Crystallisation of Wild-Type and Variant Forms of a Recombinant Plant Enzyme β-D-Glucan Glucohydrolase from Barley (Hordeum vulgare L.) and Preliminary X-ray Analysis



Abstract
:
1. Introduction
2. Results and Discussion
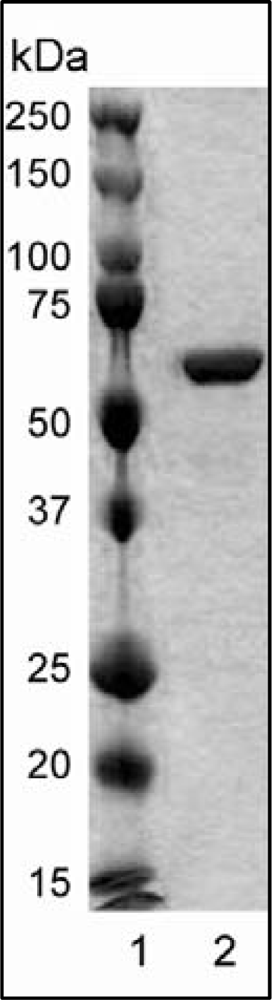
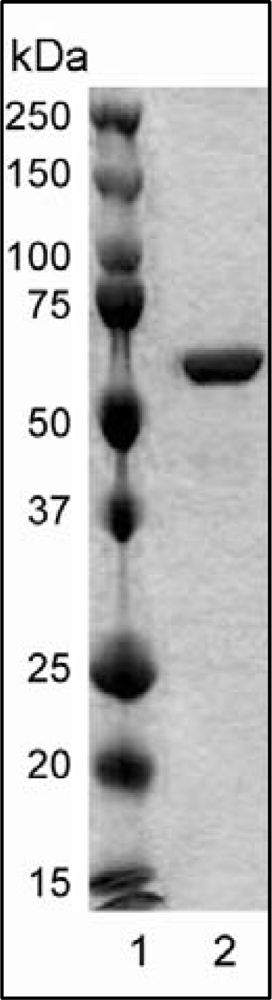
2.1. Protein Expression and Purification
2.2. Protein Crystallisation
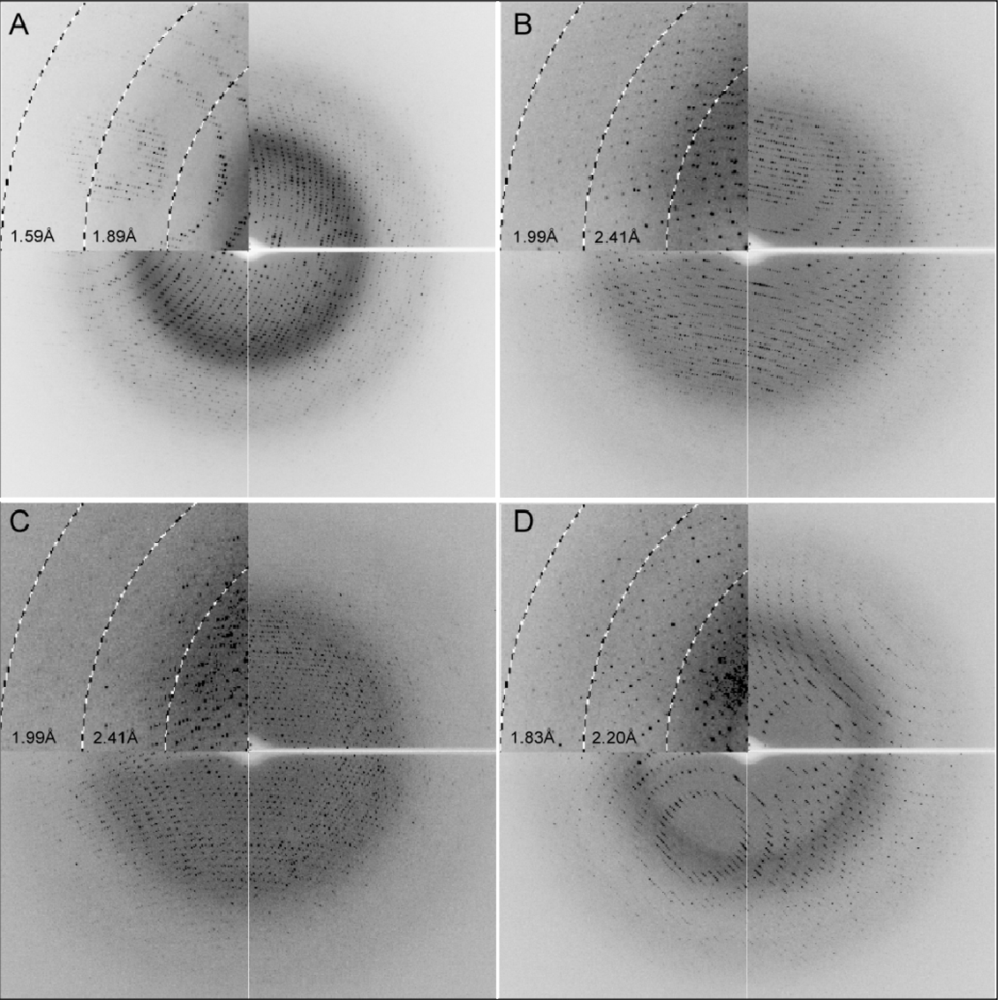
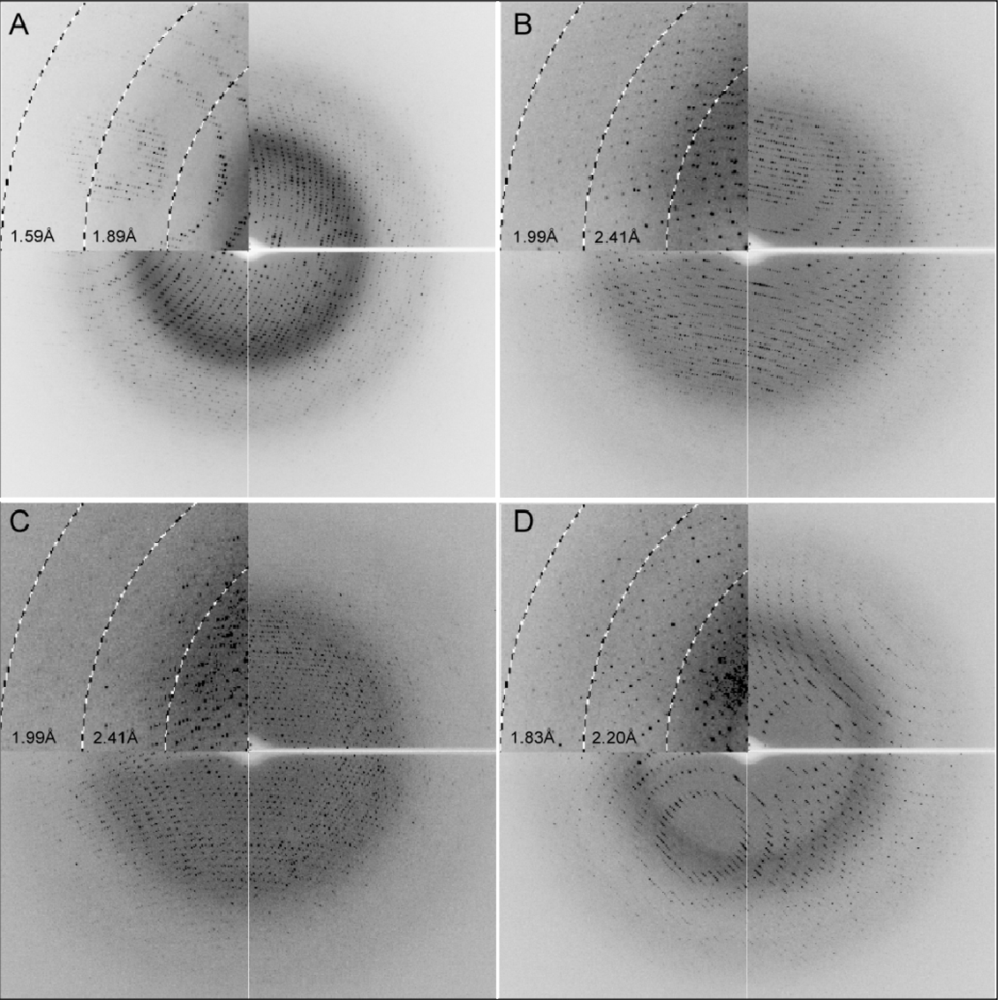
2.3. X-ray Diffraction
3. Experimental Section
3.1. Expression and Purification of Wild-Type and Variant Forms of rHvExoI
3.2. Enzyme Assays
3.3. Crystallisation of Wild-Type and Variant Forms of rHvExoI
3.4. X-ray Data Collection and Processing
4. Conclusions
Acknowledgments
References
- Cantarel, BL; Coutinho, PM; Rancurel, C; Bernard, T; Lombard, V; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res 2008, 37, D233–D238. [Google Scholar]
- Varghese, JN; Hrmova, M; Fincher, GB. Three-dimensional structure of a barley β-d-glucan exohydrolase; a family 3 glycosyl hydrolase. Struct. Fold. Des 1999, 7, 179–190. [Google Scholar]
- Pozzo, T; Pasten, JL; Karlsson, EN; Logan, DT. Structural and functional analyses of beta-glucosidase 3B from Thermotoga neapolitana: A thermostable three-domain representative of glycoside hydrolase 3. J. Mol. Biol 2010, 397, 724–739. [Google Scholar]
- Hrmova, M; Fincher, GB. Dissecting the catalytic mechanism of a plant β-d-glucan glucohydrolase through structural biology using inhibitors and substrate analogues. Carbohydr. Res 2007, 305, 209–221. [Google Scholar]
- Hrmova, M; Varghese, JN; Høj, PB; Fincher, GB. Crystallization and preliminary X-ray analysis of β-glucan exohydrolase isoenzyme ExoI from barley (Hordeum vulgare). Acta Cryst 1998, D54, 6887–689. [Google Scholar]
- Hrmova, M; Harvey, AJ; Wang, J; Shirley, NJ; Jones, GP; Høj, PB; Fincher, GB. Barley β-d-glucan exohydrolases with β-d-glucosidase activity. Purification and determination of primary structure from a cDNA clone. J. Biol. Chem 1996, 271, 5277–5286. [Google Scholar]
- Hrmova, M; Varghese, JN; DeGori, R; Smith, BJ; Driguez, H; Fincher, GB. Catalytic mechanisms and reaction intermediates along the hydrolytic pathway of plant β-d-glucan glucohydrolase. Struct. Fold. Des 2001, 9, 1005–1016. [Google Scholar]
- Hrmova, M; DeGori, R; Smith, BJ; Driguez, H; Varghese, JN; Fincher, GB. Structural basis for a broad specificity in higher plant β-d-glucan glucohydrolases. Plant Cell 2002, 14, 1033–1052. [Google Scholar]
- Hrmova, M; De Gori, R; Smith, BJ; Vasella, A; Varghese, JN; Fincher, GB. Three-dimensional structure of the barley β-d-glucan glucohydrolase in complex with a transition state mimic. J. Biol. Chem 2004, 279, 4970–4980. [Google Scholar]
- Hrmova, M; Streltsov, VA; Smith, BJ; Vasella, A; Varghese, JN; Fincher, GB. Structural rationale for low nanomolar binding of transition state mimics to a family GH3 β-d-glucan glucohydrolase from barley. Biochemistry 2005, 44, 16529–16539. [Google Scholar]
- Luang, S; Hrmova, M; Ketudat Cairns, JR. High-level expression of barley β-d-glucan exohydrolase HvExo I from a codon-optimized cDNA in Pichia pastoris. Prot. Expr. Purif 2010, 73, 90–98. [Google Scholar]
- Luang, S; Streltsov, VA; Ketudat Cairns, JR; Hrmova, M. Characterization of variant forms of β-d-glucan glucohydrolase from barley (Hordeum vulgare L). Biochem J, 2010; to be submitted. [Google Scholar]
- Stura, E; Wilson, IA. Analytical and production seeding techniques. Methods 2000, 1, 38–49. [Google Scholar]
- Hrmova, M; Fincher, GB. Barley β-d-glucan exohydrolases. Substrate specificity and kinetic properties. Carbohydr. Res 1998, 305, 209–221. [Google Scholar]
- Varghese, JN; van Donkelaar, A; Balaic, DX; Barnea, Z. CSIRO-Commonwealth Scientific Research Organization. Personal communication: Melbourne, Victoria, 2001.
- Radaev, S; Agniswamy, J; Sun, PD. A case of structure determination using pseudosymmetry. Acta Cryst 2009, D65, 1334–1340. [Google Scholar]
- Stec, B; Holtz, KM; Wojciechowski, CL; Kantrowitz, ER. Structure of the wild-type TEM-1 β-lactamase at 1.55 Å and the mutant enzyme Ser70Ala at 2.1 Å suggest the mode of noncovalent catalysis for the mutant enzyme. Acta Cryst 2005, D61, 1072–1079. [Google Scholar]
- Heaslet, H; Rosenfeld, R; Giffin, M; Lin, Y-C; Tam, K; Torbett, BE; Elder, JH; McRee, DE; Stout, CD. Conformational flexibility in the flap domains of ligand free HIV protease. Acta Cryst 2007, D63, 866–875. [Google Scholar]
- Mizutani, H; Saraboji, K; Malathy Sony, SM; Ponnuswamy, MN; Kumarevel, T; Krishna Swamy, BS; Simanshu, DK; Murthy, MR; Kunishima, N. Systematic study on crystal-contact engineering of diphthine synthase: Influence of mutations at crystal-packing regions on X-ray diffraction quality. Acta Cryst 2008, D64, 1020–1033. [Google Scholar]
- Matthews, BW. Solvent content of protein crystals. J. Mol. Biol 1968, 33, 491–497. [Google Scholar]
- Newman, J; Pham, TM; Peat, TS. Phoenito experiments: combining the strengths of commercial crystallization automation. Acta Cryst 2008, F64, 991–996. [Google Scholar]
- Newman, J. Optimization of buffer solutions for protein crystallization. Acta Cryst 2004, D60, 610–612. [Google Scholar]
- McPhillips, TM; McPhillips, SE; Chiu, HJ; Cohen, AE; Deacon, AM; Ellis, PJ; Garman, E; Gonzalez; Sauter, NK; Phizackerley, RP; Soltis, SM; Kuhn, P. Blu-ice and the distributed control system: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat 2002, 9, 401–406. [Google Scholar]
- Otwinowski, Z; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild-type | Glu220Ala | Trp434Ala | Arg158Ala/Glu161Ala | |
|---|---|---|---|---|
| Unique reflections | 119968 | 74171 | 64460 | 107601 |
| Resolution a (Å) | 1.57 (1.6−1.57) | 1.90 (1.94−1.90) | 1.95 (1.98−1.95) | 1.65 (1.68−1.65) |
| Mean multiplicity a,b | 29 (26) | 27 (16) | 27 (16) | 26 (12) |
| Completeness a,b (%) | 99 (86) | 99 (88) | 100 (100) | 99.8 (98) |
| Mean <I/σ/(I)> a | 71.2 (5.6) | 54.8 (4.3) | 37.0 (2.2) | 58.7 (2.3) |
| Rmerge a–c (%) | 6.7 (47) | 8.9 (64) | 10.1 (87) | 5.6 (82) |
| a = b (Å) d | 99.2 | 100.2 | 100.1 | 100.8 |
| c (Å) d | 183.5 | 183.2 | 183.6 | 183.2 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Luang, S.; Cairns, J.R.K.; Streltsov, V.A.; Hrmova, M. Crystallisation of Wild-Type and Variant Forms of a Recombinant Plant Enzyme β-D-Glucan Glucohydrolase from Barley (Hordeum vulgare L.) and Preliminary X-ray Analysis. Int. J. Mol. Sci. 2010, 11, 2759-2769. https://doi.org/10.3390/ijms11072759
Luang S, Cairns JRK, Streltsov VA, Hrmova M. Crystallisation of Wild-Type and Variant Forms of a Recombinant Plant Enzyme β-D-Glucan Glucohydrolase from Barley (Hordeum vulgare L.) and Preliminary X-ray Analysis. International Journal of Molecular Sciences. 2010; 11(7):2759-2769. https://doi.org/10.3390/ijms11072759
Chicago/Turabian StyleLuang, Sukanya, James R. Ketudat Cairns, Victor A. Streltsov, and Maria Hrmova. 2010. "Crystallisation of Wild-Type and Variant Forms of a Recombinant Plant Enzyme β-D-Glucan Glucohydrolase from Barley (Hordeum vulgare L.) and Preliminary X-ray Analysis" International Journal of Molecular Sciences 11, no. 7: 2759-2769. https://doi.org/10.3390/ijms11072759