Computer-Based De Novo Designs of Tripeptides as Novel Neuraminidase Inhibitors



Abstract
:1. Introduction
2. Results and Discussion
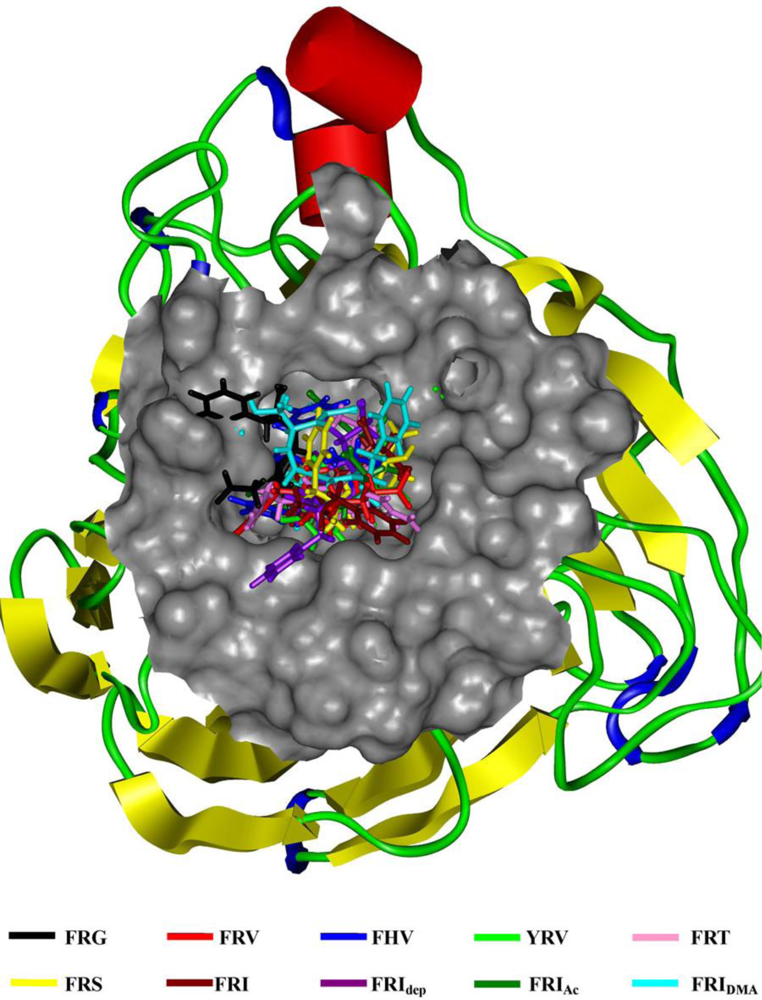
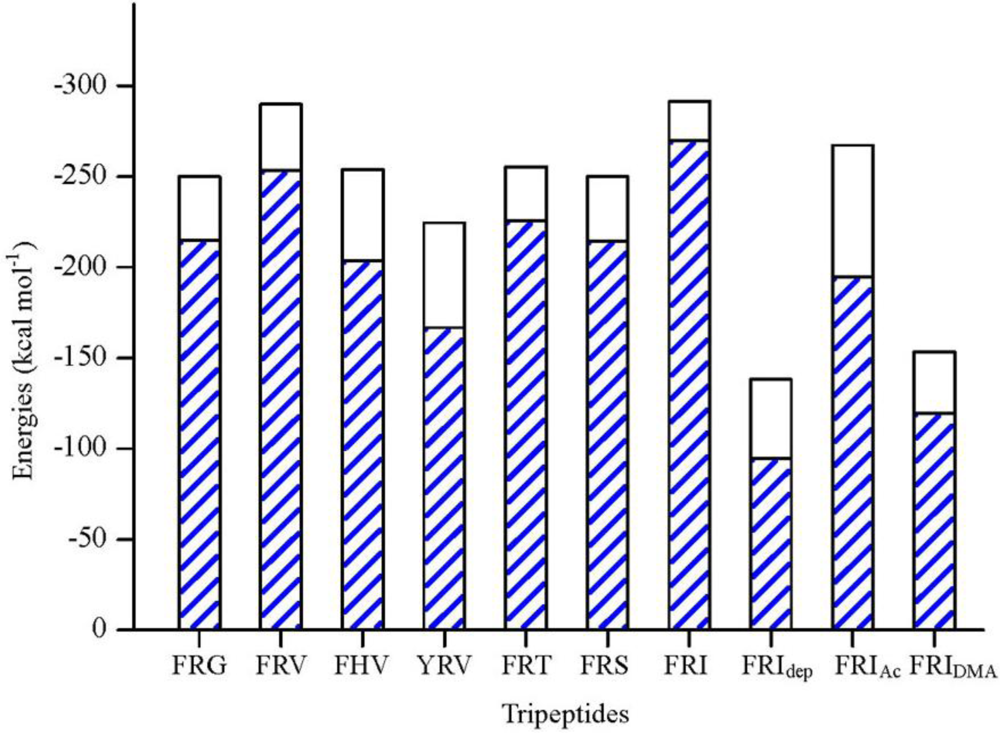
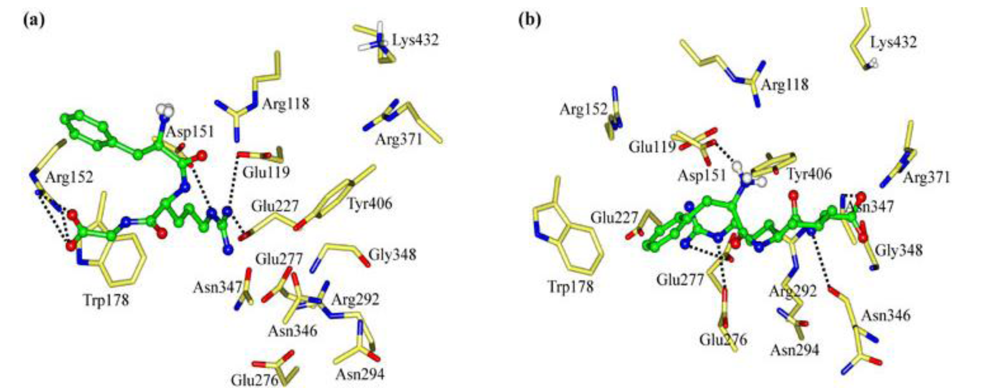
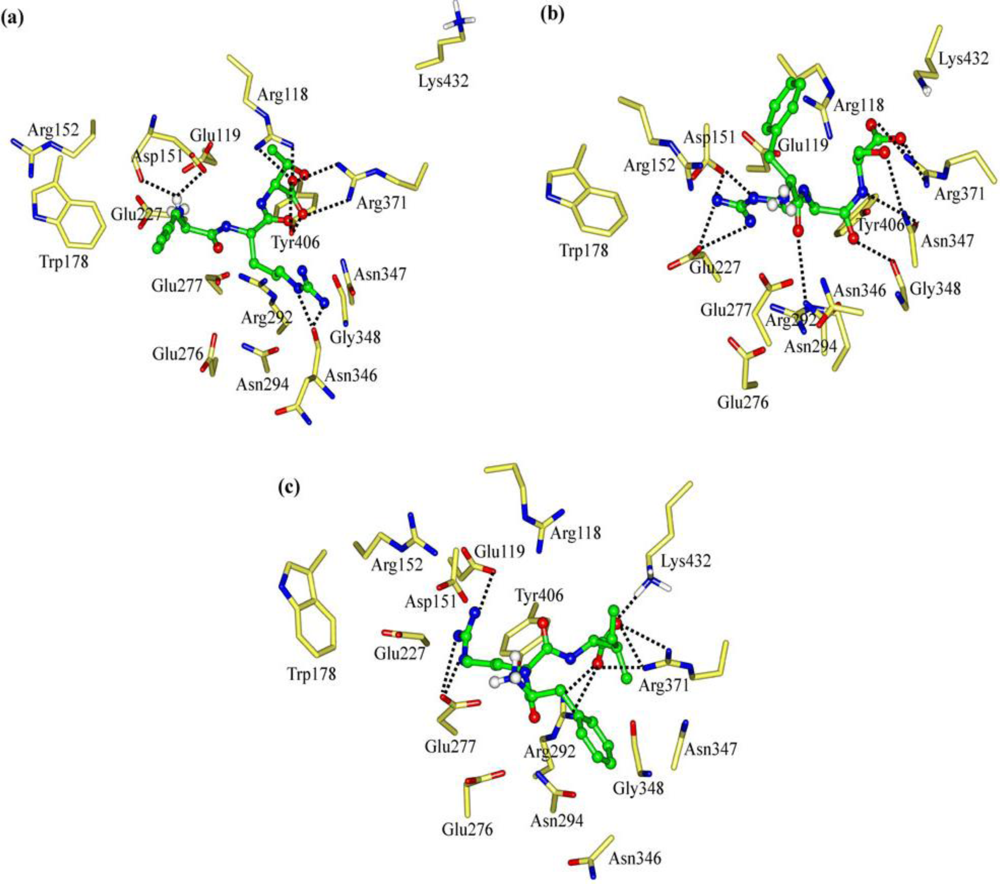
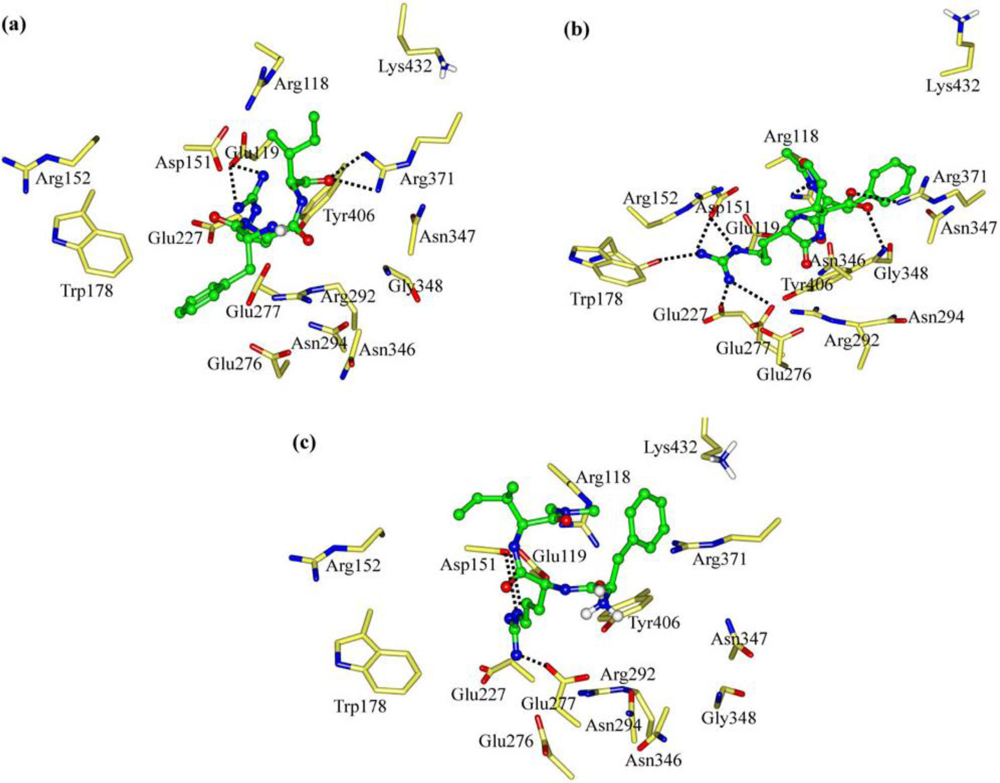
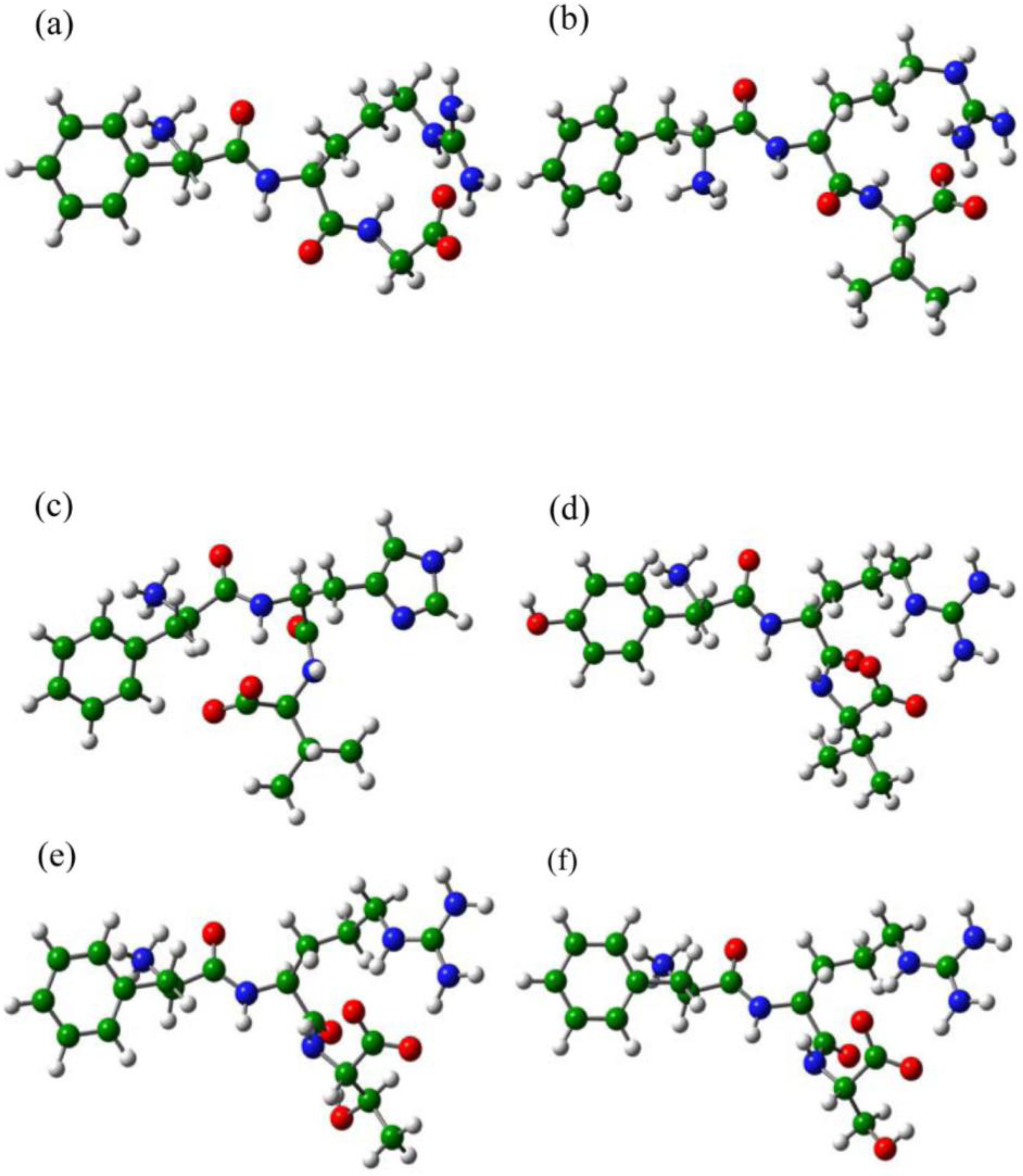
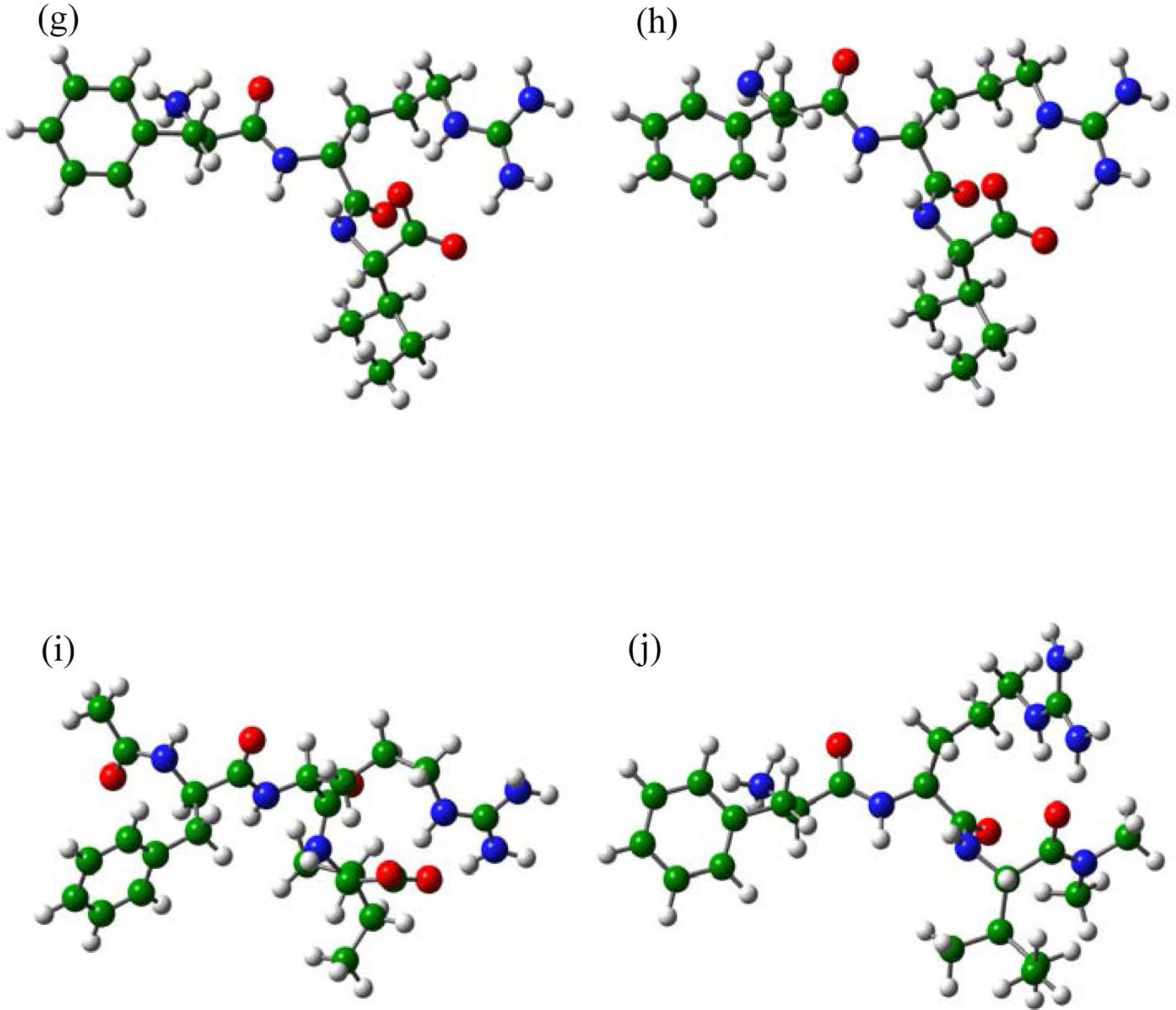
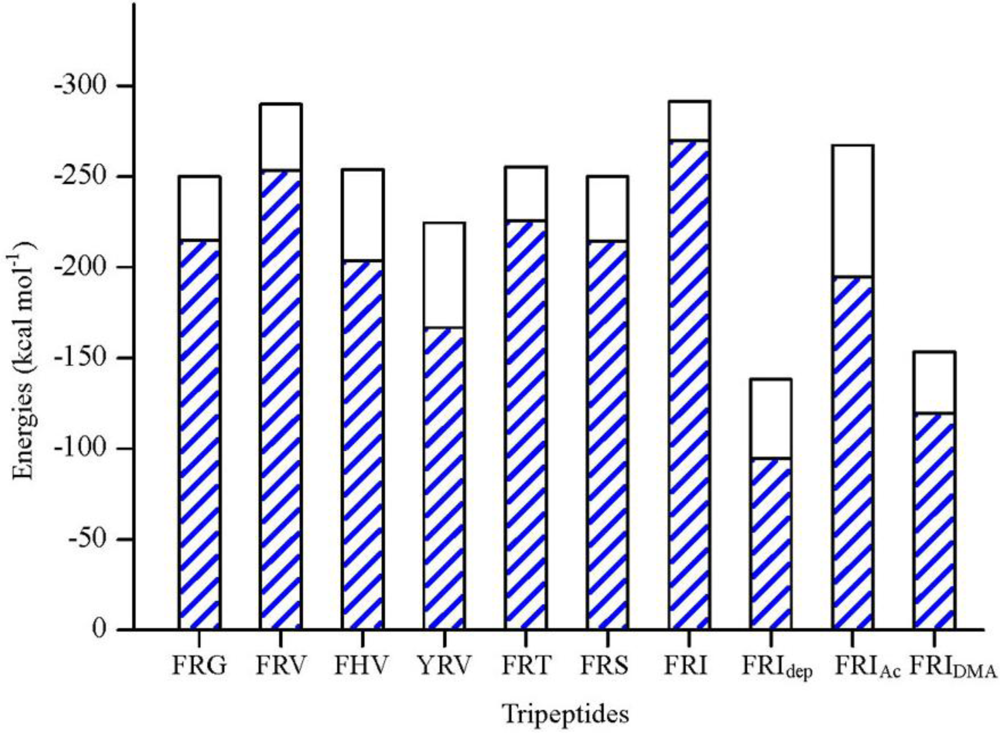
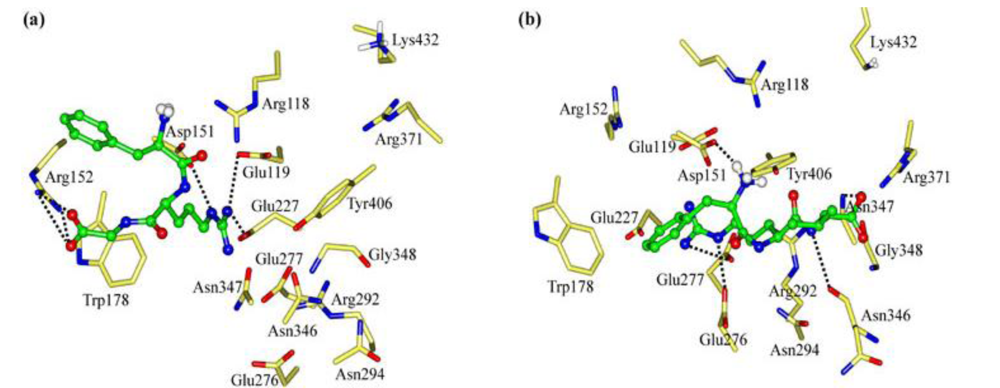
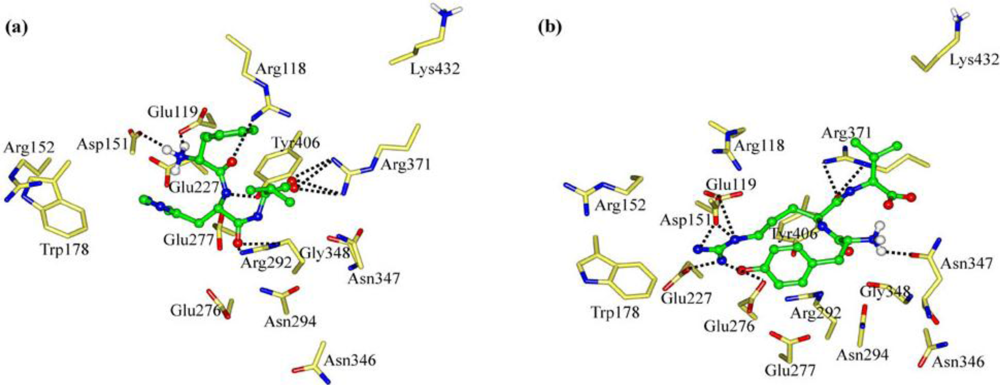
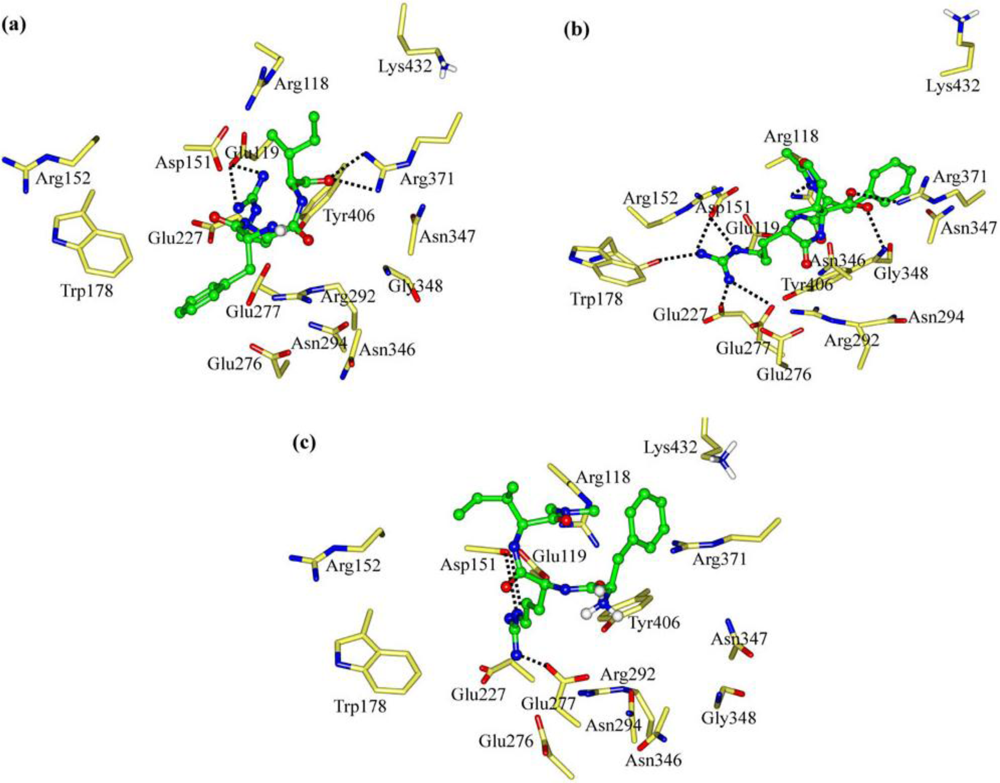
2.1. The Tripeptides FRG and FRV as the NA Inhibitors
2.2. The Roles of the Arg and Phe Portions in the Tripeptides
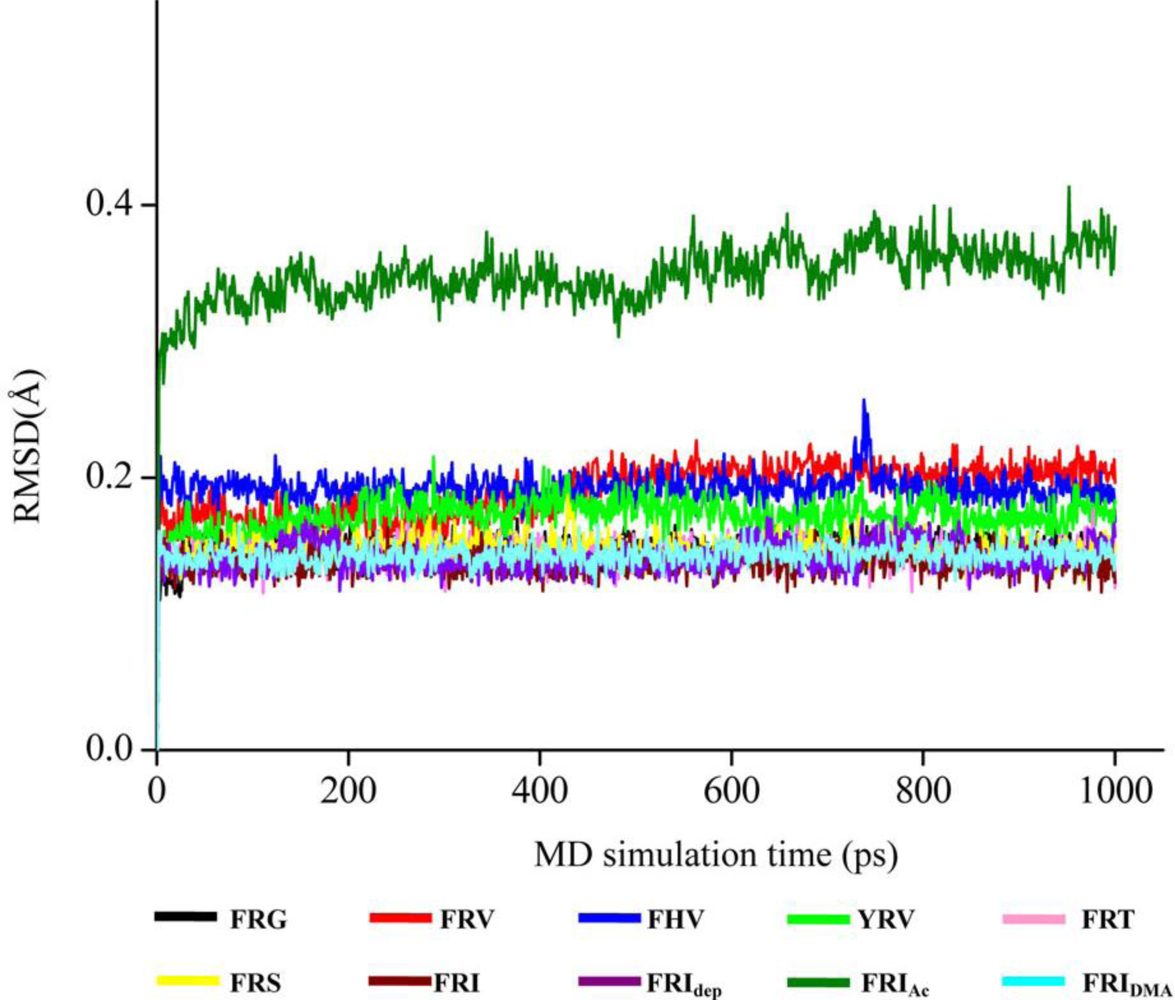
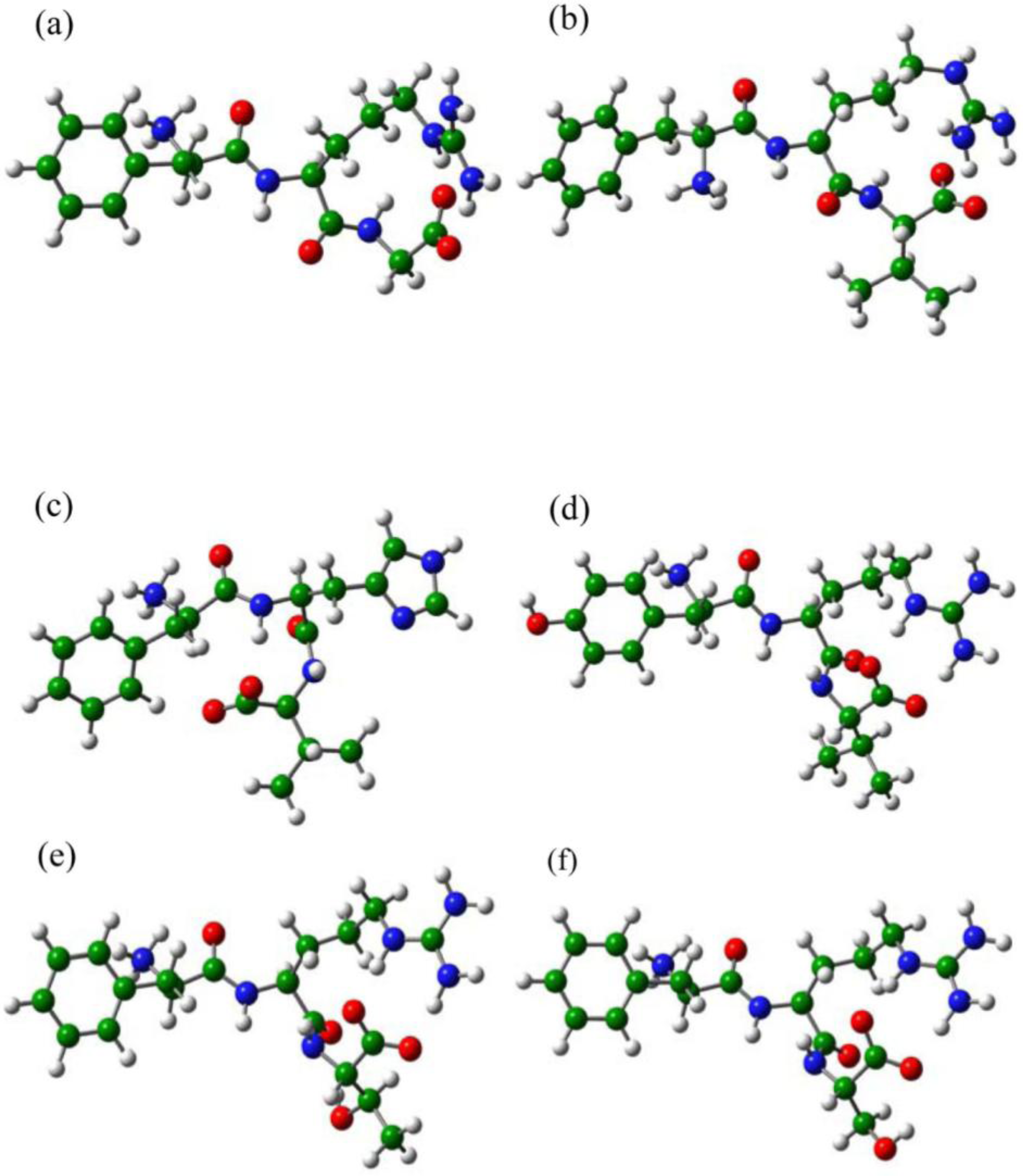
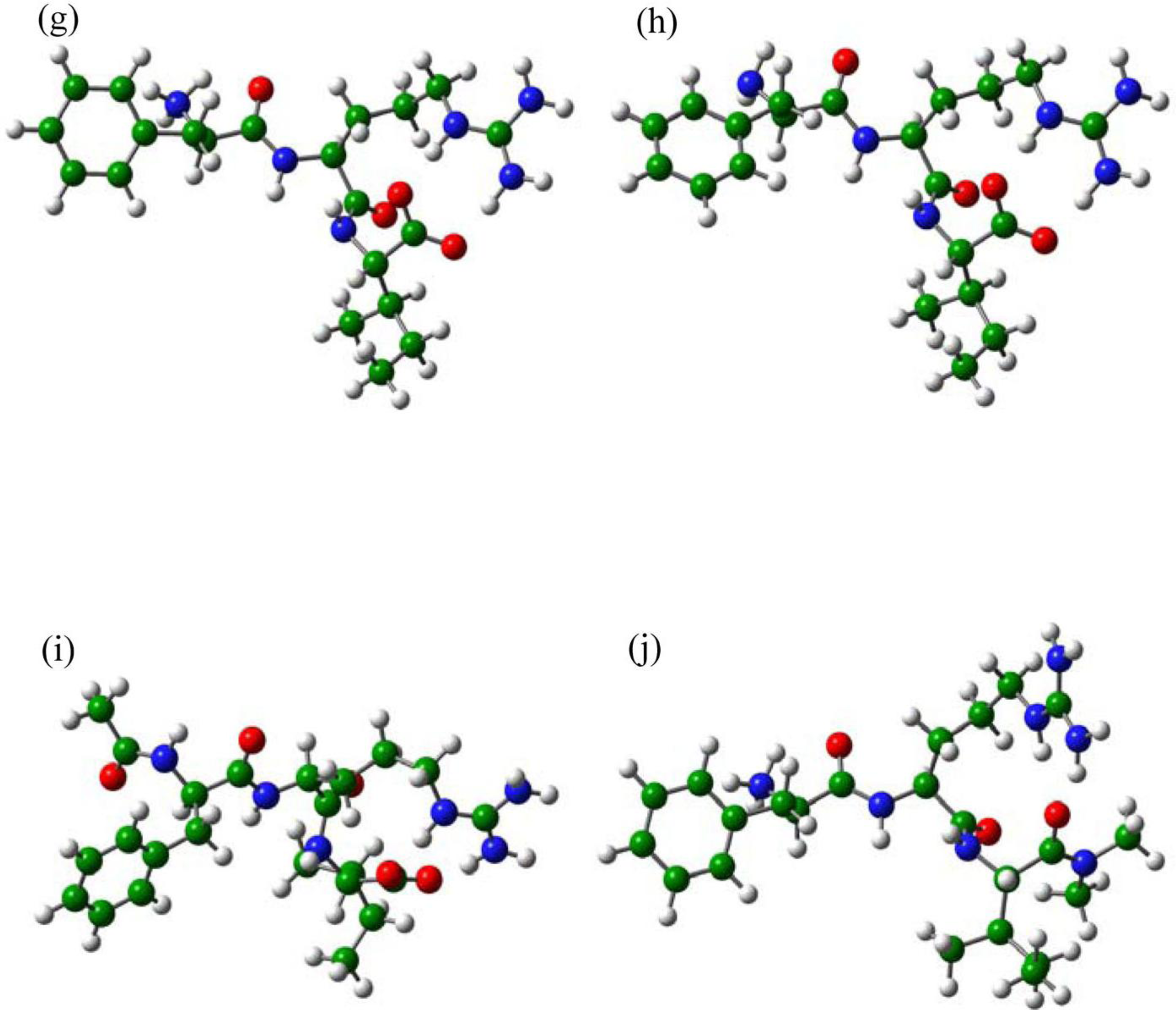
2.3. The Improvement of FRV-Based NA Inhibitors
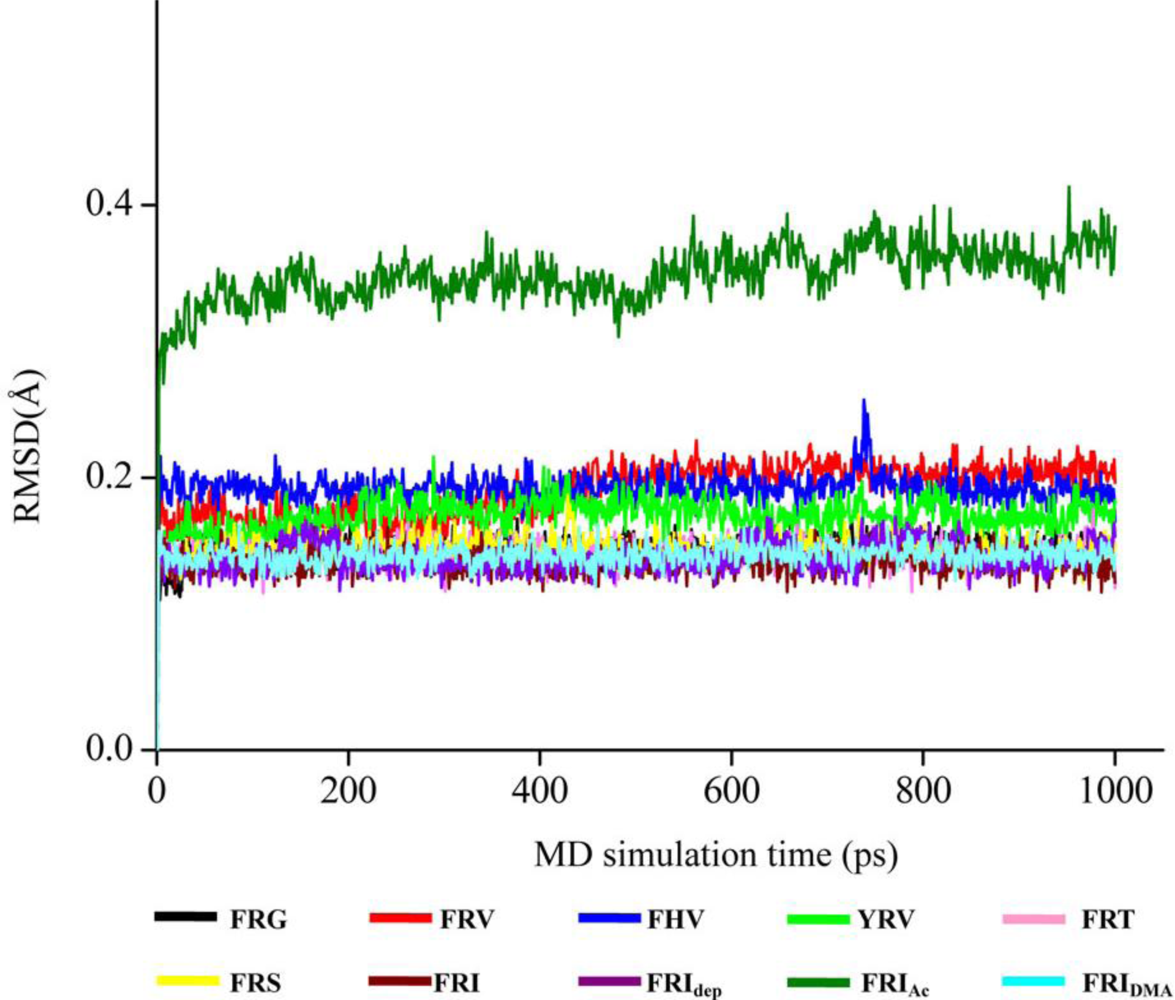
2.4. The Chemical States of the Termini in Tripeptide FRI
3. Computational Methods
3.1. System Preparations
3.2. Flexible Docking
3.3. Molecular Dynamics (MD)
4. Conclusions
Supplemental Material
Supplementary Materials
1. Density Functional Calculations
Figure S1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FRG-NA | FRV-NA | |||||
|---|---|---|---|---|---|---|
| Residue | EvdW | Eele | Einter | EvdW | Eele | Einter |
| Glu119 | −1.23 | −93.23 | −94.46 | −1.39 | −73.28 | −74.67 |
| Ile149 | −1.18 | −9.19 | −10.37 | − | − | − |
| His150 | −8.32 | −6.59 | −14.91 | − | − | − |
| Asp151 | −2.86 | −132.39 | −135.25 | −5.32 | −125.36 | −130.68 |
| Arg152 | −2.55 | −37.21 | −39.76 | − | − | − |
| Trp178 | − | − | − | −3.11 | −1.33 | −4.44 |
| Glu227 | −1.02 | −63.39 | −66.41 | −0.97 | −68.08 | −69.05 |
| Ala246 | − | − | − | −5.62 | −0.30 | −5.92 |
| Thr247 | − | − | − | −3.07 | −2.93 | −6.00 |
| Glu276 | −0.18 | −37.08 | −37.26 | −0.24 | −103.29 | −103.53 |
| Glu277 | −1.32 | −66.47 | −67.79 | −0.02 | −101.73 | −101.75 |
| Asn347 | −2.34 | −6.17 | −8.51 | −4.11 | −20.27 | −24.38 |
| FHV-NA | YRV-NA | |||||
|---|---|---|---|---|---|---|
| Residue | EvdW | Eele | Einter | EvdW | Eele | Einter |
| Arg118 | −2.58 | −13.71 | −16.29 | − | − | − |
| Glu119 | −0.07 | −57.24 | −57.31 | − | − | − |
| Ile149 | −4.16 | −1.51 | −5.67 | − | − | − |
| Asp151 | −5.74 | −57.83 | −63.57 | −0.48 | −126.92 | −127.40 |
| Glu227 | −0.36 | −14.15 | −14.51 | −0.88 | −63.89 | −64.77 |
| Glu276 | − | − | − | −2.33 | −52.17 | −54.50 |
| Glu277 | −2.80 | −5.07 | −7.87 | −1.12 | −69.65 | −70.77 |
| Arg292 | −3.49 | −28.50 | −31.99 | − | − | − |
| Asn347 | − | − | − | −2.70 | −6.33 | −9.03 |
| Arg371 | 1.72 | −84.84 | −83.12 | − | − | − |
| Lys432 | −0.09 | −4.02 | −4.11 | − | − | − |
| FRT-NA | FRS-NA | FRI-NA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Residue | EvdW | Eele | Einter | EvdW | Eele | Einter | EvdW | Eele | Einter |
| Arg118 | −1.04 | −20.01 | −21.05 | − | − | − | − | − | − |
| Glu119 | −0.40 | −30.38 | −30.78 | −2.75 | −65.75 | −68.50 | −1.82 | −56.25 | −58.07 |
| Asp151 | 3.55 | −113.02 | −109.47 | 3.39 | −124.73 | −121.34 | −1.22 | −70.93 | −72.15 |
| Glu227 | −0.05 | −22.80 | −22.85 | −1.47 | −67.11 | −68.58 | −0.70 | −48.77 | −49.47 |
| Ala246 | − | − | − | −1.64 | −3.41 | −5.05 | − | − | − |
| Thr247 | −4.46 | −1.39 | −5.85 | − | − | − | −4.35 | −0.46 | −4.81 |
| Glu276 | −0.32 | −36.13 | −36.45 | −0.40 | −45.66 | −46.06 | −0.69 | −45.56 | −46.25 |
| Glu277 | −0.27 | −21.88 | −22.15 | −1.99 | −72.05 | −74.04 | 3.25 | −108.43 | −105.18 |
| Arg292 | − | − | − | − | − | − | −1.34 | −6.50 | −7.84 |
| Asn346 | 0.20 | −16.38 | −16.18 | − | − | − | − | − | − |
| Asn347 | −4.89 | −15.91 | −20.80 | −3.90 | −1.07 | −4.97 | − | − | − |
| Arg371 | 1.05 | −48.40 | −47.35 | 0.94 | −30.40 | −29.46 | 1.18 | −43.88 | −42.70 |
| Tyr406 | −1.52 | −14.99 | −16.51 | − | − | − | − | − | − |
| Pro431 | −1.78 | −2.29 | −4.07 | − | − | − | − | − | − |
| Lys432 | − | − | − | 1.11 | −34.44 | −33.33 | 0.83 | −29.62 | −28.79 |
| FRIdep-NA | FRIAc-NA | FRIDMA-NA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Residue | EvdW | Eele | Einter | EvdW | Eele | Einter | EvdW | Eele | Einter |
| Glu119 | −1.16 | −34.45 | −35.61 | −4.14 | −41.99 | −46.13 | −1.28 | −97.43 | −98.71 |
| Ile149 | − | − | − | − | − | − | −3.03 | −2.78 | −5.81 |
| Asp151 | −5.76 | −19.80 | −25.56 | −1.52 | −74.80 | −76.32 | −7.84 | −155.98 | −163.82 |
| Trp178 | − | − | − | −2.33 | −3.54 | −5.87 | − | − | − |
| Glu227 | −0.20 | −12.64 | −12.84 | −0.49 | −38.62 | −39.11 | −0.24 | −65.95 | −66.19 |
| Ala246 | −5.69 | 0.15 | −5.54 | − | − | − | − | − | − |
| Glu276 | −0.55 | −7.29 | −7.84 | − | − | − | −0.90 | −82.62 | −83.52 |
| Glu277 | −1.37 | −19.89 | −21.26 | −1.83 | −32.30 | −34.13 | −1.33 | −94.14 | −95.47 |
| Asn346 | − | − | − | − | − | − | −0.50 | −3.53 | −4.03 |
| Asn347 | − | − | − | −3.01 | −1.15 | −4.16 | − | − | − |
| Gly348 | − | − | − | −4.61 | −7.14 | −11.75 | − | − | − |
| Arg371 | 0.91 | −48.85 | −47.94 | −7.50 | −62.66 | −70.16 | − | − | − |
| Pro431 | − | − | − | −2.97 | −2.64 | −5.61 | − | − | − |
| Lys432 | − | − | − | −0.61 | −9.71 | −10.32 | − | − | − |
Acknowledgments
References
- Colman, PM; Varghese, JN; Laver, WG. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar]
- von Itzstein, M. The war against influenza: discovery and development of sialidase inhibitors. Nat. Rev. Drug. Discov 2007, 6, 967–974. [Google Scholar]
- Itoh, Y; Shinya, K; Kiso, M; Watanabe, T; Sakoda, Y; Hatta, M; Muramoto, Y; Tamura, D; Sakai-Tagawa, Y; Noda, T; et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 2009, 460, 1021–1025. [Google Scholar]
- Moscona, A. Medical management of influenza infection. Annu. Rev. Med 2008, 59, 397–413. [Google Scholar]
- Moscona, A. Neuraminidase inhibitors for influenza. N. Engl. J. Med 2005, 353, 1363–1373. [Google Scholar]
- Ward, P; Small, I; Smith, J; Suter, P; Dutkowski, R. Oseltamivir (Tamiflu) and its potential for use in the event of an influenza pandemic. J. Antimicrob. Chemother 2005, 55(Suppl. S1), i5–i21. [Google Scholar]
- Beigel, J; Bray, M. Current and future antiviral therapy of severe seasonal and avian influenza. Antivir. Res 2008, 78, 91–102. [Google Scholar]
- Gubareva, LV. Molecular mechanisms of influenza virus resistance to neuraminidase inhibitors. Virus Res 2004, 103, 199–203. [Google Scholar]
- de Jong, MD; Tran, TT; Truong, HK; Vo, MH; Smith, GJ; Nguyen, VC; Bach, VC; Phan, TQ; Do, QH; Guan, Y; Peiris, JS; Tran, TH; Farrar, J. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N. Engl. J. Med 2005, 353, 2667–2672. [Google Scholar]
- Bautista, E; Chotpitayasunondh, T; Gao, Z; Harper, SA; Shaw, M; Uyeki, TM; Zaki, SR; Hayden, FG; Hui, DS; Kettner, JD; Kumar, A; Lim, M; Shindo, N; Penn, C; Nicholson, KG. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N. Engl. J. Med 2010, 362, 1708–1719. [Google Scholar]
- de Clercq, E; Neyts, J. Avian influenza A (H5N1) infection: targets and strategies for chemotherapeutic intervention. Trends Pharmacol. Sci 2007, 28, 280–285. [Google Scholar]
- Garman, E; Laver, G. Controlling influenza by inhibiting the virus’s neuraminidase. Curr. Drug Targets 2004, 5, 119–136. [Google Scholar]
- Russell, RJ; Haire, LF; Stevens, DJ; Collins, PJ; Lin, YP; Blackburn, GM; Hay, AJ; Gamblin, SJ; Skehel, JJ. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49. [Google Scholar]
- Aruksakunwong, O; Malaisree, M; Decha, P; Sompornpisut, P; Parasuk, V; Pianwanit, S; Hannongbua, S. On the lower susceptibility of oseltamivir to influenza neuraminidase subtype N1 than those in N2 and N9. Biophys. J 2007, 92, 798–807. [Google Scholar]
- Das, K; Aramini, JM; Ma, LC; Krug, RM; Arnold, E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol 2010, 17, 530–538. [Google Scholar]
- Varghese, JN; Epa, VC; Colman, PM. Three-dimensional structure of the complex of 4-guanidino-Neu5Ac2en and influenza virus neuraminidase. Protein Sci 1995, 4, 1081–1087. [Google Scholar]
- Varghese, JN; Smith, PW; Sollis, SL; Blick, TJ; Sahasrabudhe, A; McKimm-Breschkin, JL; Colman, PM. Drug design against a shifting target: A structural basis for resistance to inhibitors in a variant of influenza virus neuraminidase. Structure 1998, 6, 735–746. [Google Scholar]
- Smith, BJ; Colman, PM; von Itzstein, M; Danylec, B; Varghese, JN. Analysis of inhibitor binding in influenza virus neuraminidase. Protein Sci 2001, 10, 689–696. [Google Scholar]
- Smith, BJ; McKimm-Breshkin, JL; McDonald, M; Fernley, RT; Varghese, JN; Colman, PM. Structural Studies of the Resistance of Influenza Virus Neuramindase to Inhibitors. J. Med. Chem 2002, 45, 2207–2212. [Google Scholar]
- von Itzstein, M; Wu, WY; Kok, GB; Pegg, MS; Dyason, JC; Jin, B; van Phan, T; Smythe, ML; White, HF; Oliver, SW. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar]
- Kim, CU; Lew, W; Williams, MA; Liu, H; Zhang, L; Swaminathan, S; Bischofberger, N; Chen, MS; Mendel, DB; Tai, CY. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc 1997, 119, 681–690. [Google Scholar]
- Jones, JC; Turpin, EA; Bultmann, H; Brandt, CR; Schultz-Cherry, S. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J. Virol 2006, 80, 11960–11967. [Google Scholar]
- Kuang, ZZ; Zheng, LS; Li, S; Duan, ZJ; Qi, ZY; Qu, XW; Liu, WP; Zhang, WJ; Li, DD; Gao, HC; Hou, YD. Screening of peptides as broad-spectrum neuraminidase inhibitors against influenza viruses. Chin. J. Virol 2007, 23, 165–171. [Google Scholar]
- Matsubara, T; Sumi, M; Kubota, H; Taki, T; Okahata, Y; Sato, T. Inhibition of influenza virus infections by sialylgalactose-binding peptides selected from a phage library. J. Med. Chem 2009, 52, 4247–4256. [Google Scholar]
- Rajik, M; Jahanshiri, F; Omar, AR; Ideris, A; Hassan, SS; Yusoff, K. Identification and characterisation of a novel anti-viral peptide against avian influenza virus H9N2. Virol. J 2009, 6, 74. [Google Scholar]
- Bray, BL. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat. Rev. Drug Discov 2003, 2, 587–593. [Google Scholar]
- Owens, J. Building blocks for peptide drugs. Nat. Rev. Drug Discov 2004, 3, 476–476. [Google Scholar]
- Rappocciolo, E. Antimicrobial peptides as carriers of drugs. Drug Discov. Today 2004, 9, 470. [Google Scholar]
- Watt, PM. Screening for peptide drugs from the natural repertoire of biodiverse protein folds. Nat. Biotechnol 2006, 24, 177–183. [Google Scholar]
- Haine, ER; Moret, Y; Siva-Jothy, MT; Rolff, J. Antimicrobial defense and persistent infection in insects. Science 2008, 322, 1257–1259. [Google Scholar]
- Bender, FC; Whitbeck, JC; Lou, H; Cohen, GH; Eisenberg, RJ. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol 2005, 79, 11588–11597. [Google Scholar]
- Townsend, DM; Tew, KD; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother 2003, 57, 145–155. [Google Scholar]
- Shen, GX. Development and current applications of thrombin-specific inhibitors. Curr. Drug Targets Cardiovasc. Haematol. Disord 2001, 1, 41–49. [Google Scholar]
- Kiso, Y. Design and synthesis of substrate-based peptidomimetic human immunodeficiency virus protease inhibitors containing the hydroxymethylcarbonyl isostere. Biopolymers 1996, 40, 235–244. [Google Scholar]
- Njoroge, FG; Chen, KX; Shih, NY; Piwinski, JJ. Challenges in modern drug discovery: a case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection. Acc. Chem. Res 2008, 41, 50–59. [Google Scholar]
- Luger, TA; Scholzen, TE; Brzoska, T; Bohm, M. New insights into the functions of alpha-MSH and related peptides in the immune system. Ann. N.Y. Acad. Sci 2003, 994, 133–140. [Google Scholar]
- Yang, ZW; Yang, G; Zu, YG; Fu, YJ; Zhou, LJ. The conformational analysis and proton transfer of the neuraminidase inhibitors: a theoretical study. Phys. Chem. Chem. Phys 2009, 11, 10035–10041. [Google Scholar]
- Yang, Z; Nie, Y; Yang, G; Zu, Y; Fu, Y; Zhou, L. Synergistic effects in the designs of neuraminidase ligands: Analysis from docking and molecular dynamics studies. J. Theor. Biol 2010, 267, 363–374. [Google Scholar]
- Masukawa, KM; Kollman, PA; Kuntz, ID. Investigation of Neuraminidase-Substrate Recognition Using Molecular Dynamics and Free Energy Calculations. J. Med. Chem 2003, 46, 5628–5637. [Google Scholar]
- Bonnet, P; Bryce, RA. Molecular dynamics and free energy analysis of neuraminidase-ligand interactions. Protein Sci 2004, 13, 946–957. [Google Scholar]
- Bonnet, P; Bryce, RA. Scoring binding affinity of multiple ligands using implicit solvent and a single molecular dynamics trajectory: Application to Influenza neuraminidase. J. Mol. Graph. Model 2005, 24, 147–156. [Google Scholar]
- Stoll, V; Stewart, KD; Maring, CJ; Muchmore, S; Giranda, V; Gu, Y-GY; Wang, G; Chen, Y; Sun, M; Zhao, C; Kennedy, AL; Madigan, DL; Xu, Y; Saldivar, A; Kati, W; Laver, G; Sowin, T; Sham, HL; Greer, J; Kempf, D. Influenza neuraminidase inhibitors: structure-based design of a novel inhibitor series. Biochemistry 2003, 42, 718–727. [Google Scholar]
- Yang, G; Yang, ZW; Zu, YG; Wu, XM; Fu, YJ. The calcium ion and conserved water molecules in neuraminidases: roles and implications for substrate binding. Internet. Electron. J. Mol. Des 2008, 7, 97–113. [Google Scholar]
- Amaro, RE; Cheng, X; Ivanov, I; Xu, D; McCammon, JA. Characterizing Loop Dynamics and Ligand Recognition in Human- and Avian-Type Influenza Neuraminidases via Generalized Born Molecular Dynamics and End-Point Free Energy Calculations. J. Am. Chem. Soc 2009, 131, 4702–4709. [Google Scholar]
- Colman, PM. Influenza virus neuraminidase: structure, antibodies, and inhibitors. Protein Sci 1994, 3, 1687–1696. [Google Scholar]
- Yang, ZW; Zu, YG; Wu, XM; Liu, CB; Yang, G. A computational investigation on the interaction mechanisms of neuraminidases and 3-(3-pentyloxy)benzoic acid. Acta. Chimica. Sinica 2010, 14, 1370–1378. [Google Scholar]
- InisghtII, 2005; Accelrys Inc: San Diego, CA, USA, 2005.
- Wall, ID; Leach, AR; Salt, DW; Ford, MG; Essex, JW. Binding constants of neuraminidase inhibitors: An investigation of the linear interaction energy method. J. Med. Chem 1999, 42, 5142–5152. [Google Scholar]
- Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Montgomery, JA; Vreven, T, Jr; Kudin, KN; Burant, JC; et al. Gaussian 03, Revision D01; Gaussian, Inc: Wallingford, CT, USA, 2004. [Google Scholar]
- Yang, ZW; Wu, XM; Zhou, LJ; Yang, G. A proline-based neuraminidase inhibitor: DFT studies on the zwitterion conformation, stability and formation. Int. J. Mol. Sci 2009, 10, 3918–3930. [Google Scholar]
- Affinity User Guide; Accelrys Inc: San Diego, CA, USA, 2005.
- Jorgensen, WL; Chandrasekhar, J; Madura, JD; Impey, RW; Klein, ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- Adelman, SA; Doll, JD. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J. Chem. Phys 1976, 64, 2375–2388. [Google Scholar]
- Shamovsky, IL; Ross, GM; Riopelle, RJ. Theoretical studies on the origin of β-sheet twisting. J. Phys. Chem. B 2000, 104, 11296–11307. [Google Scholar]
- Bouř, P; Buděšínský, M; Špirko, V; Kapitán, J; Šebestík, J; Sychrovský, V. A complete set of NMR chemical shifts and spin−spin coupling constants for l-alanyl-l-alanine zwitterion and analysis of its conformational behavior. J. Am. Chem. Soc 2005, 127, 17079–17089. [Google Scholar]
- Kang, YK; Scheraga, HA. An efficient method for calculating atomic charges of peptides and proteins from electronic populations. J. Phys. Chem. B 2008, 112, 5470–5478. [Google Scholar]
- LaPointe, SM; Farrag, S; Bohórque, HJ; Boyd, RJ. QTAIM study of an α-helix hydrogen bond network. J. Phys. Chem. B 2009, 113, 10957–10964. [Google Scholar]
- Yang, G; Zu, Y; Fu, Y; Zhou, L; Zhu, R; Liu, C. Assembly and stabilization of multi-amino acid zwitterions by the Zn(II) ion: a computational exploration. J. Phys. Chem. B 2009, 113, 4899–4906. [Google Scholar]
- Yang, G; Xing, C; Liu, C-B; Fu, Y-J; Zhou, L-J; Zu, Y-G. First-principle conformational analysis of glycine residues in the αβ-tubulin dimer. Interdiscip. Sci.: Comput. Life Sci 2009, 1, 196–203. [Google Scholar]
- Yang, G; Zu, Y; Liu, C; Fu, Y; Zhou, L. Stabilization of amino acid zwitterions with varieties of anionic species: the intrinsic mechanism. J. Phys. Chem. B 2008, 112, 7104–7110. [Google Scholar]


© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, Z.; Yang, G.; Zu, Y.; Fu, Y.; Zhou, L. Computer-Based De Novo Designs of Tripeptides as Novel Neuraminidase Inhibitors. Int. J. Mol. Sci. 2010, 11, 4932-4951. https://doi.org/10.3390/ijms11124932
Yang Z, Yang G, Zu Y, Fu Y, Zhou L. Computer-Based De Novo Designs of Tripeptides as Novel Neuraminidase Inhibitors. International Journal of Molecular Sciences. 2010; 11(12):4932-4951. https://doi.org/10.3390/ijms11124932
Chicago/Turabian StyleYang, Zhiwei, Gang Yang, Yuangang Zu, Yujie Fu, and Lijun Zhou. 2010. "Computer-Based De Novo Designs of Tripeptides as Novel Neuraminidase Inhibitors" International Journal of Molecular Sciences 11, no. 12: 4932-4951. https://doi.org/10.3390/ijms11124932