Theoretical Study for High-Energy-Density Compounds Derived from Cyclophosphazene. IV. DFT Studies on 1,1-Diamino-3,3,5,5,7,7-hexaazidocyclotetraphosphazene and Its Isomers

Abstract
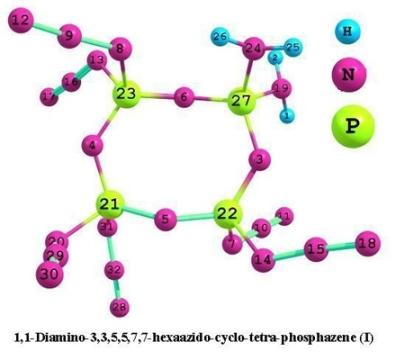
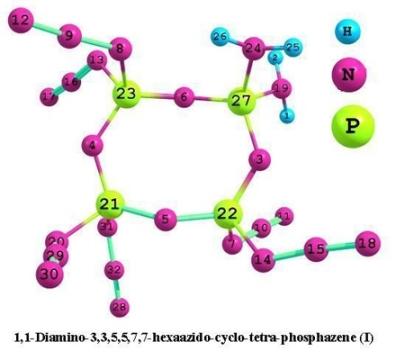
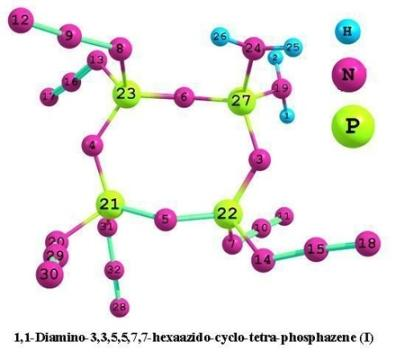
:
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Structural analysis
3.2. Vibrational analysis
3.3. NBO analysis
3.4. Mulliken overlaps population analysis and the atomic charges
3.5. Heats of formation from computed atomization energies
3.6. Relative total energies and the frontier orbital energies for the compounds I–V
4. Conclusions
Acknowledgments
References
- Huynh, MV; Hiskey, MA; Hartline, EL; Montoyo, DP; Gilardi, R. Polyazido high nitrogen compounds: Hydrazo- and azo-1,3,5-triazine. Angew. Chem. Int. Ed 2004, 43, 4924–4928. [Google Scholar]
- Krause, HH. Energetic Materials; Teipel, U, Ed.; Wiley-VCH. Weinheim: Berlin, Germany, 2005; pp. 1–25. [Google Scholar]
- Hammerl, A; Klapotke, TM. Tetrazolylpentazoles: Nitrogen-rich compounds. Inorg. Chem 2002, 41, 906–912. [Google Scholar]
- Chang, MS; Matuszko, AJ. Synthesis of triphosphonitrilic diamidetetraazide. J. Am. Chem 1960, 82, 5756–5757. [Google Scholar]
- Shaw, RA. Aspects of cyclic and acyclic phosphorus-nitrogen compounds. Pure Appl. Chem 1980, 22, 1063–1097. [Google Scholar]
- Allcock, HR. Recent advances in phosphazene (phosphonitrilic) chemistry. Chem. Rev 1972, 72, 315–356. [Google Scholar]
- Fincham, JK; Hursthouse, MB; Parker, HG; Shaw, RA; Shaw, LS. Phosphorus–nitrogen compounds. Part 54. J Chem Soc Dalton Trans 1988, 1169–1178. [Google Scholar]
- Chaplin, AB; Harrison, JA; Dyson, PJ. Revisiting the electronic structure of phosphazenes. Inorg. Chem 2005, 44, 8407–8417. [Google Scholar]
- Forohar, F; Dave, PR; Iyer, S. Substituted cyclotetraphophazene compound and method of producing the same. 2001. [Google Scholar]
- Zhang, JG; Zheng, HH; Bi, YG; Zhang, TL; Yang, L; Feng, LN. High energy density compounds from cyclophospazene. II. The preparation, structural characterization, and theoretical studies of 1,1-spiro(ethylenediamino)-3,3,5,5-tetrachlorocyclotriphosphazene and its nitration product. Struc. Chem 2008, 19, 297–305. [Google Scholar]
- Zhang, JG; Zheng, HH; Zhang, TL; Yang, L; Feng, LN. Theoretical study for high energy density compounds from cyclophosphazene. III. A quantum chemistry study: High nitrogen-contented energetic compound of 1,1,3,3,5,5,7,7-octaazidocyclotetraphosphazene: N4P4(N3)8. Inorg. Chim. Acta 2008, 361, 4143–4147. [Google Scholar]
- Huang, HS; Zhang, JG; Zhang, TL; Yang, L; Zheng, HH. High energy density compounds from cyclophospazene. I. Ab initio study of electronic structure and properties in crystalline 1,1,3,3,5,5-hexaazidocyclotriphosphazene. Chin. J. Chem 2008, 26, 845–858. [Google Scholar]
- Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Zakrzewski, VG; Montgomery, JA; Stratmann, RE; Burant, JC; Dapprich, S; Millam, JM; Daniels, AD; Kudin, KN; Strain, MC; Farkas, O; Tomasi, J; Barone, V; Cossi, M; Cammi, R; Mennucci, B; Pomelli, C; Adamo, C; Clifford, S; Ochterski, J; Petersson, GA; Ayala, PY; Cui, Q; Morokuma, K; Malick, DK; Rabuck, AD; Raghavachari, K; Foresman, JB; Cioslowski, J; Ortiz, JV; Baboul, AG; Stefanov, BB; Liu, G; Liashenko, A; Piskorz, P; Komaromi, I; Gomperts, R; Martin, RL; Fox, DJ; Keith, T; Al-Laham, MA; Peng, CY; Nanayakkara, A; Challacombe, M; Gill, PMW; Johnson, B; Chen, W; Wong, MW; Andres, JL; Gonzalez, C; Head-Gordon, M; Replogle, ES; Pople, JA. Gaussian 03, Revision D4; Gaussian, Inc: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Becke, AD. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- Reed, AE; Weinstock, RB; Weinhold, FJ. Natural population analysis. J. Chem. Phys 1985, 83, 735–746. [Google Scholar]
- Enlow, M. Ab-initio studies of cyclic phosphazine systems (NPX2)n. A study of the structure and bonding in such systems and a search for model systems for the polymer. Polyhedron 2003, 22, 473–482. [Google Scholar]
- Gall, M; Breza, M. On the structure of hexahydroxocyclotriphosphazene. J. Mol. Struct. (Theohem) 2008, 861, 33–38. [Google Scholar]
- Allcock, HR. Phosphorus-Nitrogen Compounds; Academic Press: New York, NY, USA, 1972. [Google Scholar]
- Sabzyan, H; Kalantar, Z. Ab initio RHF and density functional B3LYP and B3PW91 study of (NPF2)n; n=2; 3; 4 and (NPX2)3; X=H, Cl, Br cyclic phosphazenes. J. Mol. Struct. (Theohem) 2003, 663, 149–157. [Google Scholar]
- Lee, C; Yang, W; Parr, RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Curtiss, LA; Raghavachari, K; Redfern, PC; Pople, JA. Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J. Chem. Phys 1997, 106, 1063–1079. [Google Scholar]
- Chase, MW, Jr; Davies, CA; Downey, JR, Jr; Frurip, DJ; McDonald, RA; Syverud, AN. JANAF Thermochemical Tables, Third Edition. J. Phys. Chem. Ref. Data 1985, 14, 1–1856. [Google Scholar]
- Gobel, M; Karaghiosoff, K; Klapotke, TM. The first structural characterization of a binary P-N molecule: The highly energetic compound P3N21. Angew. Chem. Int. Ed 2006, 45, 6037–6040. [Google Scholar]
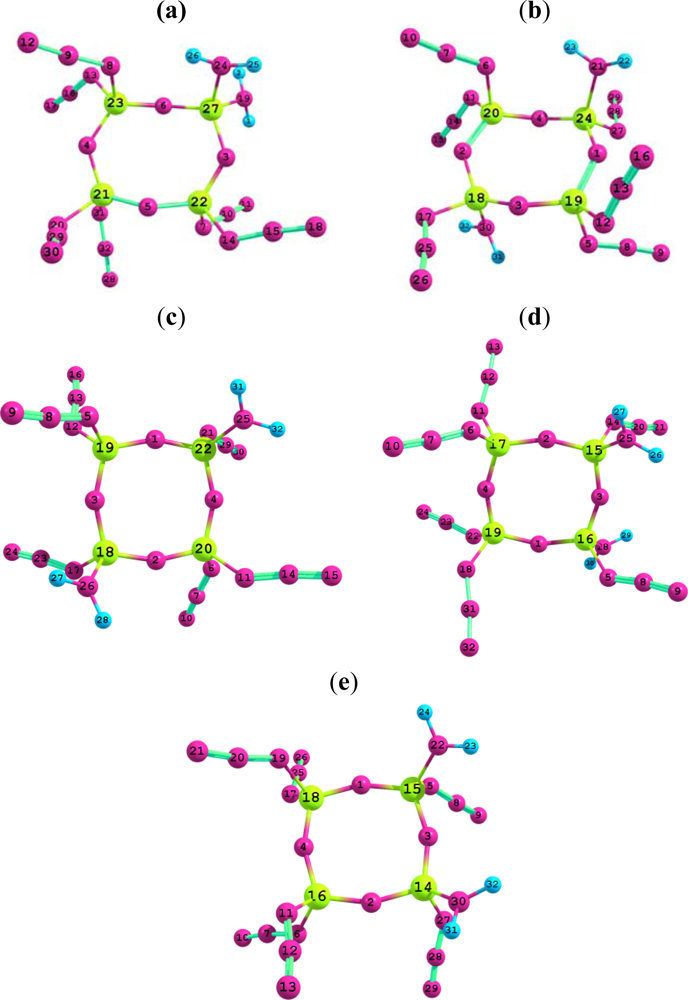

{kind=link}
{kind=link}
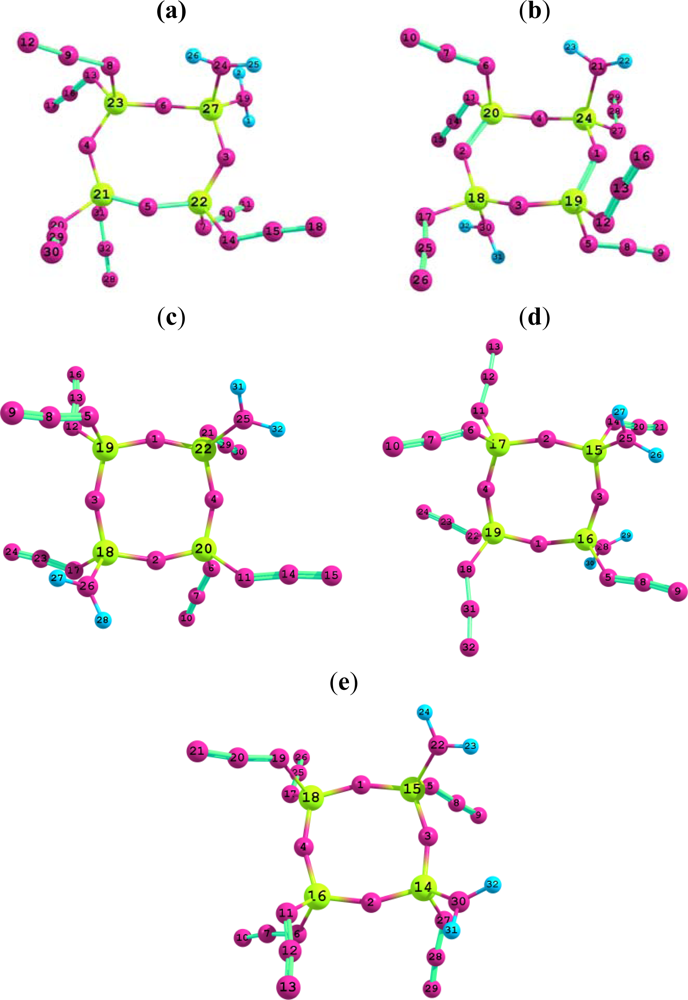{kind=link}
| Structure parameter | B3LYP/6-31G* | B3LYP/6-31G** | B3PW91/6-31G* | B3PW91/6-31G** |
|---|---|---|---|---|
| Bond lengths | ||||
| N4-P23 | 1.599 | 1.599 | 1.596 | 1.596 |
| N3-P27 | 1.621 | 1.621 | 1.619 | 1.619 |
| N3-P22 | 1.580 | 1.579 | 1.577 | 1.578 |
| N6-P27 | 1.609 | 1.610 | 1.606 | 1.605 |
| N5-P22 | 1.583 | 1.583 | 1.580 | 1.581 |
| P21-N4 | 1.592 | 1.592 | 1.589 | 1.589 |
| P21-Nα | 1.709 | 1.723 | 1.716 | 1.716 |
| Nα-Nβ | 1.240 | 1.240 | 1.235 | 1.235 |
| P27-NH2 | 1.667 | 1.666 | 1.663 | 1.661 |
| Nβ-Nγ | 1.135 | 1.135 | 1.133 | 1.133 |
| N19-H1 | 1.015 | 1.013 | 1.014 | 1.012 |
| Bond angles | ||||
| P21-N4-P23 | 135.9 | 135.9 | 135.4 | 135.4 |
| N4-P21-N5 | 120.7 | 120.6 | 120.5 | 120.5 |
| Nα-P21-Nα | 101.4 | 101.4 | 101.5 | 101.5 |
| P21-Nα-Nβ | 118.3 | 118.3 | 118.3 | 118.3 |
| Nα-Nβ-Nγ | 174.2 | 173.7 | 174.2 | 174.2 |
| H1-N19-H2 | 113.2 | 113.6 | 113.4 | 113.9 |
| N3-P22-N5-P21 | 68.9 | 68.8 | 68.5 | 68.4 |
| P22-N5-P21-N4 | −70.1 | −70.0 | −70.3 | −70.3 |
| N5-P21-N4-P23 | 26.1 | 26.1 | 26.3 | 26.4 |
| P21-N4-P23-N6 | 20.8 | 20.7 | 20.3 | 20.5 |
| N4-P23-N6-P27 | −87.1 | −87.2 | −88.0 | −87.9 |
| P23-N6-P27-N3 | 75.9 | 75.7 | 75.3 | 75.7 |
| ν | I | II | III | IV | V | Assignment |
|---|---|---|---|---|---|---|
| 1 | 390.2 (96) | 406.4 (57) | 478.4(129) | P-N ring in-plane twist | ||
| 2 | 540.9 (184)
550.2 (211) | 542.2 (215)
552.4 (76) | P-N ring twist
N-H bending, –N3 twist | |||
| 3 | 563.6 (294) | 576.1 (106) | 563.1(214) | 556.5(188) | −N3 torsion, N-H bending | |
| 4 | 578.8 (22)
612.5 (234) | 590.9 (238)
608.8 (230) | 610.8(418) | −N3 twist, torsion | ||
| 5 | 689.6 (266) | 754.4 (210) | 709.4(275) | 758.7(397) | Nα-Nβ in plane twist | |
| 6 | 802.2 (176) | 821.1 (196) | N-H unsymmetrical twist | |||
| 7 | 939.6 (126) | 923.4 (217) | 920.3(149) | 913.7(170) | 915.9(129) | P-NH2 in-plane stretching |
| 8 | 1,034.3 (68)
1,049.9 (75) | 1,020.1 (25)
1,014.2 (53) | 941.7(148) | N-H in-plane unsymmetrical wag | ||
| 9 | 1,266.3 (1689) | 1,281.8 (1775) | 1,270.4(1847) | 1,293.2(1980) | 1,296.6(2017) | P-N-P in-plane stretching |
| 10 | 1,313.8 (298)
1,318.7 (380) 1,326.1 (337) 1,341.8 (104) | 1,314.8 (134)
1,321.0 (461) 1,336.8 (431) 1,349.2 (128) | 1,338.3(318) | 1,315.5(1282)
1,322.9(479) | 1,319.0(380) | Nα-Nγ symmetrical stretching |
| 11 | 1,330.3 (1155) | 1,307.8 (1553) | 1,311.0(1096) | 1,326.7(389) | 1,299.3(2080) | P-N-P in-plane stretching |
| 12 | 1,371.5 (748) | 1,364.5 (586) | 1,367.2(632) | 1,380.8(915) | P-N-P symmetrical stretching | |
| 13 | 1,592.4 (100) | 1,587.5 (95) | −NH2 in-plane bending | |||
| 14 | 2,277.9 (533)
2,299.4 (607) 2,309.1 (1046) | 2,295.1 (213)
2,295.4 (1004) 2,300.0 (1078) | 2,292.2(359)
2,295.8(588) 2,308.1(858) | 2,286.9(781)
2,296.4(1006) 2,300.1(730) | 2,286.7(730)
2,294.7(496) 2,300.8(631) | Nβ-Nγ unsymmetrical stretching |
| 15 | 3,542.0 (52)
3,566.2 (37) | 3,539.0 (47)
3,562.6 (41) | 3,562.9(66) | 3,530.4(61) | 3,574.3(56) | N-H symmetrical stretching |
| 16 | 3,660.2 (66)
3,679.4 (39) | 3,651.2 (57)
3,680.7 (36) | 3,681.1(35) | 3,683.9(38) | 3,694.5(40) | N-H unsymmetrical stretching |
| Donor NBO (i) | Acceptor NBO (j) | E(2)/(kcal·mol−1) |
|---|---|---|
| LP(2)N7 | BD*(2)N10-N11 | 113.83 |
| LP(2)N7 | BD*(1)N5-P22 | 2.30 |
| LP(2)N7 | BD*(1)N3-P22 | 0.87 |
| LP(1)N7 | BD*(3)N10-N11 | 5.77 |
| LP(2)N8 | BD*(2)N9-N12 | 106.13 |
| LP(2)N8 | BD*(1)N4-P23 | 5.65 |
| LP(2)N8 | BD*(1)N6-P23 | 2.50 |
| LP(2)N13 | BD*(2)N16-N17 | 106.72 |
| LP(2)N14 | BD*(2)N15-N18 | 107.55 |
| LP(2)N20 | BD*(2)N29-N30 | 107.18 |
| LP(2)N31 | BD*(2)N28-N32 | 104.22 |
| BD(1)N20-P21 | BD*(1)N5-P22 | 2.63 |
| BD(1)N20-P21 | BD*(1)N4-P23 | 1.77 |
| BD(1)N19-P27 | BD*(1)N6-P23 | 1.76 |
| BD(1)N19-P27 | BD*(1)N3-P22 | 1.49 |
| BD(1)N3-P22 | BD*(1)N5-P21 | 0.88 |
| BD(1)N4-P21 | BD*(1)N5-P21 | 1.01 |
| BD(1)N7-P22 | BD*(3)N10-N11 | 17.97 |
| BD(1)N8-P23 | BD*(3)N9-N12 | 16.35 |
| LP(2)N3 | BD*(1)N7-P22 | 23.17 |
| LP(2)N3 | BD*(1)N24-P27 | 12.98 |
| LP(2)N4 | BD*(1)N8-P23 | 19.13 |
| LP(2)N4 | BD*(1)P21-N31 | 15.62 |
| LP(1)N19 | BD*(1)N6-P27 | 12.95 |
| LP(1)N24 | BD*(1)N3-P27 | 12.40 |
| BD*(1)N4-P23 | BD*(1)N6-P23 | 24.07 |
| BD*(1)N6-P27 | BD*(1)N6-P23 | 53.73 |
| BD*(1)N3-P27 | BD*(1)N3-P22 | 46.89 |
| Compounds | Donor NBO (i) | Acceptor NBO (j) | E(2)/(kcal·mol−1) |
|---|---|---|---|
| trans-1,5-diamino-1,3,3,5,7,7-hexaazidocyclotetraphosphazene (II) | |||
| BD(1)N1-P19 | BD*(2)N5-P19 | 47.76 | |
| BD(1)N1-P19 | BD*(1)N12-P19 | 30.31 | |
| BD(1)N3-P19 | BD*(2)N12-P19 | 82.96 | |
| BD(1)N3-P19 | BD*(1)N5-P19 | 20.46 | |
| BD(1)N2-P20 | BD*(2)N6-N20 | 50.05 | |
| BD(1)N2-P20 | BD*(1)N11-P20 | 32.63 | |
| BD(1)N2-P18 | BD*(1)N17-P18 | 2.08 | |
| BD(1)N1-P22 | BD*(1)N21-P22 | 4.97 | |
| BD(1)N4-P22 | BD*(1)N21-P22 | 1.44 | |
| BD(1)N4-P22 | BD*(2)N21-P22 | 94.77 | |
| BD*(1)N5-P19 | BD*(2)N12-P19 | 109.85 | |
| BD*(1)N25-P22 | BD*(2)N21-P22 | 34.99 | |
| LP(2)N17 | BD*(2)N23-N24 | 111.66 | |
| cis-1,5-diamino-1,3,3,5,7,7-hexaazidocycl-tetraphosphazene (III) | |||
| BD(1)N2-P20 | BD*(2)N6-N20 | 49.58 | |
| BD(1)N2-P20 | BD*(1)N11-P20 | 32.61 | |
| BD(1)N4-P20 | BD*(2)N11-P20 | 80.24 | |
| BD(1)N3-P19 | BD*(2)N12-P19 | 84.57 | |
| BD(1)N1-P19 | BD*(2)N5-P19 | 50.43 | |
| BD(1)P18-N3 | BD*(2)N17-P18 | 11.84 | |
| BD(1)P18-N3 | BD*(1)N30-P18 | 11.37 | |
| BD(1)N1-P24 | BD*(1)N21-P24 | 3.80 | |
| BD(1)N4-P24 | BD*(2)N27-P24 | 55.88 | |
| BD(2)N5-P19 | BD*(2)N8-N9 | 95.82 | |
| BD*(2)N17-P18 | BD*(1)N30-P18 | 72.58 | |
| BD*(2)P24-N27 | BD*(1)N21-P24 | 116.83 | |
| Compounds | Donor NBO (i) | Acceptor NBO (j) | E(2)/(kcal·mol−1) |
|---|---|---|---|
| trans-1,3-diamino-1,3,5,5,7,7-hexaazidocyclotetraphosphazene (IV) | |||
| LP(2)N5 | BD*(2)N8-N9 | 110.94 | |
| LP(2)N18 | BD*(2)N31-N32 | 108.18 | |
| LP(2)N6 | BD*(2)N7-N10 | 105.75 | |
| BD*(1)N1-P16 | BD*(1)N1-P19 | 102.54 | |
| BD*(1)N2-P15 | BD*(1)N2-P17 | 97.4 | |
| BD*(1)N5-P16 | BD*(1)P16-N28 | 2.28 | |
| BD*(1)N14-P15 | BD*(1)P15-N25 | 3.95 | |
| BD*(1)P16-N28 | BD*(1)N3-P15 | 3.28 | |
| BD*(1)N18-P19 | BD*(1)P19-N22 | 4.11 | |
| BD*(1)P19-N22 | BD*(1)N1-P16 | 2.08 | |
| BD*(1)P19-N22 | BD*(1)N4-P17 | 3.27 | |
| BD*(3)N23-N24 | BD*(1)N23-N24 | 9.52 | |
| BD*(3)N23-N24 | BD*(1)P19-N22 | 6.03 | |
| cis-1,3-diamino-1,3,5,5,7,7-hexaazidocyclotetraphosphazene (V) | |||
| BD(1)N17-P18 | BD*(1)N19-P18 | 3.20 | |
| BD(1)P14-N27 | BD*(1)N30-P14 | 3.21 | |
| BD(1)N5-P15 | BD*(1)N22-P15 | 3.22 | |
| BD(1)N5-P15 | BD*(1)N5-P19 | 20.46 | |
| BD(1)N5-P15 | BD*(3)N8-N9 | 16.04 | |
| LP(2)N19 | BD*(2)N20-N21 | 105.23 | |
| LP(1)N22 | BD*(1)N5-P15 | 12.77 | |
| LP(2)N27 | BD*(2)N28-N29 | 112.74 | |
| LP(2)N27 | BD*(1)N30-P14 | 10.01 | |
| LP(1)N30 | BD*(1)N27-P14 | 14.36 | |
| BD*(1)N1-P15 | BD*(1)N1-P18 | 99.55 | |
| BD*(1)N2-P14 | BD*(1)N2-P16 | 90.33 | |
| BD*(3)N25-N26 | BD*(1)N17-P18 | 7.12 | |
| BD*(3)N28-N29 | BD*(1)N28-N29 | 10.3 | |
| Isomers | P-N(ring) | P-Nα | Nα-Nβ | Nβ-Nγ | P-N(amino) | N-H |
|---|---|---|---|---|---|---|
| I | 0.383 ~ 0.510 | 0.201 ~ 0.275 | 0.265 ~ 0.301 | 0.586 ~ 0.601 | 0.350 ~ 0.353 | 0.341 ~ 0.345 |
| II | 0.442 ~ 0.501 | 0.210 ~ 0.271 | 0.264 ~ 0.294 | 0.597 ~ 0.601 | 0.306 ~ 0.352 | 0.341 ~ 0.344 |
| III | 0.445 ~ 0.498 | 0.219 ~ 0.276 | 0.266 ~ 0.303 | 0.595 ~ 0.597 | 0.310 ~ 0.353 | 0.341 ~ 0.347 |
| IV | 0.454 ~ 0.495 | 0.217 ~ 0.273 | 0.272 ~ 0.313 | 0.594 ~ 0.600 | 0.295 ~ 0.343 | 0.338 ~ 0.344 |
| V | 0.438 ~ 0.493 | 0.207 ~ 0.268 | 0.264 ~ 0.327 | 0.596 ~ 0.599 | 0.320 ~ 0.342 | 0.343 ~ 0.346 |
| Isomers | I | II | III | IV | V |
|---|---|---|---|---|---|
| P (ring) | 1.070 ~ 1.089
1.078 | 1.062 ~ 1.072
1.068 | 1.061 ~ 1.071
1.066 | 1.057 ~ 1.068
1.063 | 1.062 ~ 1.070
1.067 |
| N (ring) | −0.683 ~ −0.602
−0.640 | −0.639 ~ −0.630
−0.635 | −0.649 ~ −0.622
−0.635 | −0.683 ~ −0.619
−0.636 | −0.645 ~ −0.621
−0.633 |
| Nα | −0.537 ~ −0.472
−0.491 | −0.530 ~ −0.449
−0.476 | −0.475 ~ −0.471
−0.473 | −0.472 ~ −0.470
−0.471 | −0.472 ~ −0.469
−0.478 |
| Nβ | 0.458 ~ 0.480
0.465 | 0.437 ~ 0.460
0.444 | 0.438 ~ 0.452
0.447 | 0.435 ~ 0.453
0.447 | 0.437 ~ 0.453
0.448 |
| Nγ | −0.257 ~ −0.216
−0.228 | −0.233 ~ −0.211
−0.224 | −0.234 ~ −0.214
−0.228 | −0.251 ~ −0.213
−0.226 | −0.238 ~ −0.212
−0.223 |
| N (amino) | −0.861 ~ −0.818
−0.840 | −0.829 ~ −0.813
−0.821 | −0.821 ~ −0.821
−0.821 | −0.823 ~ −0.823
−0.823 | −0.823 ~ −0.820
−0.822 |
| H (amino) | 0.348 ~ 0.379
0.361 | 0.355 ~ 0.375
0.362 | 0.359 ~ 0.362
0.361 | 0.359 ~ 0.360
0.360 | 0.355 ~ 0.361
0.357 |
| Compounds | B3LYP/6-31G** | B3PW91/6-31G** | Expt [24] |
|---|---|---|---|
| I | 424.03 | 434.93 | ----- |
| II | 423.12 | 433.92 | ----- |
| III | 422.97 | 433.85 | ----- |
| IV | 422.86 | 433.65 | ----- |
| V | 421.74 | 432.47 | ----- |
| Hexaazidocyclotriphosphazene | 444.43 | 451.71 | 455.16 |
| Compounds | Etotal | ELUMO | EHOMO | ΔEL-H |
|---|---|---|---|---|
| I | 2.12 | −1.56 | −6.01 | 4.46 |
| II | 0.00 | −1.46 | −6.18 | 4.72 |
| III | 1.01 | −1.50 | −6.19 | 4.69 |
| IV | 1.15 | −1.47 | −6.06 | 4.59 |
| V | 1.29 | −1.54 | −6.12 | 4.58 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, J.; Zheng, H.; Zhang, T.; Wu, M. Theoretical Study for High-Energy-Density Compounds Derived from Cyclophosphazene. IV. DFT Studies on 1,1-Diamino-3,3,5,5,7,7-hexaazidocyclotetraphosphazene and Its Isomers. Int. J. Mol. Sci. 2009, 10, 3502-3516. https://doi.org/10.3390/ijms10083502
Zhang J, Zheng H, Zhang T, Wu M. Theoretical Study for High-Energy-Density Compounds Derived from Cyclophosphazene. IV. DFT Studies on 1,1-Diamino-3,3,5,5,7,7-hexaazidocyclotetraphosphazene and Its Isomers. International Journal of Molecular Sciences. 2009; 10(8):3502-3516. https://doi.org/10.3390/ijms10083502
Chicago/Turabian StyleZhang, Jianguo, Huihui Zheng, Tonglai Zhang, and Man Wu. 2009. "Theoretical Study for High-Energy-Density Compounds Derived from Cyclophosphazene. IV. DFT Studies on 1,1-Diamino-3,3,5,5,7,7-hexaazidocyclotetraphosphazene and Its Isomers" International Journal of Molecular Sciences 10, no. 8: 3502-3516. https://doi.org/10.3390/ijms10083502