Computational Study on the Conformation and Vibration Frequencies of β-Sheet of ε-Polylysine in Vacuum

Abstract
:1. Introduction
2. Results and Discussion
2.1. Geometry and peptide combination of the ɛ-PLL double chains
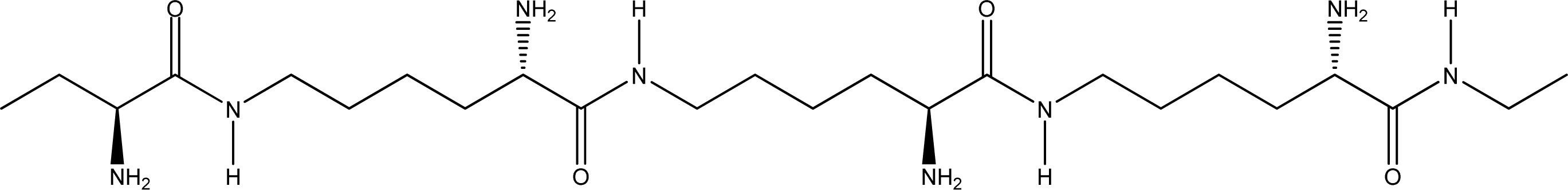
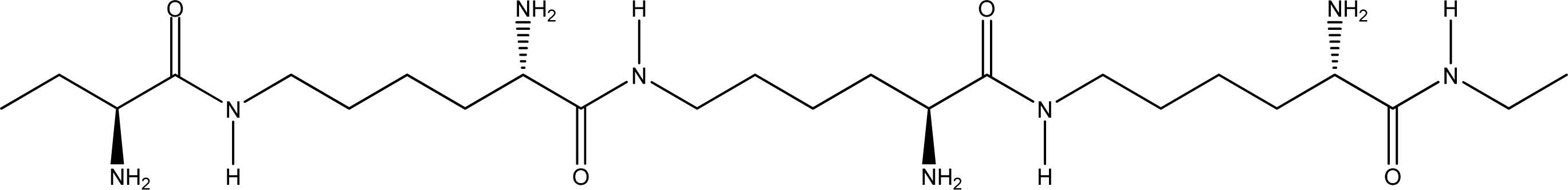
- Random coiled form - there is no order of the hydrogen bond combinations between the two ɛ-PLL molecules and the two chains are random coiled.
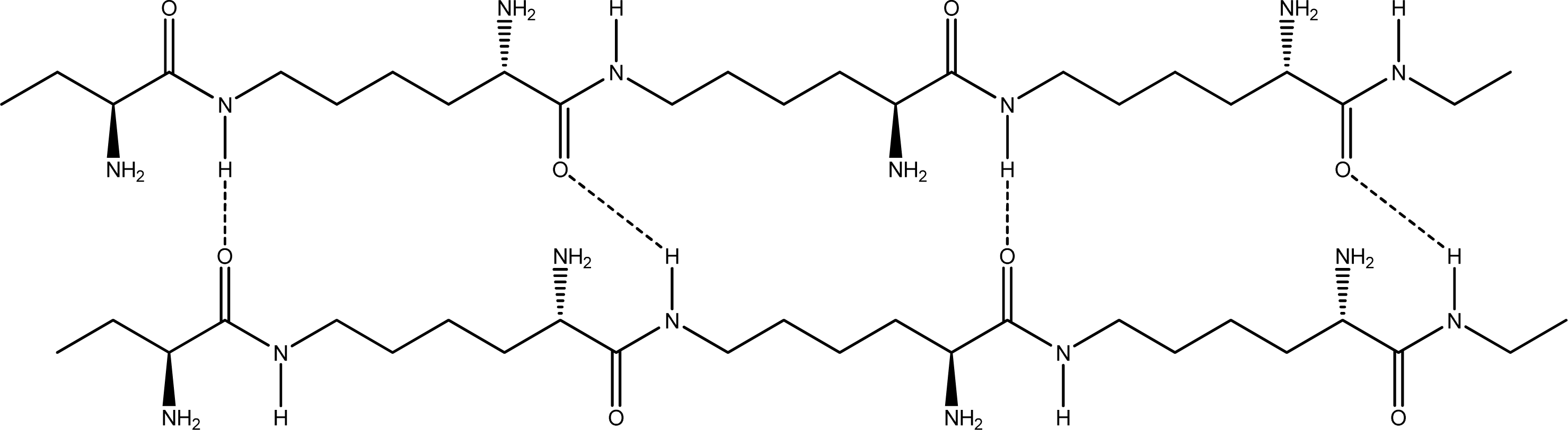
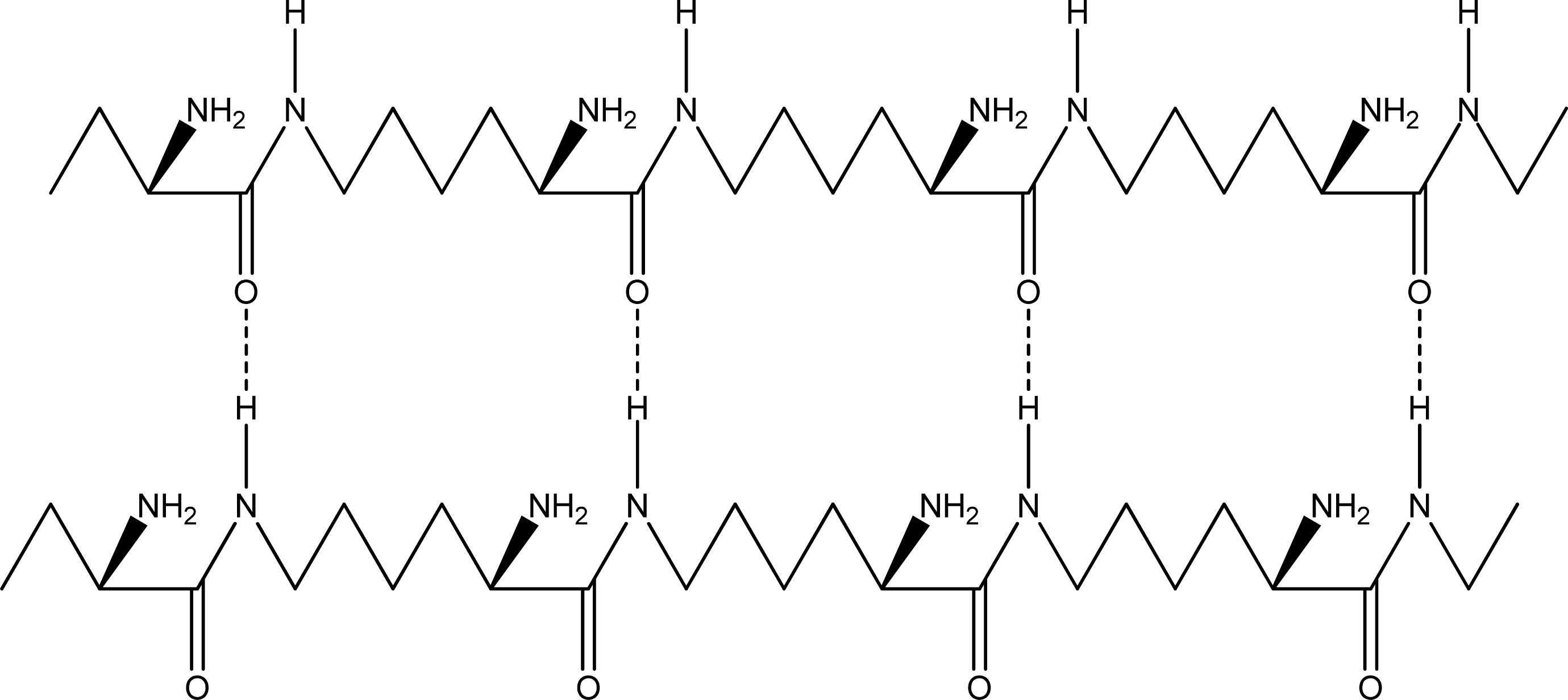
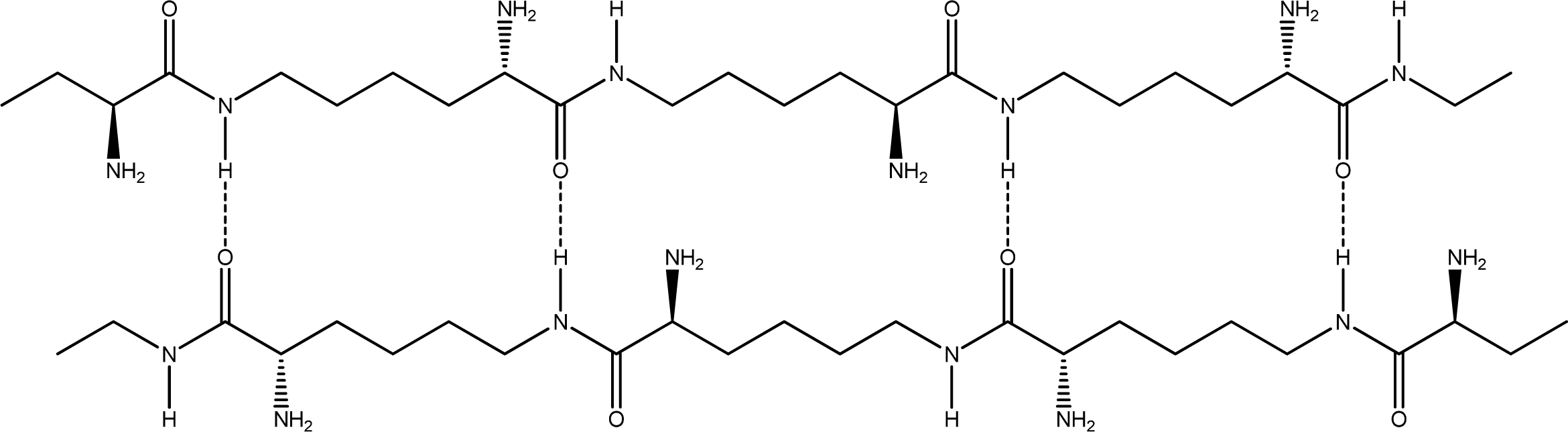
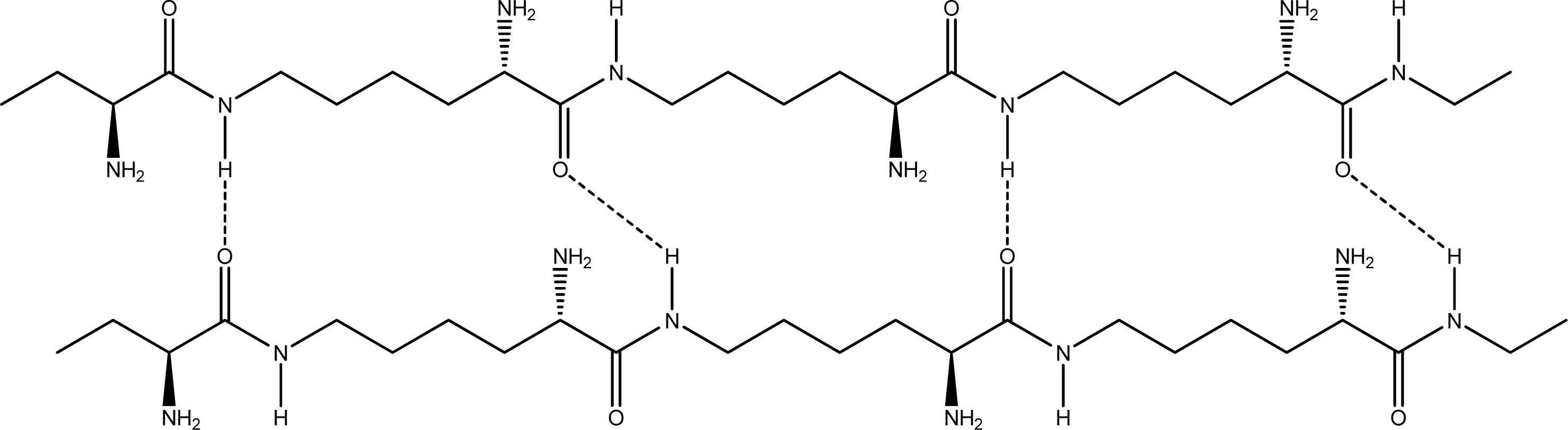
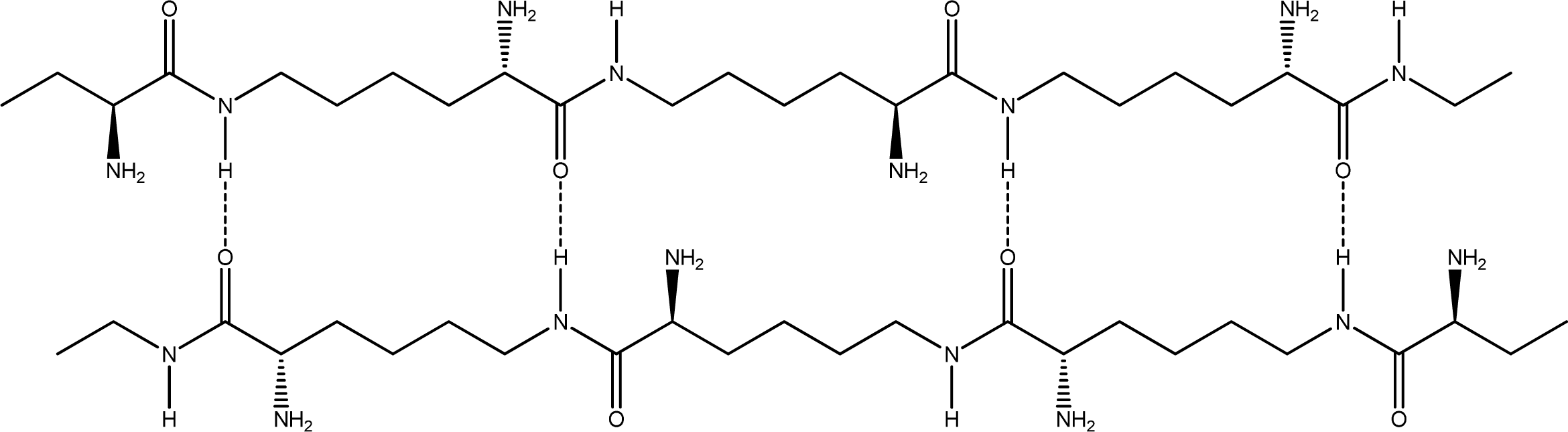
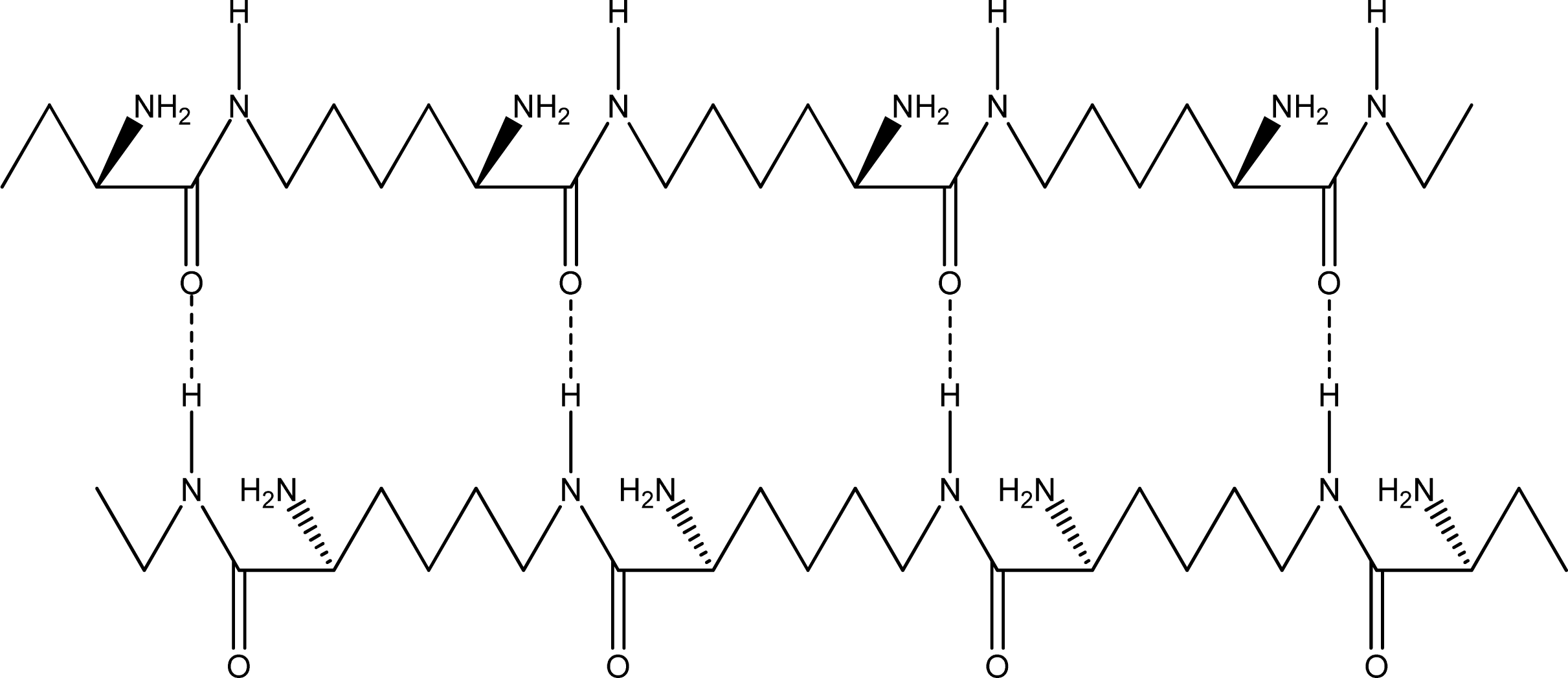
- Parallel β-sheet form - the two molecular chains are arranged in parallel. The C terminals of the double chains are at one end, while the N terminals are at the opposite [Figures 2 (a) and (b)].
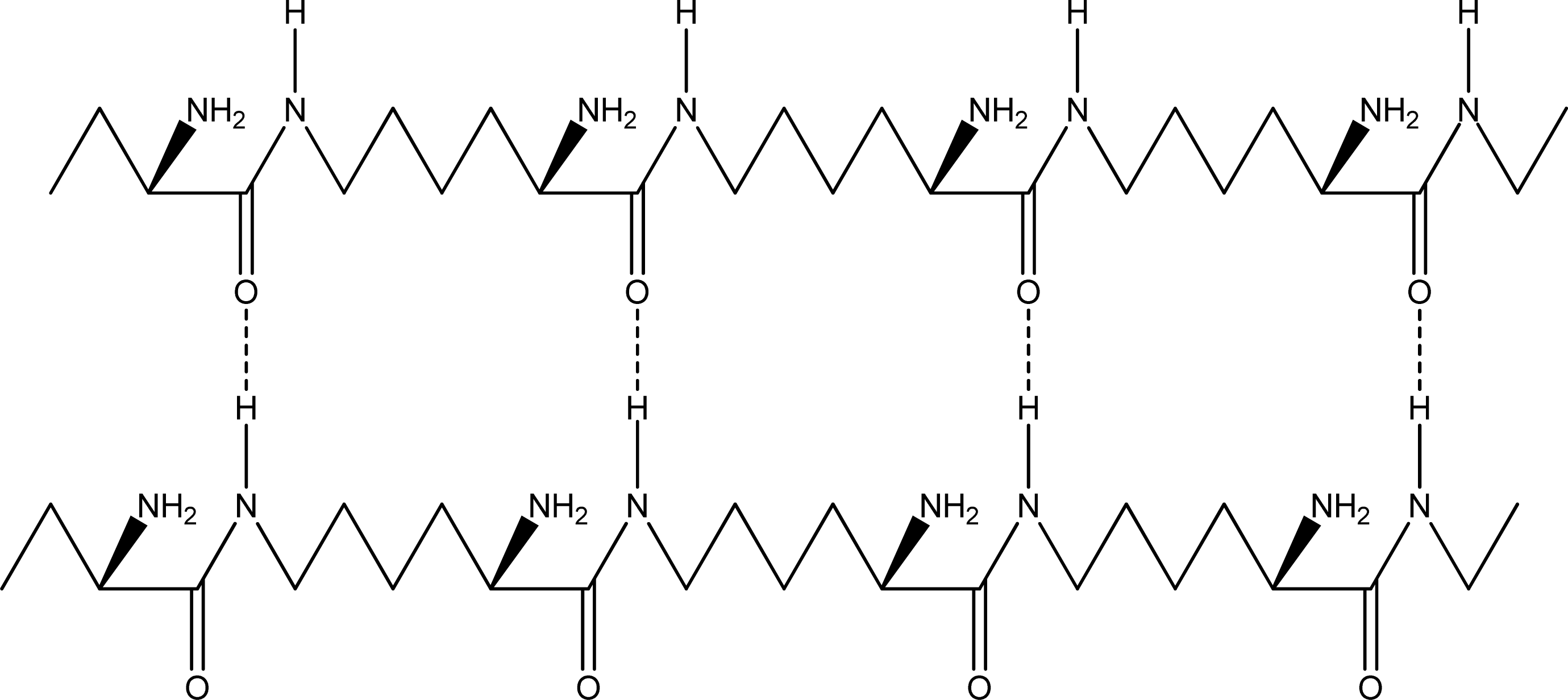
- Anti-parallel β-sheet form - the C terminal of one chain combines with the N terminal of the other one, and the two mono-chains are arranged in the opposite direction [Figures 3 (a) and (b)].
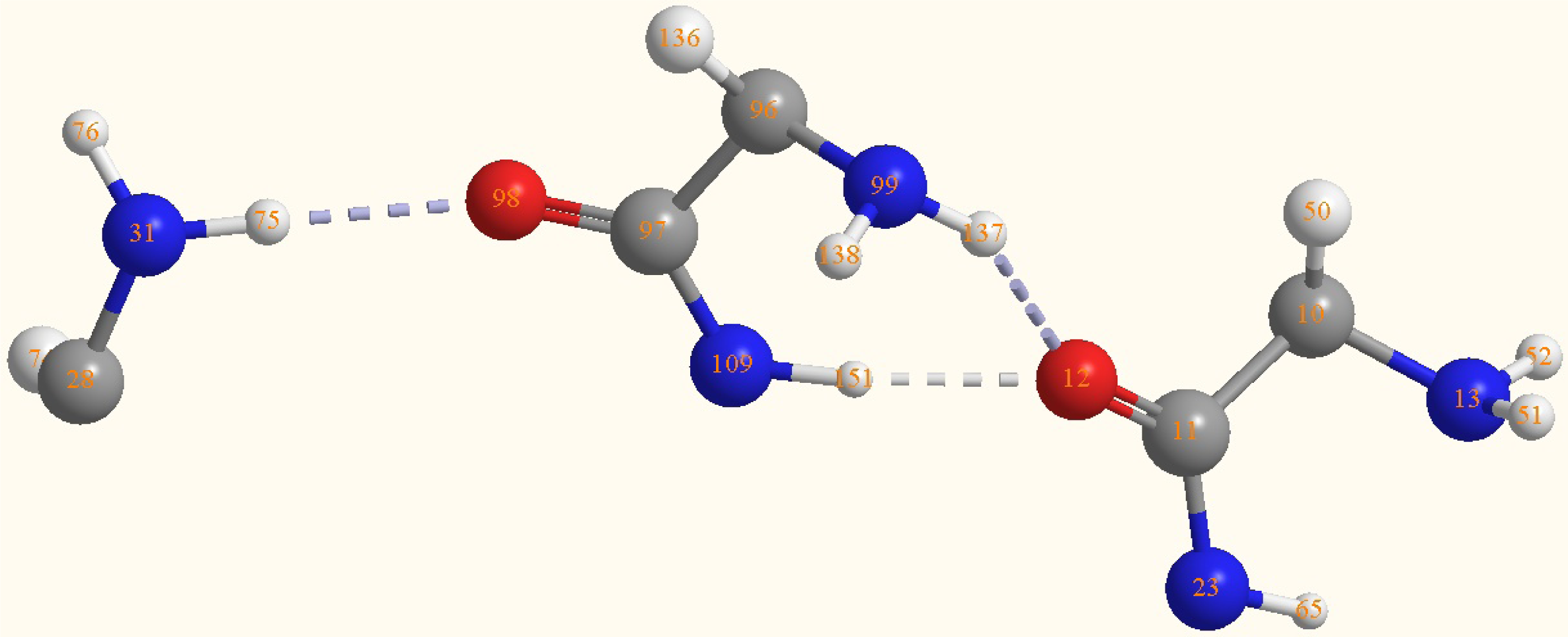
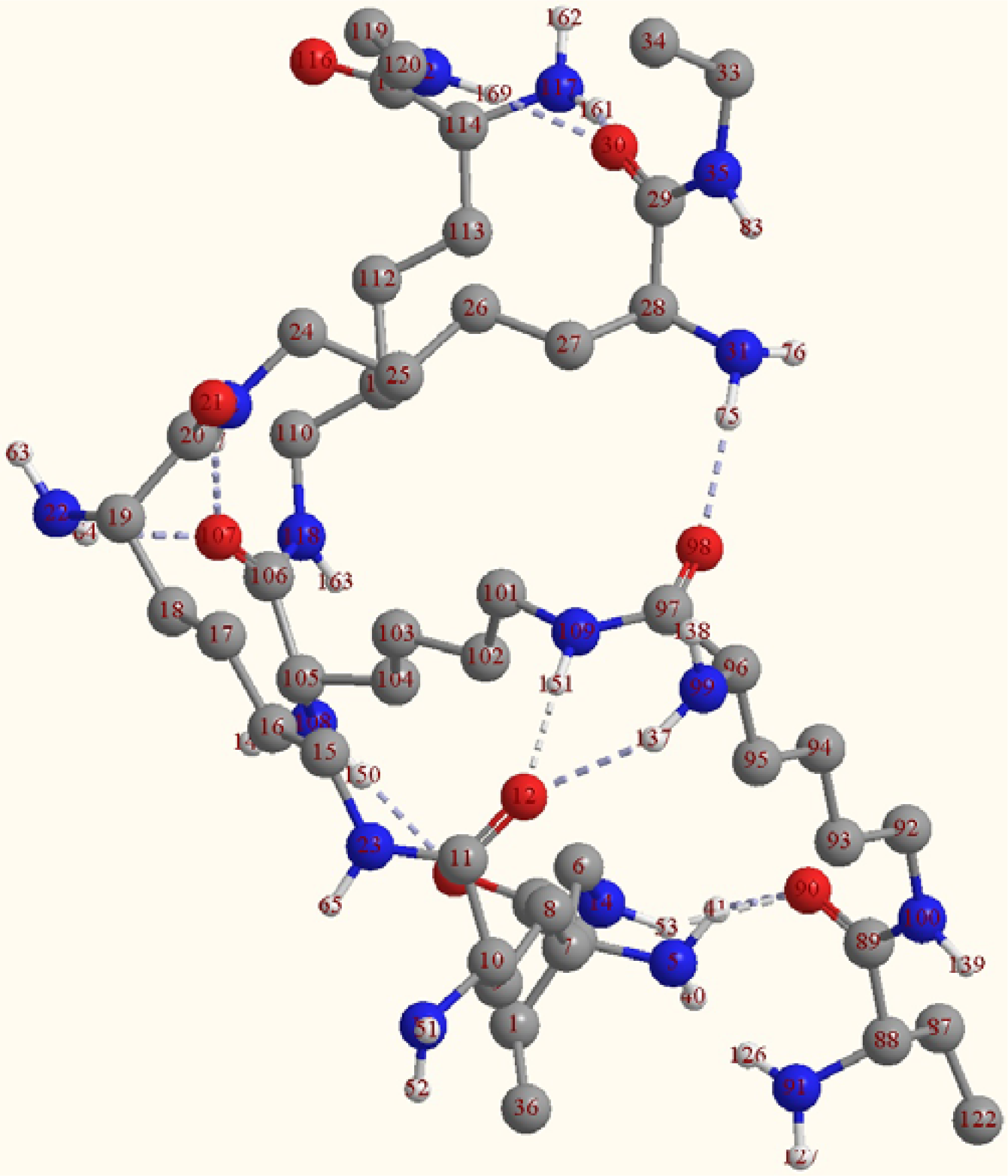
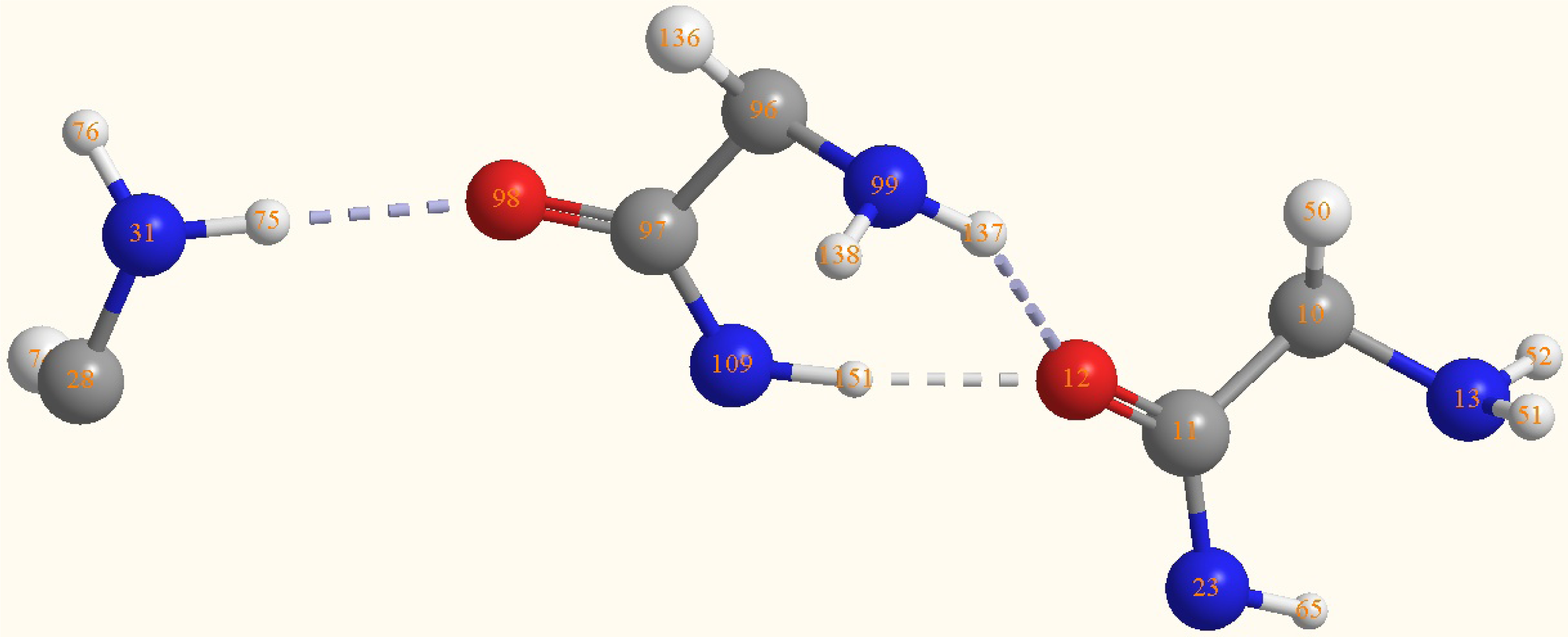
2.2. Structure of the cyclohepta bifurcated hydrogen bonds
2.3. Frequency analysis and IR spectra
3. Experimental Section
- The polar functional groups such as the amide, amino and carbonyl groups are at the high quantum chemical level of RB3LYP/6-31G for the structure optimization.
- All other atoms or groups such as methylene are at a semi-empirical PM3 level.
4. Conclusions
Acknowledgments
References
- Ho, YT; Ishizaki, S; Tanaka, M. Improving emulsifying activity of ɛ-polylysine by conjugation with dextran through the Maillard reaction. Food Chem 2000, 68, 449–455. [Google Scholar]
- Park, SM; Tran, HD; Kim, MS; Yu, RN; Yoo, H. Dendritic α, ɛ-poly(l-lysine)s as delivery agents for antisense oligonucleotides. Pharm. Res 2007, 24, 1581–1589. [Google Scholar]
- Tsujita, T; Sumiyoshi, M; Takaku, T; Momsen, WE; Lowe, ME; Brockman, HL. Inhibition of lipases by ɛ-polylysine. J. Lipid Res 2003, 44, 2278–2286. [Google Scholar]
- Salick, DA; Kretsinger, JK; Pochan, DJ; Schneider, JP. Inherent antibacterial activity of a peptide-based β-hairpin hydrogel. J. Am. Chem. Soc 2007, 129, 14793–14799. [Google Scholar]
- Sant, V; Leroux, J-C. pH-sensitive block copolymers for pharmaceutical compositions.
- Hiraki, J; Ichikawa, T; Ninomiya, S; Seki, H; Uohama, K; Seki, H; Kimura, S; Yanagimoto, Y; Barnett, JW, Jr. Use of ADME studies to confirm the safety of epsilon-polylysine as a preservative in food. Regul. Toxicol. Pharmacol 2003, 37, 328–340. [Google Scholar]
- Kakiuchi, K; Tsuboi, A. Association of poly-(l-lysine) homologues in sodium carbonate solution. Colloid. Polym. Sci 1990, 268, 544–551. [Google Scholar]
- Dzwolak, W; Muraki, T; Kato, M; Taniguchi, Y. Chain-length dependence of α-helix to β-sheet transition in polylysine: Model of protein aggregation studied by temperature-tuned FTIR spectroscopy. Biopolym 2004, 73, 463–469. [Google Scholar]
- Hayakawa, K; Murata, H; Satake, I. Conformational change of poly(l-lysine) and poly(l-ornithine) and cooperative binding of sodium alkanesulfonate surfactants with different chain length. Colloid. Polym. Sci 1990, 268, 1044–1051. [Google Scholar]
- Jurgensen, VW; Jalkanen, K. The VA, VCD, Raman and ROA spectra of tri-l-serine in aqueous solution. Phys. Biol 2006, 3, S63–S79. [Google Scholar]
- Shima, S; Sakai, H. Polylysine produced by Streptomyces. Agric. Biol. Chem 1977, 41, 1807–1809. [Google Scholar]
- Shima, S; Matsuoka, H; Iwamoto, T; Sakai, H. Antimicrobial action of ɛ-poly-l-lysine. J. Antibiot 1984, 37, 1449–1455. [Google Scholar]
- Hiraki, J. ɛ-Polylysine: Its development and utilization. Fine Chem 2000, 29, 18–25. [Google Scholar]
- Geornaras, I; Sofos, JN. Activity of ɛ-polylysine against Escherichia coli O157:H7, Salmonella Typhimurium, and Listeria monocytogenes. J. Food Sci 2005, 70, M404–M408. [Google Scholar]
- Yoshida, T; Nagasawa, T. ɛ-Poly-l-lysine: Microbial production, biodegradation and application potential. Appl. Microbiol. Biotechnol 2003, 62, 21–26. [Google Scholar]
- Shih, IL; Shen, MH; Van, YT. Microbial synthesis of poly (ɛ-lysine) and its various applications. Bioresour. Technol 2006, 97, 1148–1159. [Google Scholar]
- Johansson, J; Gudmundsson, GH; Rottenberg, ME; Berndt, KD; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem 1998, 273, 3718–3724. [Google Scholar]
- Lee, H; Yamaguchi, H; Fujimori, D; Ohnishi, T; Nishida, A; Yamamoto, H. Synthesis and conformation of monodispersed oligo(ɛ-l-lysine)mers studied by CD and IR spectroscopies. Spectrosc. Lett 1995, 28, 177–190. [Google Scholar]
- Maeda, S; Kunimoto, K; Sasaki, C; Kuwae, A; Hanai, K. Characterization of microbial poly (ɛ-l-lysine) by FT-IR, Raman and solid state 13C NMR spectroscopies. J. Mol. Struct 2003, 655, 149–155. [Google Scholar]
- Xiong, YZ; Chen, PY. ONIOM DFT/PM3 calculation on the interaction between STI-571 and abelson tyrosine kinase. J. Mol. Model 2008, 14, 1083–1086. [Google Scholar]
- Liang, YH; Chen, FE. ONIOM DFT/PM3 calculations on the interaction between dapivirine and HIV-1 reverse transcriptase, a theoretical study. Drug. Discov. Ther 2007, 1, 57–60. [Google Scholar]
- Pinisakul, A; Kritayakornupong, C; Ruangpornvisuti, V. Molecular modeling of nitrosamines adsorbed on H-ZSM-5 zeolite: An ONIOM study. J. Mol. Model 2008, 14, 1035–1041. [Google Scholar]
- Foresman, JB; Frisch, A. Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian, 2nd ed; Gaussian Inc: Pittsburgh, PA, USA, 1996; pp. 122–124. [Google Scholar]
- Huang, N; MacKerell, AD, Jr. An ab initio quantum mechanical study of hydrogen-bonded complexes of biological interest. J. Phys. Chem. A 2002, 106, 7820–7827. [Google Scholar]
- Antonczak, S; Monard, G; Ruiz-Lopez, MF; Rivail, J-L. Modeling of peptide hydrolysis by thermolysin. A semiempirical and QM/MM study. J. Am. Chem. Soc 1998, 120, 8825–8833. [Google Scholar]
- Szilagyi, RK; Musaev, DG; Morokuma, K. Theoretical studies of biological nitrogen fixation. Part II. Hydrogen bonded networks as possible reactant and product channels. J. Mol. Struct. Theochem 2000, 506, 131–146. [Google Scholar]
- Franzen, S. Use of periodic boundary conditions to calculate accurate β-sheet frequencies using density functional theory. J. Phys. Chem. A 2003, 107, 9898–9902. [Google Scholar]
- Vener, MV; Egorova, AN; Fomin, DP; Tsirelson, VG. Hierarchy of the non-covalent interactions in the alanine-based secondary structures. DFT study of the frequency shifts and electron-density features. J. Phys. Org. Chem 2009, 22, 177–185. [Google Scholar]
- Friesner, RA; Dunietz, BD. Large-scale ab-initio quantum chemical calculations on biological systems. Accounts Chem. Res 2001, 34, 351–358. [Google Scholar]
- Mlinsek, G; Novic, M; Hodoscek, M; Solmajer, T. Prediction of enzyme binding: Human thrombin inhibition study by quantum chemical and artificial intelligence methods based on x-ray structures. J. Chem. Info. Comput. Sci 2001, 41, 1286–1294. [Google Scholar]
- Nishihira, J; Tachikawa, H. Theoretical evaluation of a model of the catalytic triads of serine and cysteine proteases by ab initio molecular orbital calculation. J. Theor. Biol 1999, 196, 513–519. [Google Scholar]
- Liang, YH; Chen, FE. ONIOM DFT/PM3 calculations on the interaction between dapivirine and HIV-1 reverse transcriptase, a theoretical study. Drug. Discov. Ther 2007, 1, 57–60. [Google Scholar]
- Wieczorek, R; Dannenberg, JJ. Enthalpies of hydrogen-bonds in α-helical peptides. An ONIOM DFT/AM1 study. J. Am. Chem. Soc 2005, 127, 14534–14535. [Google Scholar]
- Senn, HM; Thiel, W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed 2009, 48, 1198–1229. [Google Scholar]
- Matsubara, T; Dupuis, M; Aida, M. Ab initio ONIOM-molecular dynamics (MD) study on the deamination reaction by cytidine deaminase. J. Phys. Chem. B 2007, 111, 9965–9974. [Google Scholar]
- Ferrante, F; la Manna, G. ONIOM study on the equilibrium geometries of some cyclopeptides. J. Mol. Struct. (Theochem) 2003, 634, 181–186. [Google Scholar]
- Wieczorek, R; Dannenberg, JJ. Amide I vibrational frequencies of α-helical peptides based upon ONIOM and density functional theory (DFT) studies. J. Phys. Chem. B 2008, 112, 1320–1328. [Google Scholar]
- Lutz, HD. Structure and strength of hydrogen bonds in inorganic solids. J. Mol. Struct 2003, 646, 227–236. [Google Scholar]
- Rozenberg, M; Shoham, G. FTIR spectra of solid poly-l-lysine in the stretching NH mode range. Biophysical. Chem 2007, 125, 166–171. [Google Scholar]
- Szyc, L; Pilorz, S; Czarnik-Matusewicz, B. FTIR-ATR investigations of an α-helix to β-sheet conformational transition in poly (l-lysine). J. Mol. Liq 2008, 141, 155–159. [Google Scholar]
- Mauerer, A; Lee, G. Changes in the amide I FT-IR bands of poly-l-lysine on spray-drying from α-helix, β-sheet or random coil conformations. Eur. J. Pharm. Biopharm 2006, 62, 131–142. [Google Scholar]
- DeFlores, LP; Ganim, Z; Nicodemus, RA; Tokmakoff, A. Amide I′- II′ 2D IR spectroscopy provides enhanced protein secondary structural sensitivity. J. Am. Chem. Soc 2009, 131, 3385–3391. [Google Scholar]
- Vaden, TD; Boer, TSJA; Simons, JP; Snoek, LC; Suhai, S; Paizs, B. Vibrational spectroscopy and conformational structure of protonated polyalanine peptides isolated in the gas phase. J. Phys. Chem. A 2008, 112, 4608–4616. [Google Scholar]
- Liang, YH; Chen, FE. ONIOM DFT/PM3 calculations on the interaction between dapivirine and HIV-1 reverse transcriptase, a theoretical study. Drug Discov. Ther 2007, 1, 57–60. [Google Scholar]
- Schweitzer-Stenner, R; Eker, F; Huang, Q. Structure analysis of dipeptides in water by exploring and utilizing the structural sensitivity of amide III by polarized visible Raman, FTIR-spectroscopy and DFT based normal coordinate analysis. J. Phys. Chem. B 2002, 106, 4294–4304. [Google Scholar]
- Vaden, TD; Gowers, SAN; Boer, TSJA; Steill, JD; Oomens, J; Snoek, LC. Conformational preferences of an amyloidogenic peptide: IR spectroscopy of ac-VQIVYK-NHMe. J. Am. Chem. Soc 2008, 130, 14640–14650. [Google Scholar]
- Chem3D Ultra, Version 10.0; CambridgeSoft: Cambridge, MA, USA, 2006.
- Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Montgomery, JA; Vreven, T; Kudin, KN; Burant, JC; Millam, JM; Iyengar, SS; Tomasi, J; Barone, V; Mennucci, B; Cossi, M; Scalmani, G; Rega, N; Petersson, GA; Nakatsuji, H; Hada, M; Ehara, M; Toyota, K; Fukuda, R; Hasegawa, J; Ishida, M; Nakajima, T; Honda, Y; Kitao, O; Nakai, H; Klene, M; Li, X; Knox, JE; Hratchian, HP; Cross, JB; Adamo, C; Jaramillo, J; Gomperts, R; Stratmann, RE; Yazyev, O; Austin, AJ; Cammi, R; Pomelli, C; Ochterski, JW; Ayala, PY; Morokuma, K; Voth, GA; Salvador, P; Dannenberg, JJ; Zakrzewski, GV; Dapprich, S; Daniels, AD; Strain, MC; Farkas, O; Malick, DK; Rabuck, AD; Raghavachari, K; Foresman, JB; Ortiz, JV; Cui, Q; Baboul, AG; Clifford, S; Cioslowski, J; Stefanov, BB; Liu, G; Liashenko, A; Piskorz, P; Komaromi, I; Martin, RL; Fox, DJ; Keith, T; Al-Laham, MA; Peng, CY; Nanayakkara, A; Challacombe, M; Gill, PMW; Johnson, B; Chen, W; Wong, MW; Gonzalez, C; Pople, JA. Gaussian 03, Revision B01; Gaussian, Inc: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Chin, W; Mons, M; Dognon, J-P; Piuzzi, F; Tardivel, B; Dimicoli, I. Competition between local conformational preferences and secondary structures in gas-phase model tripeptides as revealed by laserspectroscopy and theoretical chemistry. Phys. Chem. Chem. Phys 2004, 6, 2700–2709. [Google Scholar]
- Rice, CA; Dauster, I; Suhm, MA. Infrared spectroscopy of pyrrole-2-carboxaldehyde and its dimer: a planar beta-sheet peptide model? J. Chem. Phys 2007, 126, 134313. [Google Scholar]
- Chin, W; Piuzzi, F; Dimicoli, I; Mons, M. Probing the competition between secondary structures and local preferences in gas phase isolated peptide backbones. Phys. Chem. Chem. Phys 2006, 8, 1033–1048. [Google Scholar]
- Abo-Riziq, A; Crews, BO; Callahan, MP; Grace, L; de Vries, M. Spectroscopy of isolated gramicidin peptides. Angew. Chem. Int. Ed 2006, 45, 5166–5169. [Google Scholar]
- Sitkoff, D; Case, DA. Density functional calculations of proton chemical shifts in model peptides. J. Am. Chem. Soc 1997, 119, 12262–12273. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms | Bond lengths/Å | Atoms | Bond angles/° |
|---|---|---|---|
| C(11)-C(10) | 1.5452 | O(12)-C(11)-N(23) | 125.9887 |
| O(12)-C(11) | 1.2603 | C(10)-C(11)-N(23) | 114.1929 |
| N(13)-C(10) | 1.4818 | C(10)-C(11)-O(12) | 119.8179 |
| N(23)-C(11) | 1.3531 | H(51)-N(13)-H(52) | 112.2561 |
| N(31)-C(28) | 1.4835 | C(10)-N(13)-H(52) | 115.5022 |
| H(51)-N(13) | 1.0136 | C(10)-N(13)-H(51) | 114.2261 |
| H(52)-N(13) | 1.0133 | H(75)-N(31)-H(76) | 111.7182 |
| H(75)-N(31) | 1.0221 | C(28)-N(31)-H(76) | 113.0713 |
| H(76)-N(31) | 1.0147 | C(28)-N(31)-H(75) | 114.5283 |
| C(97)-C(96) | 1.5605 | C(97)-C(96)-N(99) | 115.5131 |
| O(98)-C(97) | 1.2585 | O(98)-C(97)-N(109) | 125.1159 |
| N(99)-C(96) | 1.4676 | C(96)-C(97)-N(109) | 114.5476 |
| N(109)-C(97) | 1.3652 | C(96)-C(97)-O(98) | 120.3339 |
| H(137)-N(99) | 1.0132 | H(137)-N(99)- | 114.2089 |
| H(138)-N(99) | 1.0105 | C(96)-N(99)-H(138) | 115.0922 |
| H(151)-N(109) | 1.0276 | C(96)-N(99)-H(137) | 115.9221 |
| O(12)-H(151) | 1.8412 | C(97)-N(109)-H(151) | 119.9472 |
| O(12)-H(137) | 2.1713 | H(151)-O(12)- | 66.6 |
| O(12)-H(151)- | 166.7 | ||
| O(12)-H(137)-N(99) | 147 | ||
| N(31)-H(75)-O(98) | 173.5 |
| −NH2 groups | Frequencies/cm− | Amide N-H | Frequencies/cm−1 | C=O groups | Frequencies/cm−1 |
|---|---|---|---|---|---|
| υasN(99)-H | 3670vw | υN(23)-H | 3446w | υC(11)-O(12) | 1639w |
| υasN(13)-H | 3646 vw | υN(109)-H | 3356m | υC(97)-O(98) | 1629m |
| υasN(31)-H | 3592 vw | ρN(109)-H | 1567s | ||
| υsN(99)-H | 3551 vw | ρN(23)-H | 1563w | ||
| υsN(13)-H | 3538 w | ||||
| υsN(31)-H | 3458m | ||||
| δN(31)-H | 1711vw | ||||
| δN(13)-H | 1686vw | ||||
| δN(99)-H | 1679vw | ||||
| δC(28)-H | 1316vw | ||||
| δC(96)-H | 1315vw | ||||
| δC(10)-H | 1328vw |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jia, S.; Mo, Z.; Dai, Y.; Zhang, X.; Yang, H.; Qi, Y. Computational Study on the Conformation and Vibration Frequencies of β-Sheet of ε-Polylysine in Vacuum. Int. J. Mol. Sci. 2009, 10, 3358-3370. https://doi.org/10.3390/ijms10083358
Jia S, Mo Z, Dai Y, Zhang X, Yang H, Qi Y. Computational Study on the Conformation and Vibration Frequencies of β-Sheet of ε-Polylysine in Vacuum. International Journal of Molecular Sciences. 2009; 10(8):3358-3370. https://doi.org/10.3390/ijms10083358
Chicago/Turabian StyleJia, Shiru, Zhiwen Mo, Yujie Dai, Xiuli Zhang, Hongjiang Yang, and Yuhua Qi. 2009. "Computational Study on the Conformation and Vibration Frequencies of β-Sheet of ε-Polylysine in Vacuum" International Journal of Molecular Sciences 10, no. 8: 3358-3370. https://doi.org/10.3390/ijms10083358