Principal Component Analysis Coupled with Artificial Neural Networks—A Combined Technique Classifying Small Molecular Structures Using a Concatenated Spectral Database

Abstract
:1. Introduction
2. Experimental
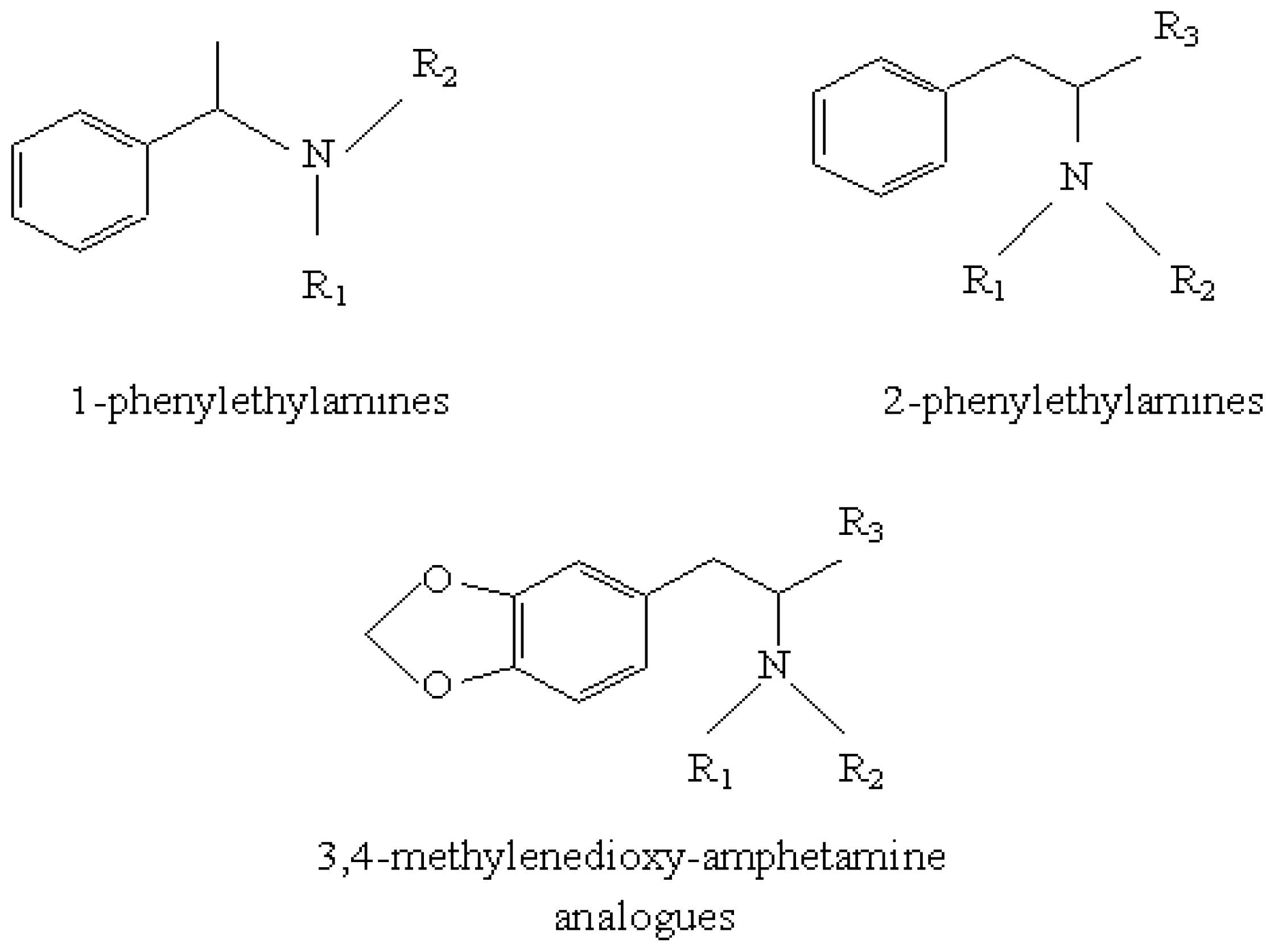
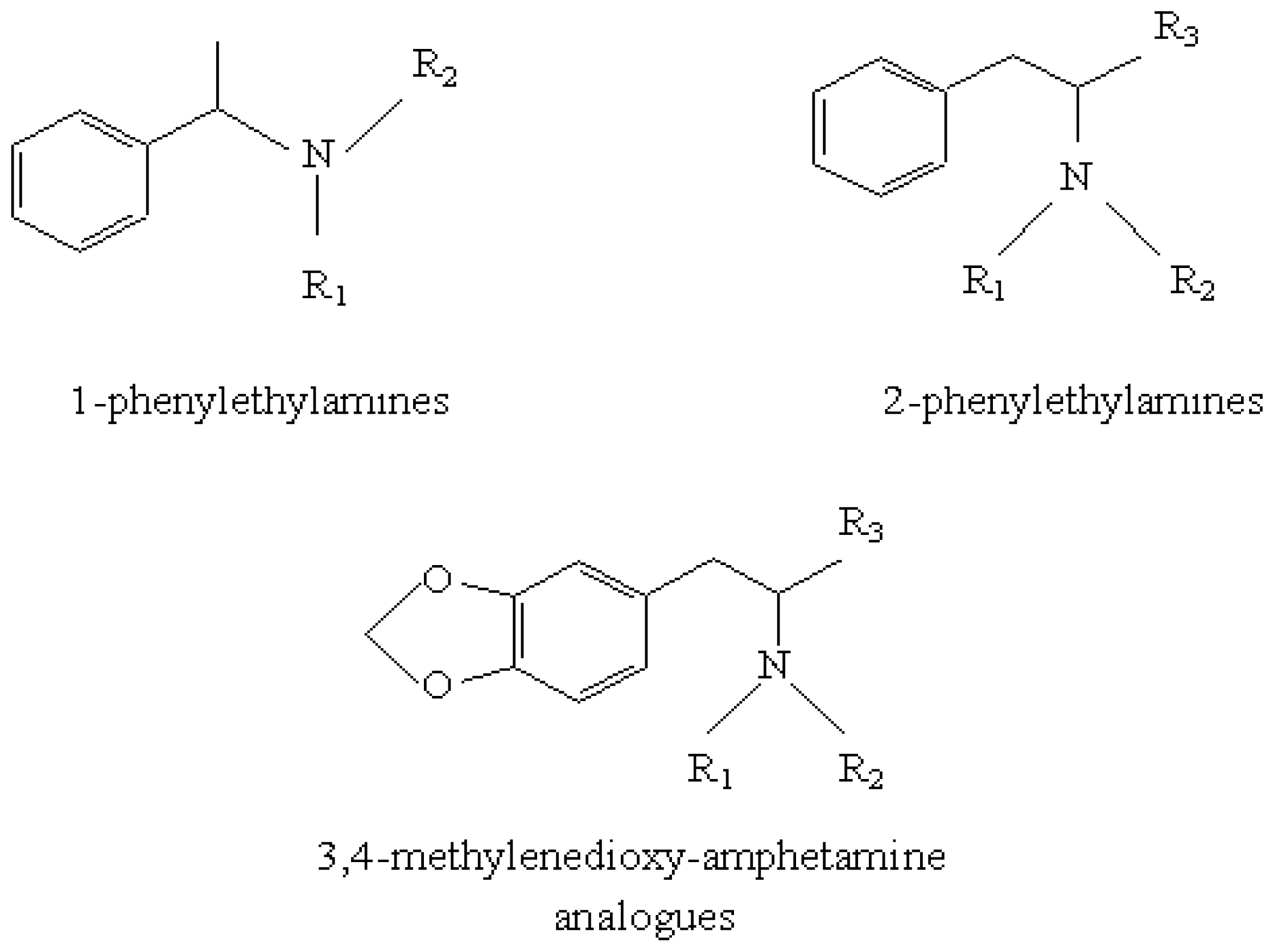
2.1. Input Database
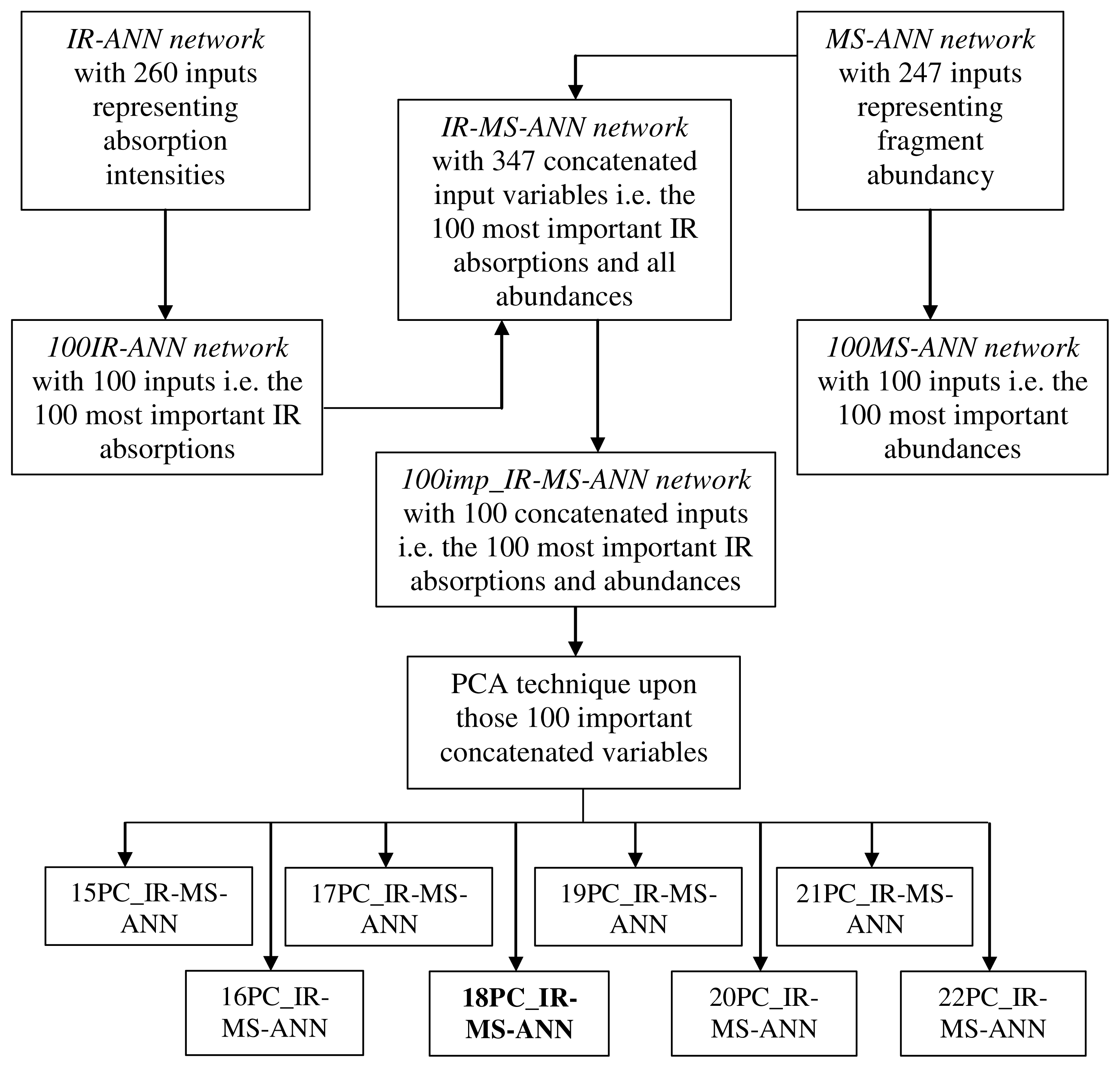
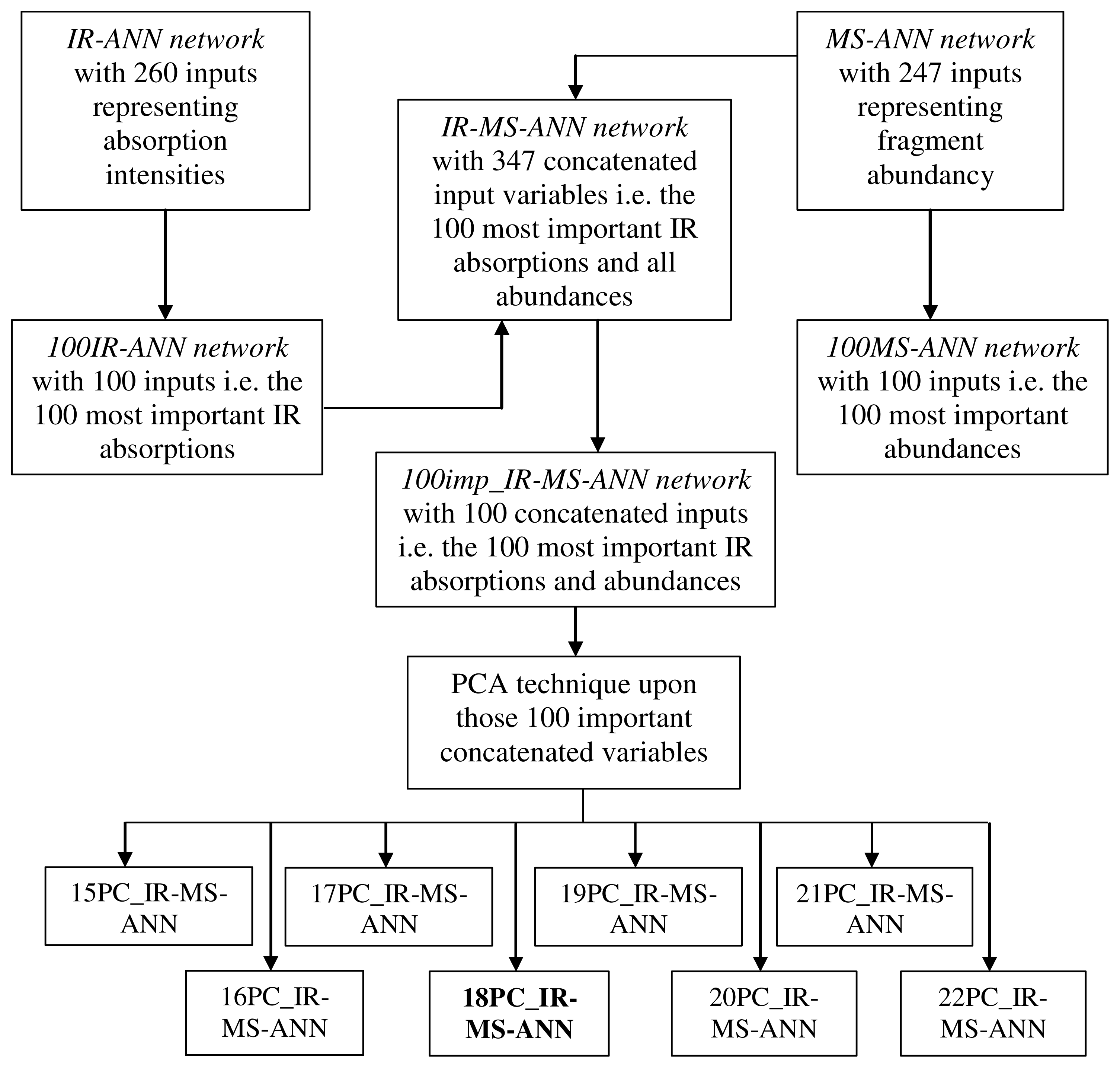
2.2. Development of the Hybrid ANN Systems Using GC-FTIR and GC-MS Data
2.3. Building the PC-ANN Systems Using GC-FTIR and GC-MS Data
3. Results and Discussion
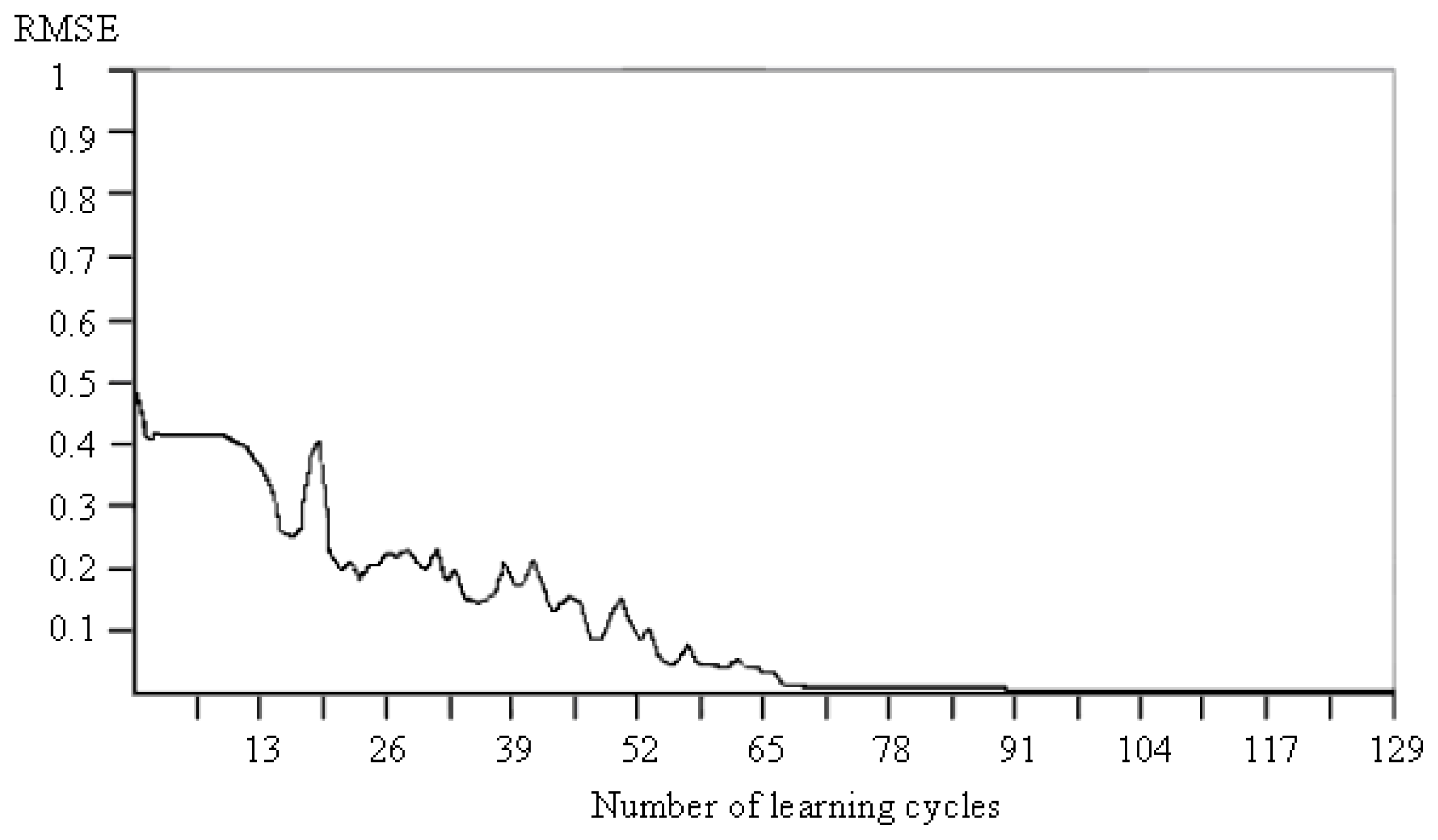
3.1. Analysis of the Validation Results of All Artificial Neural Networks
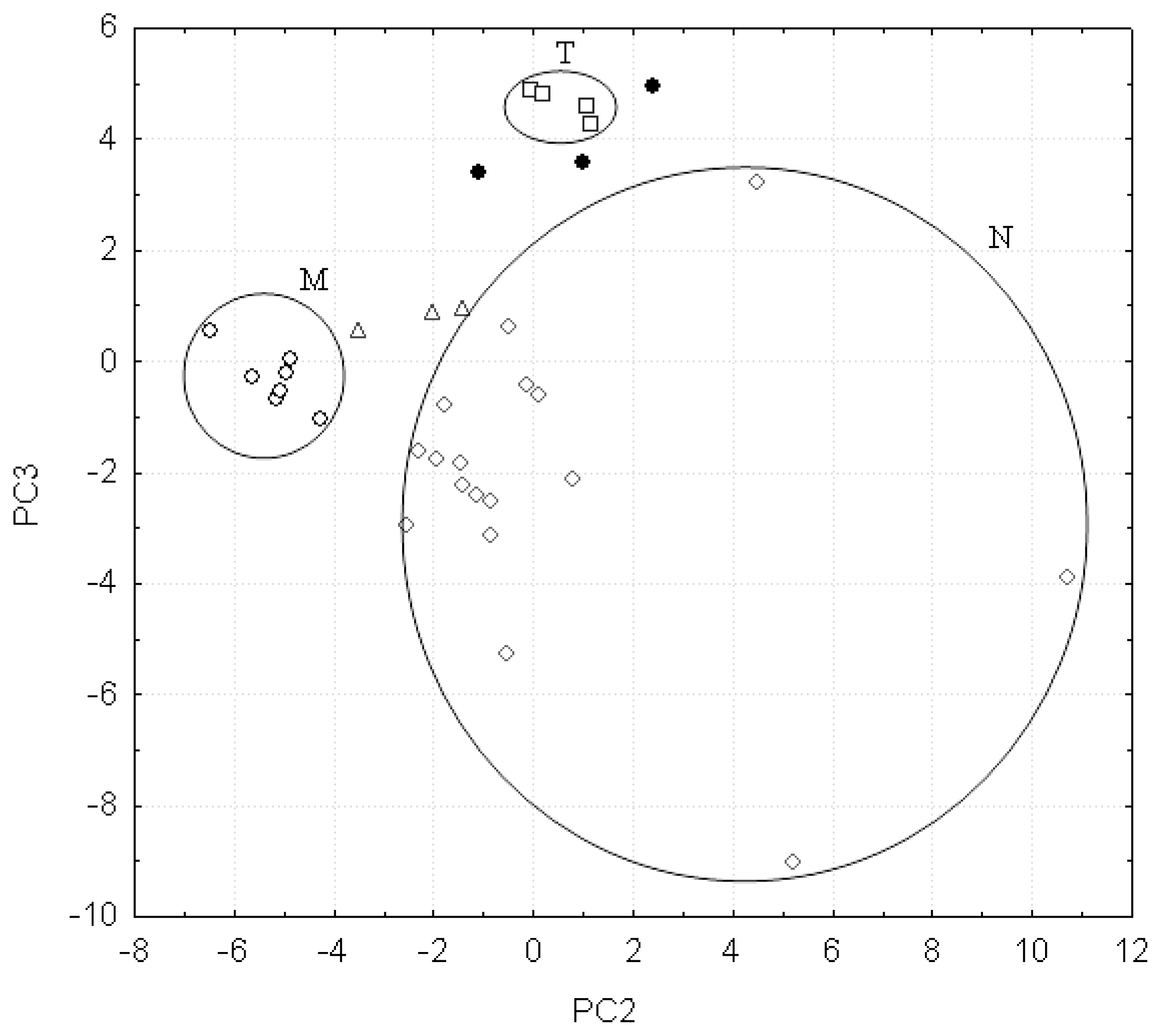
3.2. Discussion on the False Positives Obtained by 18PC_IR-MS-ANN
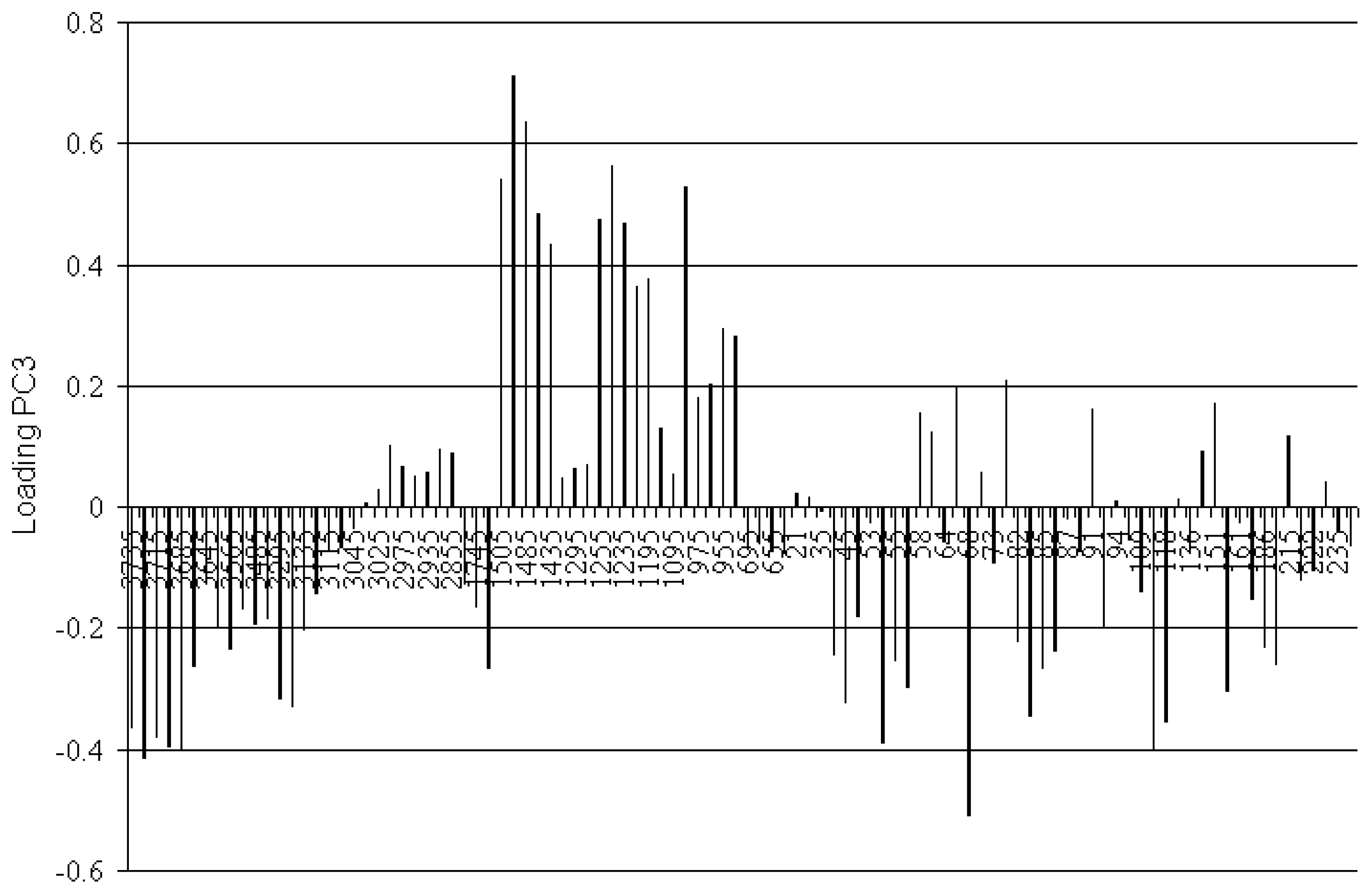
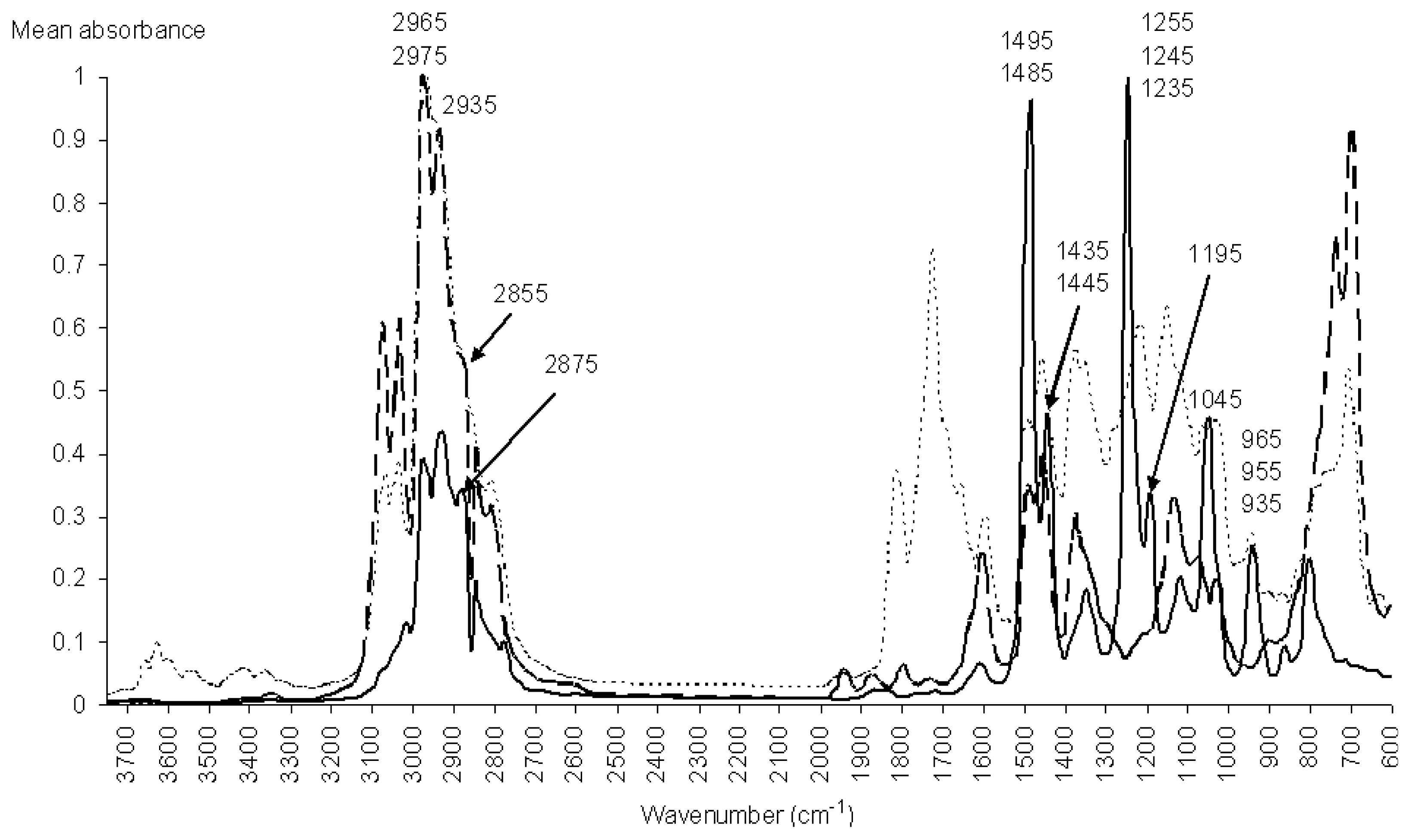
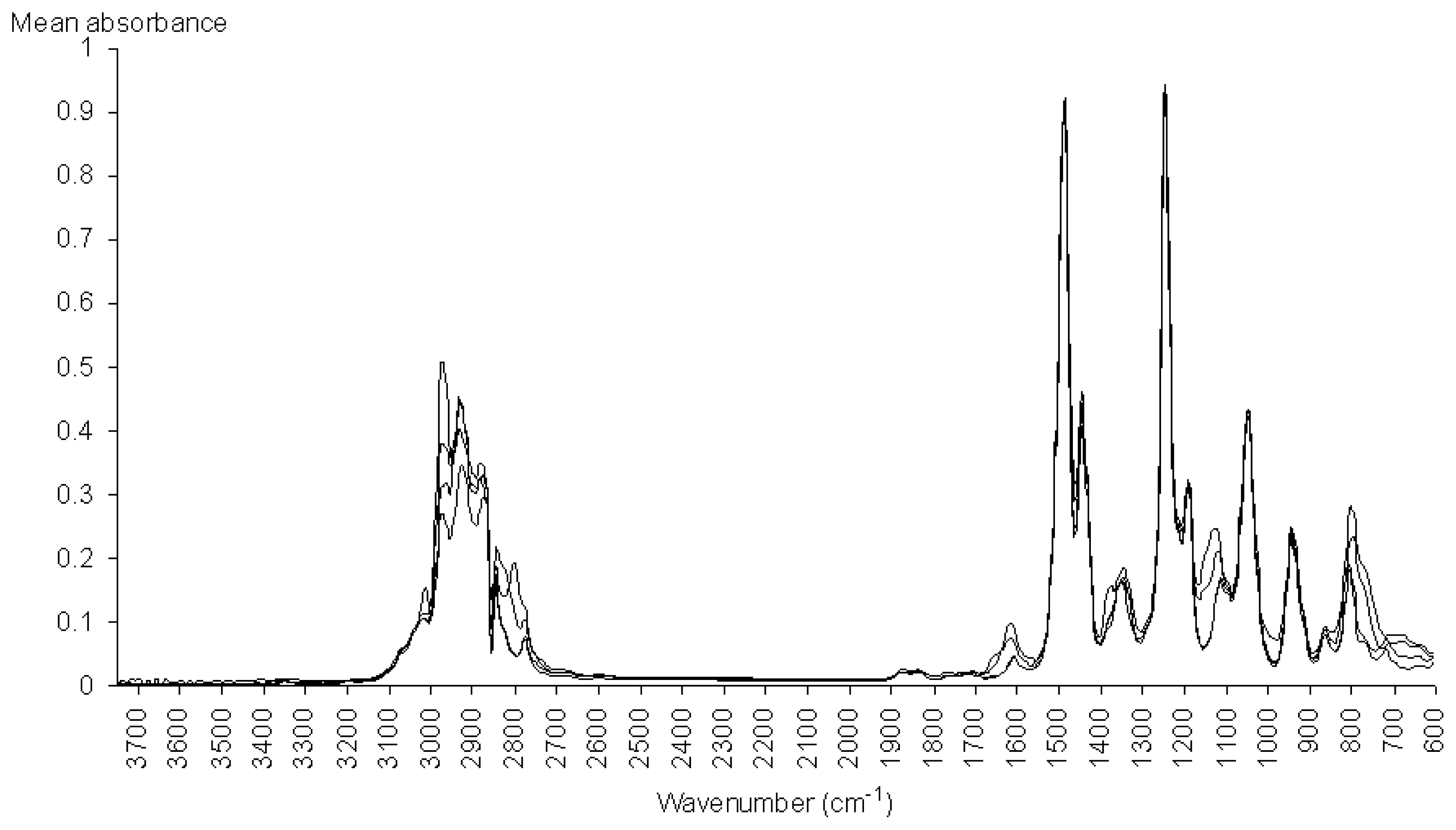
3.3. Spectroscopic Analysis of the Most Important IR Absorptions
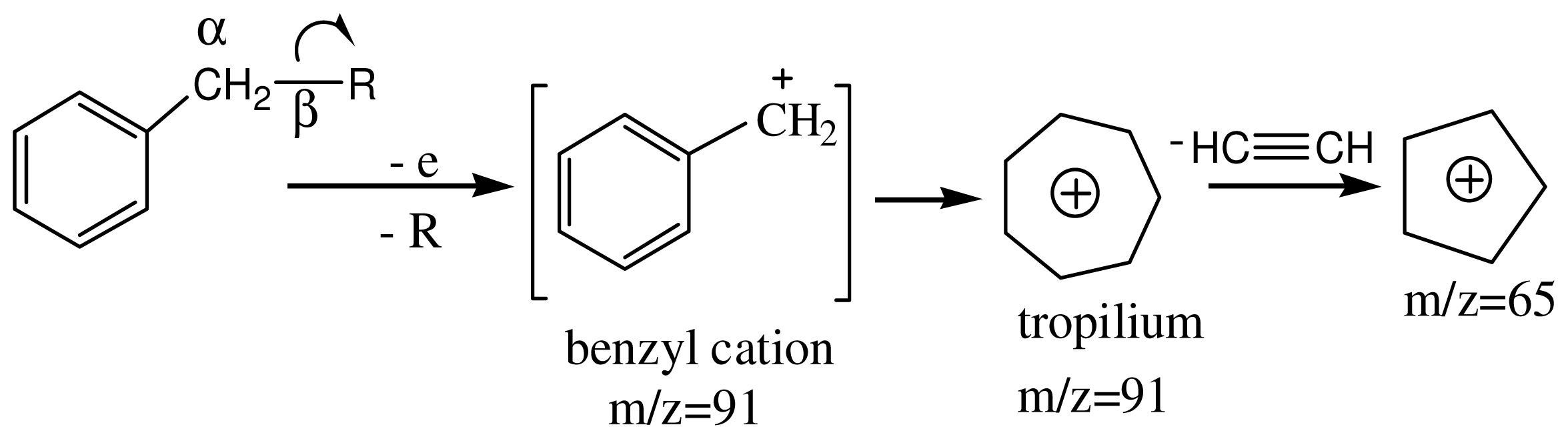
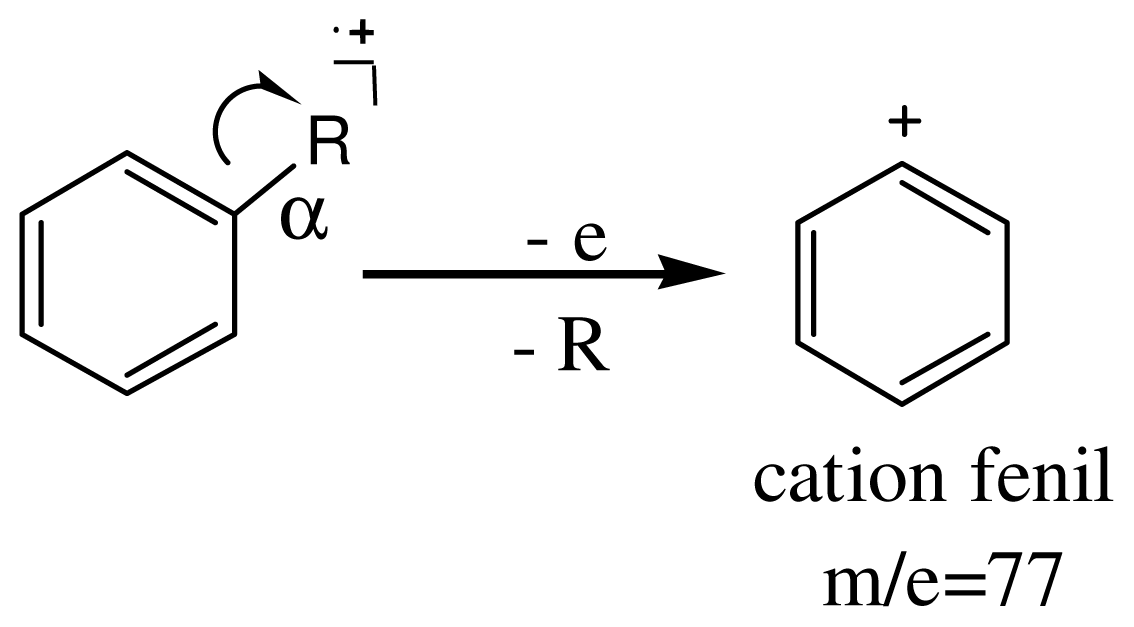
3.4. Spectroscopic Analysis of the Most Important m/z Fragment Ions
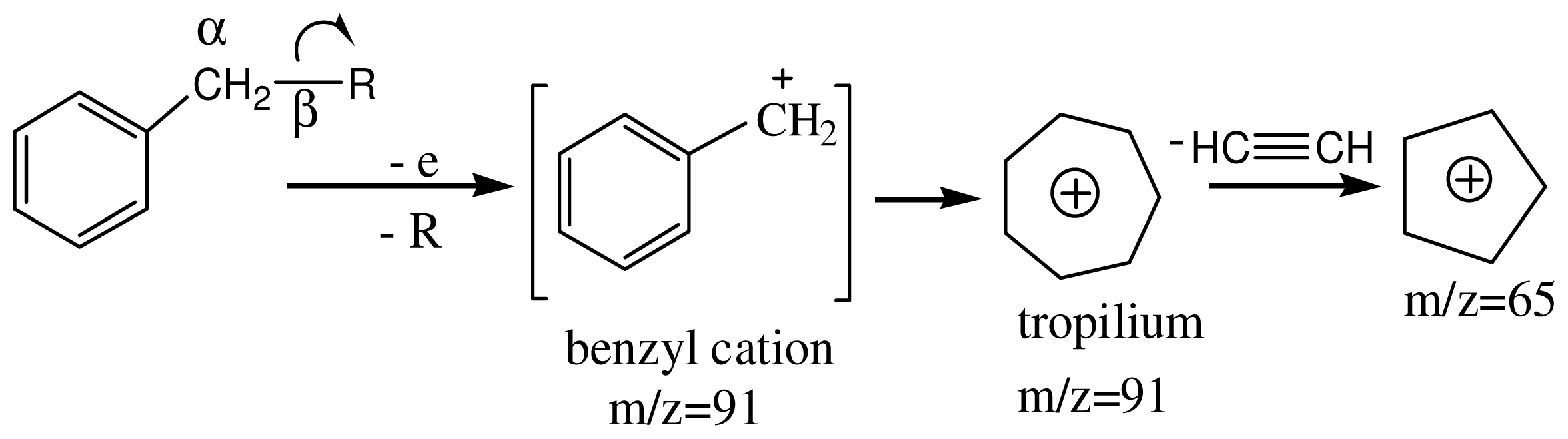
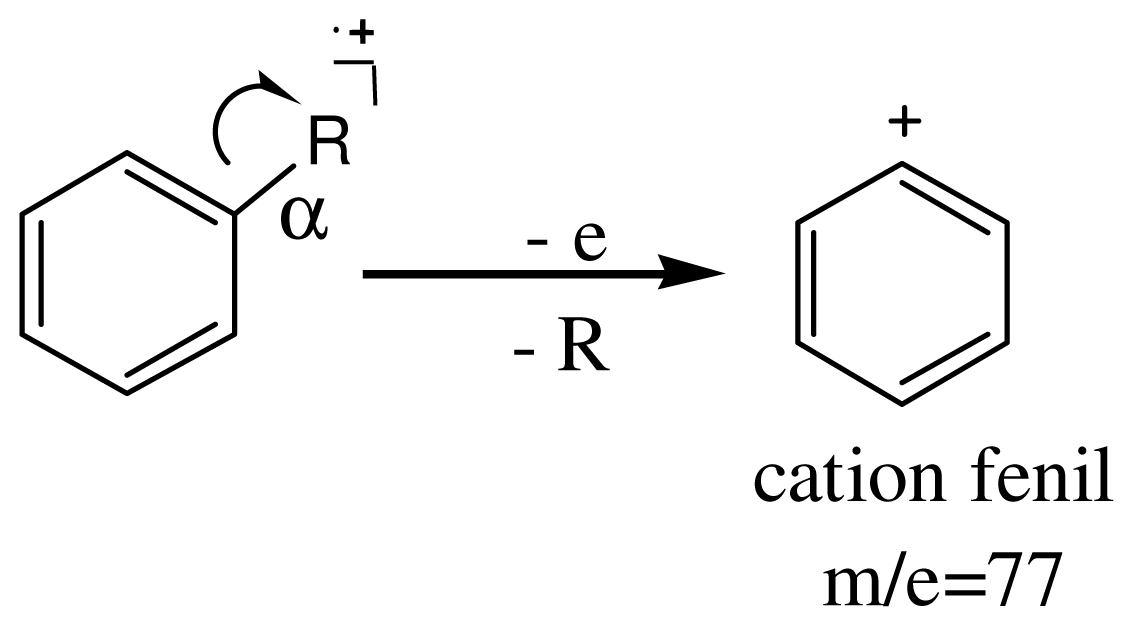
4. Conclusions
Acknowledgements
References
- Massart, DL; Vandeginste, BG; Buydens, LMC; De Jong, S; Lewi, PJ; Smeyers-Verbeke, J. Handbook of Chemometrics and Qualimetrics: Part B; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Verikas, A; Bacauskiene, M. Tutorial: Using artificial neural networks for process and system modeling. Chemom. Intel. Lab. Syst 2003, 67, 187–191. [Google Scholar]
- Zupan, J; Gasteiger, J. Neural Networks in Chemistry and Drug Design, 2nd ed; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Niculescu, SP. Tutorial: Artificial neural networks and genetic algorithms in QSAR. J. Mol. Struct. Theochem 2003, 622, 71–83. [Google Scholar]
- Wesolowski, M; Suchacz, B; Halkiewicz, J. The analysis of seasonal air pollution pattern with application of neural networks. Anal. Bioanal. Chem 2006, 384, 458–467. [Google Scholar]
- Linker, R; Shmulevich, I; Kenny, A; Shaviv, A. Soil identification and chemometrics for direct determination of nitrate in soils using FTIR-ATR mid-infrared spectroscopy. Chemosphere 2005, 61, 652–658. [Google Scholar]
- Li, HZ; Tao, W; Gao, T; Li, H; Lu, YH; Su, ZM. Improving the Accuracy of Density Functional Theory (DFT) calculation for homolysis bond dissociation energies of Y–NO bond: Generalized regression neural network based on grey relational analysis and principal component analysis. Int. J. Mol. Sci 2011, 12, 2242–2261. [Google Scholar]
- Rezzi, S; Axelson, DE; Héberger, K; Reniero, F; Mariani, C; Guillou, C. Classification of olive oils using high throughput flow ^1H NMR fingerprinting with principal component analysis, linear discriminant analysis and probabilistic neural networks. Anal. Chim. Acta 2005, 552, 13–24. [Google Scholar]
- Elhallaoui, M; Elasri, M; Ouazzani, F; Mechaqrane, A; Lakhlifi, T. Quantitative structure-activity relationships of noncompetitive antagonists of the NMDA receptor: A study of a series of mk801 derivative molecules using statistical methods and neural network. Int. J. Mol. Sci 2003, 4, 249–262. [Google Scholar]
- Shan, Y; Zhao, R; Xu, G; Liebich, HM; Zhang, Y. Application of probabilistic neural network in the clinical diagnosis of cancers based on clinical chemistry data. Anal. Chim. Acta 2002, 471, 77–86. [Google Scholar]
- Praisler, M; Dirinck, I; van Bocxlaer, J; De Leenheer, AP; Massart, DL. Pattern recognition techniques screening for drugs of abuse with gas chromatography—Fourier transform infrared spectroscopy. Talanta 2000, 53, 177–193. [Google Scholar]
- Hemmer, MC; Gasteiger, J. Prediction of three-dimensional structure using information from infrared spectra. Anal. Chim. Acta 2000, 420, 145–154. [Google Scholar]
- Safavi, A; Abdollahi, H; Hormozi Nezhad, MR. Artificial neural networks for simultaneous spectrophotometric differential kinetic determination of Co(II) and V(IV). Talanta 2003, 59, 515–523. [Google Scholar]
- Eghbaldar, A; Forrest, TP; Cabrol-Bass, D. Development of neural networks for identification of structural features from mass spectral data. Anal. Chim. Acta 1998, 359, 283–301. [Google Scholar]
- Xu, M; Voorhees, KJ; Hadfield, TL. Repeatability and pattern recognition of bacterial fatty acid profiles generated by direct mass spectrometric analysis of in situ thermal hydrolysis/methylation of whole cells. Talanta 2003, 59, 577–589. [Google Scholar]
- Karch, SB. Drug Abuse Handbook; CRC Press: New York, NY, USA, 1998. [Google Scholar]
- Ferary, S; Auger, J; Touche, A. Trace identification of plant substances by combining gas chromatography-mass spectrometry and direct deposition gas chromatography-Fourier transform infrared spectrometry. Talanta 1996, 43, 349–357. [Google Scholar]
- Platoff, GE, Jr; Hill, DW; Koch, TR; Caplan, YH. Serial capillary gas chromatography/Fourier transform infrared spectrometry/mass spectrometry (GC/IR/MS): qualitative and quantitative analysis of amphetamine, methamphetamine, and related analogues in human urine. J. Anal. Toxicol 1992, 16, 389–397. [Google Scholar]
- Gosav, S; Praisler, M; van Bocxlaer, J; De Leenheer, AP; Massart, DL. Class identity assignment for amphetamines using neural networks and GC-FTIR data. Spectrochim. Acta Part A 2006, 64, 1110–1117. [Google Scholar]
- EasyNNplus, version 3.0i; Neural Planner Software Ltd: England, UK. Available online: http://www.easynn.com accessed on 21 September 2011.
- Gosav, S; Praisler, M. Artificial neural networks built for the recognition of illicit amphetamines using a concatenated database. Rom. J. Phys 2009, 54, 929–935. [Google Scholar]
- Gosav, S; Praisler, M; Dorohoi, DO; Popa, G. Automated identification of novel amphetamines using a pure neural network and neural networks coupled with principal component analysis. J Mol Struct 2005, 744–747, 821–825. [Google Scholar]
- Avram, M; Mateescu, GHD. The Spectroscopy in Infrared. Applications in Organic Chemistry; Technical Publishing House: Bucharest, Romania, 1966. [Google Scholar]
- Bellamy, LJ. The Infra-Red Spectra of Complex Molecules; Chapman and Hall Ltd: London, UK, 1978. [Google Scholar]
- Gosav, S; Dinica, R; Praisler, M. Choosing between GC-FTIR and GC-MS spectra for an efficient intelligent identification of illicit amphetamines. J. Mol. Struct 2008, 887, 269–278. [Google Scholar]


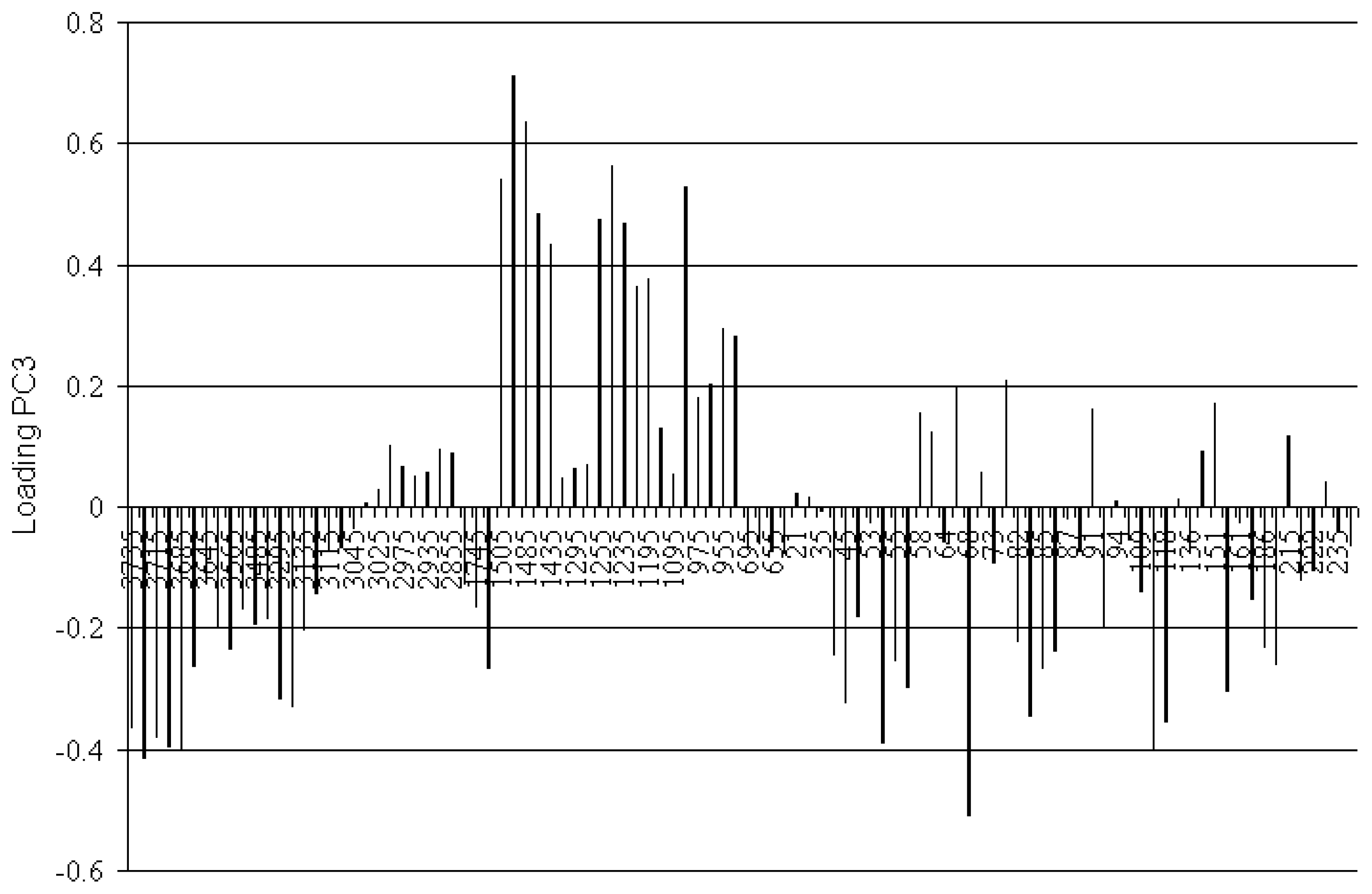
 ), from 605 to 3745 cm−1.
), from 605 to 3745 cm−1.
), from 605 to 3745 cm−1.
), from 605 to 3745 cm−1.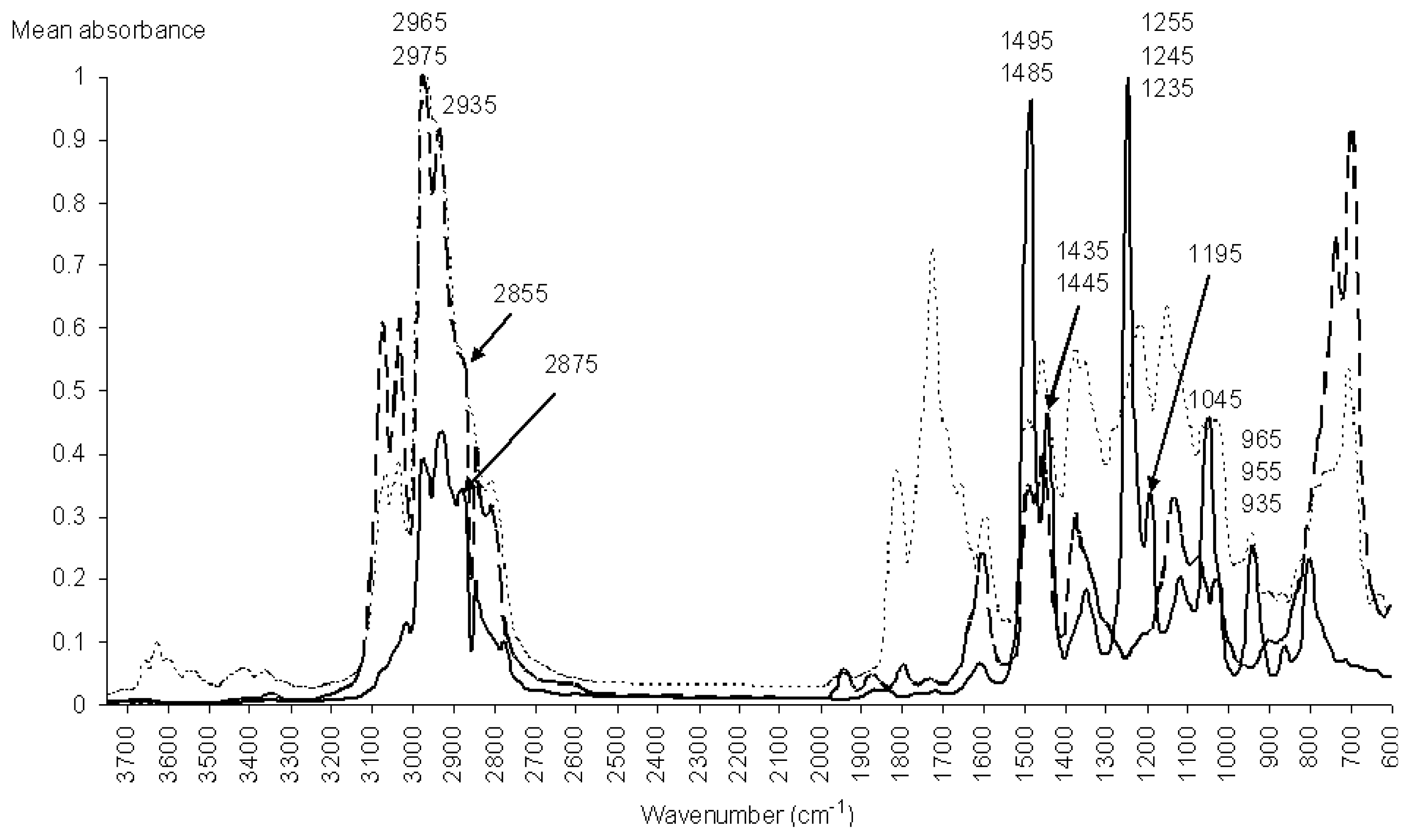


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 100imp_IR-MS-ANN | 15PC_IR-MS-ANN | 16PC_IR-MS-ANN | 17PC_IR-MS-ANN | 18PC_IR-MS-ANN | 19PC_IR-MS-ANN | 20PC_IR-MS-ANN | 21PC_IR-MS-ANN | 22PC_IR-MS-ANN | |
|---|---|---|---|---|---|---|---|---|---|
| Explained variance (%) | 72 | 73 | 75 | 77 | 78 | 79 | 81 | 82 | |
| TP (%) | 100 | 100 | 95 | 100 | 100 | 100 | 100 | 100 | 100 |
| TN (%) | 93.83 | 90.79 | 89.87 | 87.5 | 92.77 | 92.21 | 93.67 | 92.31 | 88.46 |
| FP (%) | 6.17 | 9.21 | 10.13 | 12.5 | 7.3 | 7.79 | 6.33 | 7.69 | 11.54 |
| FN (%) | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 |
| CC (%) | 95.05 | 92.71 | 90.91 | 90 | 94.17 | 93.81 | 94.95 | 93.88 | 90.82 |
| C (%) | 98.05 | 93.2 | 96.12 | 97.09 | 100 | 94.17 | 96.12 | 95.15 | 95.15 |
| 100imp_IR-MS-ANN | 15PC_IR-MS-ANN | 16PC_IR-MS-ANN | 17PC_IR-MS-ANN | 18PC_IR-MS-ANN | 19PC_IR-MS-ANN | 20PC_IR-MS-ANN | 21PC_IR-MS-ANN | 22PC_IR-MS-ANN | |
|---|---|---|---|---|---|---|---|---|---|
| Unclassified | 2 | 7 | 4 | 3 | 0 | 6 | 4 | 5 | 5 |
| False positives | 5 | 7 | 8 | 10 | 6 | 6 | 5 | 6 | 9 |
| False negatives | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 7 | 14 | 13 | 13 | 6 | 12 | 9 | 11 | 14 |
| Input name | Importance |
|---|---|
| PC3 | 40.9814 |
| PC2 | 40.8999 |
| PC4 | 29.7392 |
| PC5 | 28.8037 |
| PC1 | 25.8807 |
| PC7 | 23.7982 |
| PC12 | 23.1617 |
| PC18 | 21.7441 |
| PC10 | 16.1382 |
| PC15 | 15.9791 |
| PC8 | 15.2410 |
| PC6 | 9.1584 |
| PC14 | 8.5687 |
| PC13 | 7.5713 |
| PC17 | 6.8467 |
| PC11 | 4.4506 |
| PC16 | 3.5881 |
| PC9 | 2.2904 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gosav, S.; Praisler, M.; Birsa, M.L. Principal Component Analysis Coupled with Artificial Neural Networks—A Combined Technique Classifying Small Molecular Structures Using a Concatenated Spectral Database. Int. J. Mol. Sci. 2011, 12, 6668-6684. https://doi.org/10.3390/ijms12106668
Gosav S, Praisler M, Birsa ML. Principal Component Analysis Coupled with Artificial Neural Networks—A Combined Technique Classifying Small Molecular Structures Using a Concatenated Spectral Database. International Journal of Molecular Sciences. 2011; 12(10):6668-6684. https://doi.org/10.3390/ijms12106668
Chicago/Turabian StyleGosav, Steluţa, Mirela Praisler, and Mihail Lucian Birsa. 2011. "Principal Component Analysis Coupled with Artificial Neural Networks—A Combined Technique Classifying Small Molecular Structures Using a Concatenated Spectral Database" International Journal of Molecular Sciences 12, no. 10: 6668-6684. https://doi.org/10.3390/ijms12106668