi, R; Julien, JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat. Neurosci 2006, 9, 108–118. [Google Scholar]
- Kikuchi, H; Almer, G; Yamashita, S; Guegan, C; Nagai, M; Xu, Z; Sosunov, AA; McKhann, GM, II; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci USA 2006, 103, 6025–6030. [Google Scholar]
- Mori, A; Yamashita, S; Uchino, K; Suga, T; Ikeda, T; Takamatsu, K; Ishizaki, M; Koide, T; Kimura, E; Mita, S; et al. Derlin-1 overexpression ameliorates mutant SOD1-induced endoplasmic reticulum stress by reducing mutant SOD1 accumulation. Neurochem. Int 2011, 58, 344–353. [Google Scholar]
- Atkin, JD; Farg, MA; Walker, AK; McLean, C; Tomas, D; Horne, MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis 2008, 30, 400–407. [Google Scholar]
- Walker, AK; Atkin, JD. Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life 2011. In Press. [Google Scholar]
- Sasaki, S. Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol 2010, 69, 346–355. [Google Scholar]
- Ito, Y; Yamada, M; Tanaka, H; Aida, K; Tsuruma, K; Shimazawa, M; Hozumi, I; Inuzuka, T; Takahashi, H; Hara, H. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis 2009, 36, 470–476. [Google Scholar]
- Saxena, S; Cabuy, E; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci 2009, 12, 627–636. [Google Scholar]
- Nagata, T; Ilieva, H; Murakami, T; Shiote, M; Narai, H; Ohta, Y; Hayashi, T; Shoji, M; Abe, K. Increased ER stress during motor neuron degeneration in a transgenic mouse model of amyotrophic lateral sclerosis. Neurol. Res 2007, 29, 767–771. [Google Scholar]
- Kieran, D; Woods, I; Villunger, A; Strasser, A; Prehn, JH. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc. Natl. Acad. Sci. USA 2007, 104, 20606–20611. [Google Scholar]
- Nishitoh, H; Kadowaki, H; Nagai, A; Maruyama, T; Yokota, T; Fukutomi, H; Noguchi, T; Matsuzawa, A; Takeda, K; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev 2008, 22, 1451–1464. [Google Scholar]
- Atkin, JD; Farg, MA; Turner, BJ; Tomas, D; Lysaght, JA; Nunan, J; Rembach, A; Nagley, P; Beart, PM; Cheema, SS; et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem 2006, 281, 30152–30165. [Google Scholar]
- Turner, BJ; Atkin, JD. ER stress and UPR in familial amyotrophic lateral sclerosis. Curr. Mol. Med 2006, 6, 79–86. [Google Scholar]
- Mourelatos, Z; Adler, H; Hirano, A; Donnenfeld, H; Gonatas, JO; Gonatas, NK. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis revealed by organelle-specific antibodies. Proc. Natl. Acad. Sci. USA 1990, 87, 4393–4395. [Google Scholar]
- Lafon-Cazal, M; Adjali, O; Galéotti, N; Poncet, J; Jouin, P; Homburger, V; Bockaert, J; Marin, P. Proteomic analysis of astrocytic secretion in the mouse. Comparison with the cerebrospinal fluid proteome. J. Biol. Chem 2003, 278, 24438–24448. [Google Scholar]
- Turner, BJ; Atkin, JD; Farg, MA; Zang da, W; Rembach, A; Lopes, EC; Patch, JD; Hill, AF; Cheema, SS. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J. Neurosci 2005, 25, 108–117. [Google Scholar]
- Durham, HD; Roy, J; Dong, L; Figlewicz, DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J. Neuropathol. Exp. Neurol 1997, 56, 523–530. [Google Scholar]
- Matsumoto, G; Stojanovic, A; Holmberg, CI; Kim, S; Morimoto, RI. Structural properties and neuronal toxicity of amyotrophic lateral sclerosis-associated Cu/Zn superoxide dismutase 1 aggregates. J. Cell Biol 2005, 171, 75–85. [Google Scholar]
- Pramatarova, A; Laganière, J; Roussel, J; Brisebois, K; Rouleau, GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci 2001, 21, 3369–3374. [Google Scholar]
- Wang, LJ; Lu, YY; Muramatsu, S; Ikeguchi, K; Fujimoto, K; Okada, T; Mizukami, H; Matsushita, T; Hanazono, Y; Kume, A; et al. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J. Neurosci 2002, 22, 6920–6928. [Google Scholar]
- Gong, YH; Parsadanian, AS; Andreeva, A; Snider, WD; Elliott, JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J. Neurosci 2000, 20, 660–665. [Google Scholar]
- Clement, AM; Nguyen, MD; Roberts, EA; Garcia, ML; Boillée, S; Rule, M; McMahon, AP; Doucette, W; Siwek, D; Ferrante, RJ; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar]
- Yamanaka, K; Chun, SJ; Boillee, S; Fujimori-Tonou, N; Yamashita, H; Gutmann, DH; Takahashi, R; Misawa, H; Cleveland, DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci 2008, 11, 251–253. [Google Scholar]
- Wang, L; Gutmann, DH; Roos, RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet 2011, 20, 286–293. [Google Scholar]
- Wang, L; Sharma, K; Deng, HX; Siddique, T; Grisotti, G; Liu, E; Roos, RP. Restricted expression of mutant SOD1 in spinal motor neurons and interneurons induces motor neuron pathology. Neurobiol. Dis 2008, 29, 400–408. [Google Scholar]
- Nagai, M; Re, DB; Nagata, T; Chalazonitis, A; Jessell, TM; Wichterle, H; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci 2007, 10, 615–622. [Google Scholar]
- Di Giorgio, FP; Carrasco, MA; Siao, MC; Maniatis, T; Eggan, K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci 2007, 10, 608–614. [Google Scholar]
- Di Giorgio, FP; Boulting, GL; Bobrowicz, S; Eggan, KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 2008, 3, 637–648. [Google Scholar]
- Zhao, W; Xie, W; Xiao, Q; Beers, DR; Appel, SH. Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J. Neurochem 2006, 99, 1176–1187. [Google Scholar]
- Zhao, W; Beers, DR; Henkel, JS; Zhang, W; Urushitani, M; Julien, JP; Appel, SH. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010, 58, 231–243. [Google Scholar]
- Ezzi, SA; Larivière, R; Urushitani, M; Julien, JP. Neuronal over-expression of chromogranin A accelerates disease onset in a mouse model of ALS. J. Neurochem 2010, 115, 1102–1111. [Google Scholar]
- Liu, HN; Sanelli, T; Horne, P; Pioro, EP; Strong, MJ; Rogaeva, E; Bilbao, J; Zinman, L; Robertson, J. Lack of evidence of monomer/misfolded superoxide dismutase-1 in sporadic amyotrophic lateral sclerosis. Ann. Neurol 2009, 66, 75–80. [Google Scholar]
- Zetterström, P; Andersen, PM; Brännström, T; Marklund, SL. Misfolded superoxide dismutase-1 in CSF from amyotrophic lateral sclerosis patients. J. Neurochem 2011, 117, 91–99. [Google Scholar]
- Ezzi, SA; Urushitani, M; Julien, JP. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem 2007, 102, 170–178. [Google Scholar]
- Gruzman, A; Wood, WL; Alpert, E; Prasad, MD; Miller, RG; Rothstein, JD; Bowser, R; Hamilton, R; Wood, TD; Cleveland, DW; et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12524–12529. [Google Scholar]
- Bosco, DA; Morfini, G; Karabacak, NM; Song, Y; Gros-Louis, F; Pasinelli, P; Goolsby, H; Fontaine, BA; Lemay, N; McKenna-Yasek, D; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci 2010, 13, 1396–1403. [Google Scholar]
- Haidet-Phillips, AM; Hester, ME; Miranda, CJ; Meyer, K; Braun, L; Frakes, A; Song, S; Likhite, S; Murtha, MJ; Foust, KD; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol 2011, 29, 824–828. [Google Scholar]
- Arai, T; Hasegawa, M; Akiyama, H; Ikeda, K; Nonaka, T; Mori, H; Mann, D; Tsuchiya, K; Yoshida, M; Hashizume, Y; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun 2006, 351, 602–611. [Google Scholar]
- Neumann, M; Sampathu, DM; Kwong, LK; Truax, AC; Micsenyi, MC; Chou, TT; Bruce, J; Schuck, T; Grossman, M; Clark, CM; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar]
- Borroni, B; Bonvicini, C; Alberici, A; Buratti, E; Agosti, C; Archetti, S; Papetti, A; Stuani, C; Di Luca, M; Gennarelli, M; et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat 2009, 30, E974–E983. [Google Scholar]
- Benajiba, L; Le Ber, I; Camuzat, A; Lacoste, M; Thomas-Anterion, C; Couratier, P; Legallic, S; Salachas, F; Hannequin, D; Decousus, M; et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol 2009, 65, 470–473. [Google Scholar]
- Quadri, M; Cossu, G; Saddi, V; Simons, EJ; Murgia, D; Melis, M; Ticca, A; Oostra, BA; Bonifati, V. Broadening the phenotype of TARDBP mutations: The TARDBP Ala382Thr mutation and Parkinson’s disease in Sardinia. Neurogenetics 2011, 12, 203–209. [Google Scholar]
- Buratti, E; Dork, T; Zuccato, E; Pagani, F; Romano, M; Baralle, FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 2001, 20, 1774–1784. [Google Scholar]
- Strong, MJ; Volkening, K; Hammond, R; Yang, W; Strong, W; Leystra-Lantz, C; Shoesmith, C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci 2007, 35, 320–327. [Google Scholar]
- Rogelj, B; Briese, M; Cereda, M; Kayikci, M; König, J; Hortobágyi, T; Nishimura, AL; Zupunski, V; Patani, R; Chandran, S; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci 2011, 14, 452–458. [Google Scholar]
- Polymenidou, M; Lagier-Tourenne, C; Hutt, KR; Huelga, SC; Moran, J; Liang, TY; Ling, SC; Sun, E; Wancewicz, E; Mazur, C; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci 2011, 14, 459–468. [Google Scholar]
- Xiao, S; Sanelli, T; Dib, S; Sheps, D; Findlater, J; Bilbao, J; Keith, J; Zinman, L; Rogaeva, E; Robertson, J. RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol. Cell. Neurosci 2011, 47, 167–180. [Google Scholar]
- Ayala, YM; Misteli, T; Baralle, FE. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc. Natl. Acad. Sci. USA 2008, 105, 3785–3789. [Google Scholar]
- Fiesel, FC; Voigt, A; Weber, SS; Van den Haute, C; Waldenmaier, A; Görner, K; Walter, M; Anderson, ML; Kern, JV; Rasse, TM; et al. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J 2010, 29, 209–221. [Google Scholar]
- Iguchi, Y; Katsuno, M; Niwa, J; Yamada, S; Sone, J; Waza, M; Adachi, H; Tanaka, F; Nagata, K; Arimura, N. TDP-43 depletion induces neuronal cell damage through dysregulation of Rho family GTPases. J. Biol. Chem 2009, 284, 22059–22066. [Google Scholar]
- Anthony, K; Gallo, JM. Aberrant RNA processing events in neurological disorders. Brain Res 2010, 1338, 67–77. [Google Scholar]
- Urushitani, M; Sato, T; Bamba, H; Hisa, Y; Tooyama, I. Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. J. Neurosci. Res 2010, 88, 784–797. [Google Scholar]
- Mackenzie, IR; Bigio, EH; Ince, PG; Geser, F; Neumann, M; Cairns, NJ; Kwong, LK; Forman, MS; Ravits, J; Stewart, H; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol 2007, 61, 427–434. [Google Scholar]
- Sumi, H; Kato, S; Mochimaru, Y; Fujimura, H; Etoh, M; Sakoda, S. Nuclear TAR DNA binding protein 43 expression in spinal cord neurons correlates with the clinical course in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol 2009, 68, 37–47. [Google Scholar]
- Okamoto, Y; Ihara, M; Urushitani, M; Yamashita, H; Kondo, T; Tanigaki, A; Oono, M; Komatsu, K; Kawamata, J; Ikemoto, A; et al. An autopsy case of SOD1-related ALS with TDP-43 positive inclusions. Neurology 2011, in press. [Google Scholar]
- Wider, C; Dickson, DW; Stoessl, AJ; Tsuboi, Y; Chapon, F; Gutmann, L; Lechevalier, B; Calne, DB; Personett, DA; Hulihan, M; et al. Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat. Disord 2009, 15, 281–286. [Google Scholar]
- Nakashima-Yasuda, H; Uryu, K; Robinson, J; Xie, SX; Hurtig, H; Duda, JE; Arnold, SE; Siderowf, A; Grossman, M; Leverenz, JB; et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. (Berl. ) 2007, 114, 221–229. [Google Scholar]
- Schwab, C; Arai, T; Hasegawa, M; Yu, S; McGeer, PL. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol 2008, 67, 1159–1165. [Google Scholar]
- Weihl, CC; Temiz, P; Miller, SE; Watts, G; Smith, C; Forman, M; Hanson, PI; Kimonis, V; Pestronk, A. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1186–1189. [Google Scholar]
- Sato, T; Takeuchi, S; Saito, A; Ding, W; Bamba, H; Matsuura, H; Hisa, Y; Tooyama, I; Urushitani, M. Axonal ligation induces transient redistribution of TDP-43 in brainstem motor neurons. Neuroscience 2009, 164, 1565–1578. [Google Scholar]
- Moisse, K; Volkening, K; Leystra-Lantz, C; Welch, I; Hill, T; Strong, MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: Implications for TDP-43 in the physiological response to neuronal injury. Brain Res 2009, 1249, 202–211. [Google Scholar]
- Nishimura, AL; Zupunski, V; Troakes, C; Kathe, C; Fratta, P; Howell, M; Gallo, JM; Hortobágyi, T; Shaw, CE; Rogelj, B. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 2010, 133, 1763–1771. [Google Scholar]
- Winton, MJ; Igaz, LM; Wong, MM; Kwong, LK; Trojanowski, JQ; Lee, VM. Disturbance of nuclear and cytoplasmic Tar DNA binding protein (TDP-43) induces disease-like redistribution, sequestration and aggregate formation. J. Biol. Chem 2008, 283, 13302–13309. [Google Scholar]
- Zhang, YJ; Xu, YF; Dickey, CA; Buratti, E; Baralle, F; Bailey, R; Pickering-Brown, S; Dickson, D; Petrucelli, L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci 2007, 27, 10530–10534. [Google Scholar]
- Nishimoto, Y; Ito, D; Yagi, T; Nihei, Y; Tsunoda, Y; Suzuki, N. Characterization of alternative isoforms and inclusion body of the TAR DNA-binding protein-43. J. Biol. Chem 2010, 285, 608–619. [Google Scholar]
- Igaz, LM; Kwong, LK; Xu, Y; Truax, AC; Uryu, K; Neumann, M; Clark, CM; Elman, LB; Miller, BL; Grossman, M; et al. Enrichment of C-Terminal fragments in TAR DNA-Binding Protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol 2008, 173, 182–194. [Google Scholar]
- Nonaka, T; Kametani, F; Arai, T; Akiyama, H; Hasegawa, M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet 2009, 18, 3353–3364. [Google Scholar]
- Johnson, BS; Snead, D; Lee, JJ; McCaffery, JM; Shorter, J; Gitler, AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem 2009, 284, 20329–20339. [Google Scholar]
- Furukawa, Y; Kaneko, K; Watanabe, S; Yamanaka, K; Nukina, N. A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem 2011, 286, 18664–18672. [Google Scholar]
- Yang, C; Tan, W; Whittle, C; Qiu, L; Cao, L; Akbarian, S; Xu, Z. The C-Terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS One 2010, 5, e15878. [Google Scholar]
- Pesiridis, GS; Tripathy, K; Tanik, S; Trojanowski, JQ; Lee, VM. A “two-hit” hypothesis for inclusion formation by carboxyl-terminal fragments of TDP-43 protein linked to RNA depletion and impaired microtubule-dependent transport. J. Biol. Chem 2011, 286, 18845–18855. [Google Scholar]
- Shiina, Y; Arima, K; Tabunoki, H; Satoh, J. TDP-43 dimerizes in human cells in culture. Cell. Mol. Neurobiol 2010, 30, 641–652. [Google Scholar]
- Johnson, BS; McCaffery, JM; Lindquist, S; Gitler, AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar]
- Igaz, LM; Kwong, LK; Lee, EB; Chen-Plotkin, A; Swanson, E; Unger, T; Malunda, J; Xu, Y; Winton, MJ; Trojanowski, JQ; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest 2011, 121, 726–738. [Google Scholar]
- Ash, PE; Zhang, YJ; Roberts, CM; Saldi, T; Hutter, H; Buratti, E; Petrucelli, L; Link, CD. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet 2010, 19, 3206–3218. [Google Scholar]
- Wegorzewska, I; Bell, S; Cairns, NJ; Miller, TM; Baloh, RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar]
- Xu, YF; Gendron, TF; Zhang, YJ; Lin, WL; D’Alton, S; Sheng, H; Casey, MC; Tong, J; Knight, J; Yu, X; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci 2010, 30, 10851–10859. [Google Scholar]
- University of Tokyo. ALS Mutation Database. 2011. Available online: https://reseq.lifesciencedb.jp/resequence/GeneDetail.do?targetId=1&geneId=EG23435 accessed on 12 October 2011.
- Kabashi, E; Valdmanis, PN; Dion, P; Spiegelman, D; McConkey, BJ; Velde, CV; Bouchard, JP; Lacomblez, L; Pochigaeva, K; Salachas, F; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet 2008, 40, 572–574. [Google Scholar]
- Kabashi, E; Lin, L; Tradewell, ML; Dion, PA; Bercier, V; Bourgouin, P; Rochefort, D; Bel Hadj, S; Durham, HD; Vande Velde, C; et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet 2010, 19, 671–683. [Google Scholar]
- Sreedharan, J; Blair, IP; Tripathi, VB; Hu, X; Vance, C; Rogelj, B; Ackerley, S; Durnall, JC; Williams, KL; Buratti, E; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar]
- Rutherford, NJ; Zhang, YJ; Baker, M; Gass, JM; Finch, NA; Xu, YF; Stewart, H; Kelley, BJ; Kuntz, K; Crook, RJ; et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 2008, 4, e1000193. [Google Scholar]
- Liachko, NF; Guthrie, CR; Kraemer, BC. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci 2010, 30, 16208–16219. [Google Scholar]
- Zhou, H; Huang, C; Chen, H; Wang, D; Landel, CP; Xia, PY; Bowser, R; Liu, YJ; Xia, XG. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet 2010, 6, e1000887. [Google Scholar]
- Stallings, NR; Puttaparthi, K; Luther, CM; Burns, DK; Elliott, JL. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol. Dis 2010, 40, 404–414. [Google Scholar]
- Swarup, V; Phaneuf, D; Bareil, C; Robertson, J; Rouleau, GA; Kriz, J; Julien, JP. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 2011. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L; Bilgutay, A; Zhang, YJ; Vanderwyde, T; Citro, A; Mehta, T; Zaarur, N; McKee, A; Bowser, R; Sherman, M; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS One 2010, 5, e13250. [Google Scholar]
- Guerreiro, RJ; Schymick, JC; Crews, C; Singleton, A; Hardy, J; Traynor, BJ. TDP-43 is not a common cause of sporadic amyotrophic lateral sclerosis. PLoS One 2008, 3, e2450. [Google Scholar]
- Gijselinck, I; Sleegers, K; Engelborghs, S; Robberecht, W; Martin, JJ; Vandenberghe, R; Sciot, R; Dermaut, B; Goossens, D; van der Zee, J; et al. Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol. Aging 2009, 30, 1329–1331. [Google Scholar]
- Corrado, L; Ratti, A; Gellera, C; Buratti, E; Castellotti, B; Carlomagno, Y; Ticozzi, N; Mazzini, L; Testa, L; Taroni, F; et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum. Mutat 2009, 30, 688–694. [Google Scholar]
- Lagier-Tourenne, C; Cleveland, DW. Rethinking ALS: the FUS about TDP-43. Cell 2009, 136, 1001–1004. [Google Scholar]
- Van Deerlin, VM; Leverenz, JB; Bekris, LM; Bird, TD; Yuan, W; Elman, LB; Clay, D; Wood, EM; Chen-Plotkin, AS; Martinez-Lage, M; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet. Neurol 2008, 7, 409–416. [Google Scholar]
- Aguzzi, A; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar]
- Frost, B; Diamond, MI. Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci 2010, 11, 155–159. [Google Scholar]
- Frost, B; Ollesch, J; Wille, H; Diamond, MI. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J. Biol. Chem 2009, 284, 3546–3551. [Google Scholar]
- Desplats, P; Lee, HJ; Bae, EJ; Patrick, C; Rockenstein, E; Crews, L; Spencer, B; Masliah, E; Lee, SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar]
- Ren, PH; Lauckner, JE; Kachirskaia, I; Heuser, JE; Melki, R; Kopito, RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol 2009, 11, 219–225. [Google Scholar]
- Münch, C; O’Brien, J; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar]
- Chia, R; Tattum, MH; Jones, S; Collinge, J; Fisher, EM; Jackson, GS. Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS One 2010, 5, e10627. [Google Scholar]
- Grad, LI; Guest, WC; Yanai, A; Pokrishevsky, E; O’Neill, MA; Gibbs, E; Semenchenko, V; Yousefi, M; Wishart, DS; Plotkin, SS; et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16398–16403. [Google Scholar]
- Urushitani, M; Ezzi, SA; Julien, JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 2495–2500. [Google Scholar]
- Takeuchi, S; Fujiwara, N; Ido, A; Oono, M; Takeuchi, Y; Tateno, M; Suzuki, K; Takahashi, R; Tooyama, I; Taniguchi, N; et al. Induction of protective immunity by vaccination with wild-type apo superoxide dismutase 1 in mutant SOD1 transgenic mice. J. Neuropathol. Exp. Neurol 2010, 69, 1044–1056. [Google Scholar]
- Gros-Louis, F; Soucy, G; Larivière, R; Julien, JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J. Neurochem 2010, 113, 1188–1199. [Google Scholar]
- Che, MX; Jiang, YJ; Xie, YY; Jiang, LL; Hu, HY. Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J 2011, 25, 2344–2353. [Google Scholar]
- Noto, Y; Shibuya, K; Sato, Y; Kanai, K; Misawa, S; Sawai, S; Mori, M; Uchiyama, T; Isose, S; Nasu, S; et al. Elevated CSF TDP-43 levels in amyotrophic lateral sclerosis: Specificity, sensitivity, and a possible prognostic value. Amyotroph. Lateral Scler 2011, 12, 140–143. [Google Scholar]
- Steinacker, P; Hendrich, C; Sperfeld, AD; Jesse, S; von Arnim, CA; Lehnert, S; Pabst, A; Uttner, I; Tumani, H; Lee, VM; et al. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch. Neurol 2008, 65, 1481–1487. [Google Scholar]
- Kasai, T; Tokuda, T; Ishigami, N; Sasayama, H; Foulds, P; Mitchell, DJ; Mann, DM; Allsop, D; Nakagawa, M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. (Berl. ) 2009, 117, 55–62. [Google Scholar]


{kind=link}
{kind=link}
| Domain | Mutation | Cell death | Reference |
|---|---|---|---|
| N-terminus | D65E | [133] | |
| A66A | [133] | ||
| A90V | [126] | ||
| RRM1 | D169G | [124] | |
| RRM2 | Y214Y | [134] | |
| P225P | [133] | ||
| N267S | [135] | ||
| Glycine-rich | G287S | [136] | |
| G290A | [137] | ||
| G290S | [136] | ||
| G294A | [126] | ||
| G294V | [136] | ||
| G295R | [135] | ||
| G295S | [135] | ||
| G298S | [137] | ||
| M311V | [136] | ||
| C-terminus | A315A | [133] | |
| A315T | Tg mice, Cultured cells, Zebrafish | [124,131] | |
| Q331K | Cultured cells | [126] | |
| S332N | [135] | ||
| G335D | [136] | ||
| M337V | Tg rat, Cultured cells, Zebrafish, chick embryo | [125,126,129] | |
| Q343R | [136] | ||
| N345K | [136] | ||
| G348C | Tg mice, Cultured cells, Zebrafish | [124,131] | |
| N352N | [133] | ||
| N352S | [136] | ||
| [144] 61S | [124] | ||
| P363A | [136] | ||
| Y374X | [136] | ||
| S379C | [136] | ||
| S379P | [135] | ||
| A382P | [136] | ||
| A382T | [124] | ||
| I383V | [136] | ||
| N390D | [124] | ||
| N390S | [136] | ||
| S393L | [135] | ||
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ido, A.; Fukuyama, H.; Urushitani, M. Protein Misdirection Inside and Outside Motor Neurons in Amyotrophic Lateral Sclerosis (ALS): A Possible Clue for Therapeutic Strategies. Int. J. Mol. Sci. 2011, 12, 6980-7003. https://doi.org/10.3390/ijms12106980
Ido A, Fukuyama H, Urushitani M. Protein Misdirection Inside and Outside Motor Neurons in Amyotrophic Lateral Sclerosis (ALS): A Possible Clue for Therapeutic Strategies. International Journal of Molecular Sciences. 2011; 12(10):6980-7003. https://doi.org/10.3390/ijms12106980
Chicago/Turabian StyleIdo, Akemi, Hidenao Fukuyama, and Makoto Urushitani. 2011. "Protein Misdirection Inside and Outside Motor Neurons in Amyotrophic Lateral Sclerosis (ALS): A Possible Clue for Therapeutic Strategies" International Journal of Molecular Sciences 12, no. 10: 6980-7003. https://doi.org/10.3390/ijms12106980