Participation of Low Molecular Weight Electron Carriers in Oxidative Protein Folding

Abstract
:1. Introduction
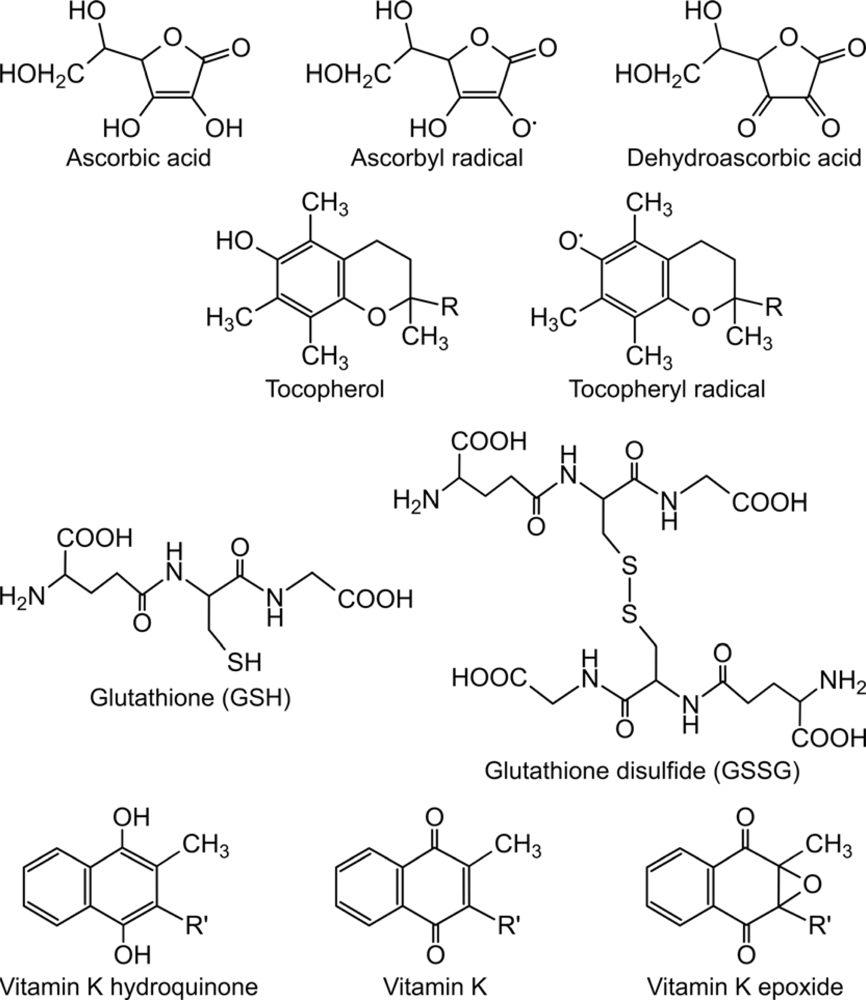
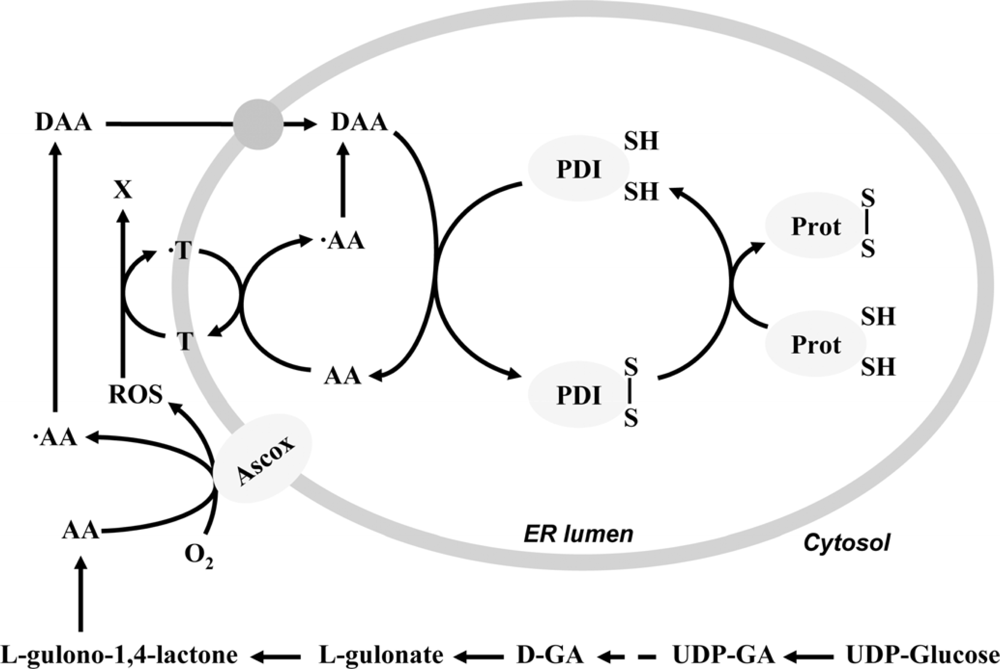
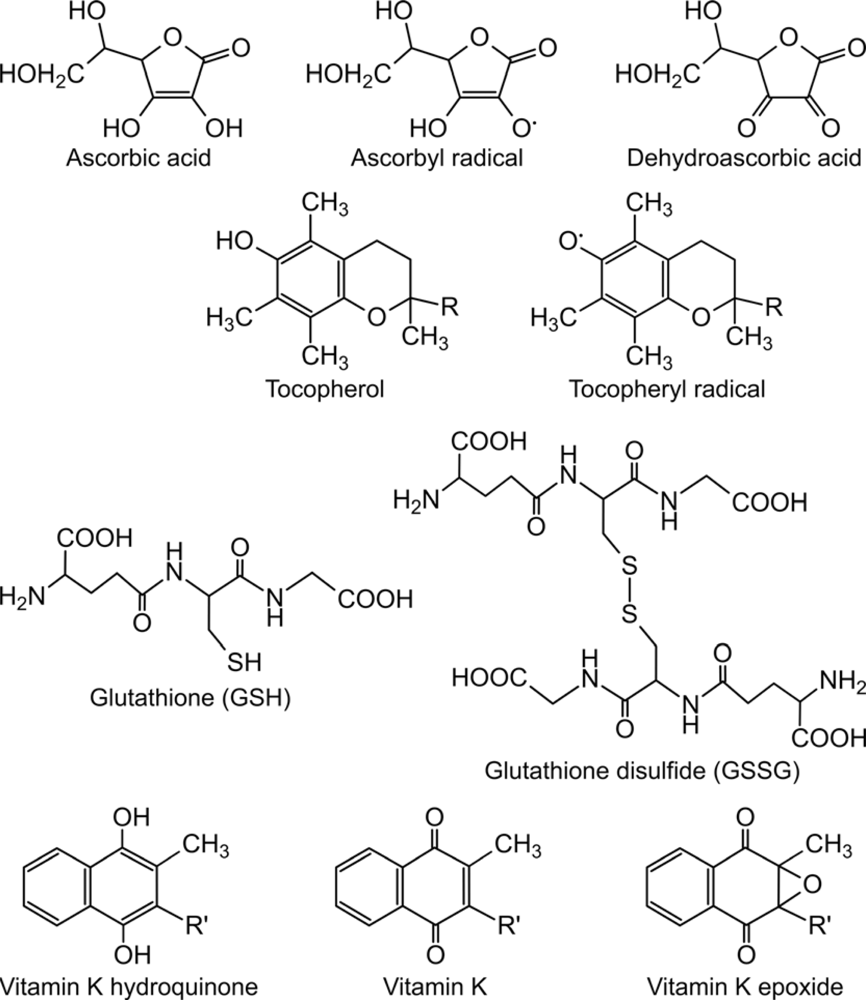
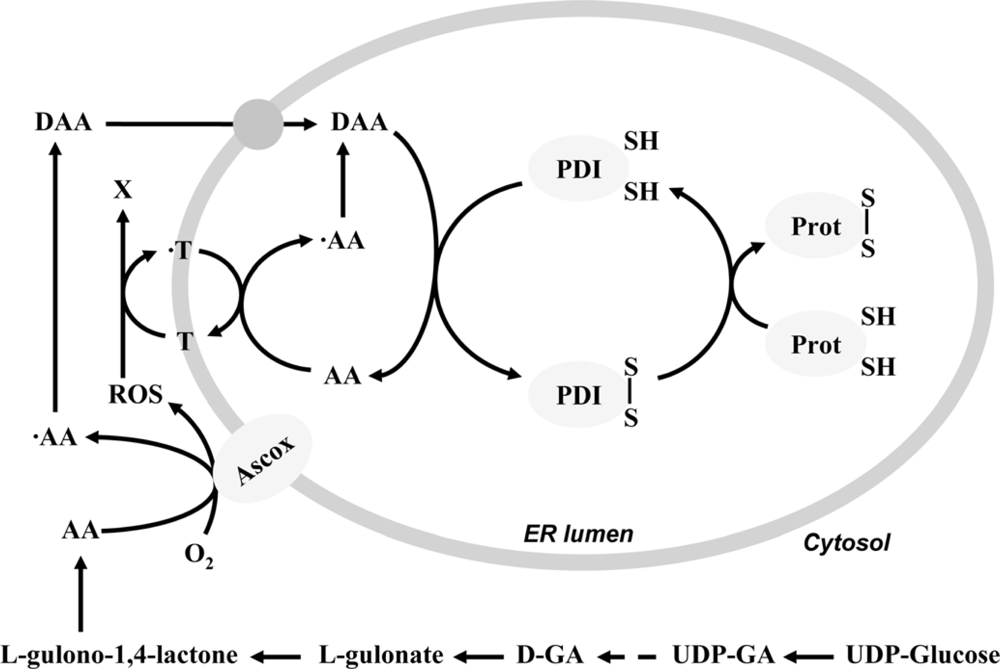
2. Ascorbate-dependent protein thiol oxidation in the ER
3. Role of lipophilic vitamins - vitamin E and K - in ascorbate-dependent protein thiol oxidation
4. Final electron acceptor(s) of the protein folding system
5. Artificial small-molecules in the catalysis of oxidative protein folding
- One is the reduction potential (E°′) of the disulfide bond which is formed between the Cys residues in the active site [33]. This value refers to the stability of the disulfide bond; the more stabile the disulfide, the lower the E°′. For PDI the E°′ is −180 mV, which corresponds to the redox potential of the ER lumen and allows PDI to remain in a nearly 50–50% mixture of its reduced and oxidized form (Table 1.).
- The other factor that governs the efficiency of thiol-disulfide oxidoreductases is the acid dissociation constant (Ka) of the N-terminal thiol group in the active site. Sulfhydryl group of free cysteine has a relatively high pKa (8,5) and as a consequence it is relatively inert for redox reactions in physiological conditions. In contrast, some structural folds in thiol-disulfide oxidoreductases provide appropriated environments for changing the pKa values of sulfhydryl groups. If this constant is close to the pH of the solution, a large part of the Cys can be deprotonated and stabilized in the anionic form called thiolate (RS−). The formed thiolate can initiate nucleophile attack to compose disulfide bonds in the substrate proteins. The pKa value of Cys in the active site depends on the proteinaceous environment; e.g. in PDI the pKa value of this Cys is 6.7 [34], which is low enough to result in a high amount of PDI-thiolate.
6. Concluding remarks
Acknowledgments
References
- Sevier, CS; Kaiser, CA. Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell. Biol 2002, 3, 836–847. [Google Scholar]
- Hwang, C; Sinskey, AJ; Lodish, HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar]
- Brigelius-Flohé, R; Traber, MG. Vitamin E: Function and metabolism. FASEB J 1999, 13, 1145–1155. [Google Scholar]
- Braun, L; Puskás, F; Csala, M; Gyorffy, E; Garzó, T; Mandl, J; Bánhegyi, G. Gluconeogenesis from ascorbic acid: ascorbate recycling in isolated murine hepatocytes. FEBS Lett 1996, 390, 183–186. [Google Scholar]
- Braun, L; Puskás, F; Csala, M; Mészáros, G; Mandl, J; Bánhegyi, G. Ascorbate as a substrate for glycolysis or gluconeogenesis: Evidence for an interorgan ascorbate cycle. Free Radic. Biol. Med 1997, 23, 804–808. [Google Scholar]
- Bánhegyi, G; Braun, L; Csala, M; Puskás, F; Mandl, J. Ascorbate metabolism and its regulation in animals. Free Radic. Biol. Med 1997, 23, 793–803. [Google Scholar]
- Braun, L; Mile, V; Schaff, Zs; Csala, M; Kardon, T; Mandl, J; Bánhegyi, G. Induction and peroxisomal appearance of gulonolactone oxidase upon clofibrate treatment in mouse liver. FEBS Lett 1999, 458, 359–362. [Google Scholar]
- Braun, L; Kardon, T; El Koulali, K; Csala, M; Mandl, J; Bánhegyi, G. Different induction of gulonolactone oxidase in aromatic hydrocarbon-responsive or -unresponsive mouse strains. FEBS Lett 1999, 463, 345–349. [Google Scholar]
- Bánhegyi, G; Braun, L; Csala, M; Puskás, F; Somogyi, A; Kardon, T; Mandl, J. Ascorbate and environmental stress. Ann. N.Y. Acad. Sci 1998, 851, 292–303. [Google Scholar]
- Cuozzo, JW; Kaiser, CA. Competition between glutathione and protein thiols for disulphide-bond formation. Nat. Cell. Biol 1999, 1, 130–135. [Google Scholar]
- Csala, M; Fulceri, R; Mandl, J; Benedetti, A; Bánhegyi, G. Glutathione transport in the endo/sarcoplasmic reticulum. Biofactors 2003, 17, 27–35. [Google Scholar]
- Horemans, N; Foyer, CH; Asard, H. Transport and action of ascorbate at the plant plasma membrane. Trends. Plant. Sci 2000, 5, 263–267. [Google Scholar]
- Szarka, A; Stadler, K; Jenei, V; Margittai, E; Csala, M; Jakus, J; Mandl, J; Bánhegyi, G. Ascorbyl free radical and dehydroascorbate formation in rat liver endoplasmic reticulum. J. Bioenerg. Biomembr 2002, 34, 317–323. [Google Scholar]
- Pinchuk, I; Gal, S; Lichtenberg, D. The dose-dependent effect of copper-chelating agents on the kinetics of peroxidation of low-density lipoprotein (LDL). Free Radic. Res 2001, 34, 349–362. [Google Scholar]
- Dawson, CR; Strothkamp, KG; Krul, KG. Ascorbate oxidase and related copper proteins. Ann. N.Y. Acad. Sci 1975, 258, 209–220. [Google Scholar]
- Csala, M; Mile, V; Benedetti, A; Mandl, J; Bánhegyi, G. Ascorbate oxidation is a prerequisite for its transport into rat liver microsomal vesicles. Biochem. J 2000, 349, 413–415. [Google Scholar]
- Wells, WW; Xu, DP; Yang, YF; Rocque, PA. Mammalian thioltransferase (glutaredoxin) and protein disulfide isomerase have dehydroascorbate reductase activity. J. Biol. Chem 1990, 265, 15361–15364. [Google Scholar]
- Nardai, G; Braun, L; Csala, M; Mile, V; Csermely, P; Benedetti, A; Mandl, J; Bánhegyi, G. Protein-disulfide isomerase- and protein thiol-dependent dehydroascorbate reduction and ascorbate accumulation in the lumen of the endoplasmic reticulum. J. Biol. Chem 2001, 276, 8825–8828. [Google Scholar]
- Schröder, M; Kaufman, RJ. ER stress and the unfolded protein response. Mutat. Res 2005, 569, 29–63. [Google Scholar]
- Margittai, E; Bánhegyi, G; Kiss, A; Nagy, G; Mandl, J; Schaff, Zs; Csala, M. Scurvy leads to endoplasmic reticulum stress and apoptosis in the liver of Guinea pigs. J. Nutr 2005, 135, 2530–2534. [Google Scholar]
- Garcia, AA; Reitsma, PH. VKORC1 and the vitamin K cycle. Vitam. Horm 2008, 78, 23–33. [Google Scholar]
- Soute, BA; Groenen-van Dooren, MM; Holmgren, A; Lundström, J; Vermeer, C. Stimulation of the dithiol-dependent reductases in the vitamin K cycle by the thioredoxin system. Strong synergistic effects with protein disulphide-isomerase. Biochem. J 1992, 281, 255–259. [Google Scholar]
- Wajih, N; Hutson, SM; Wallin, R. Disulfide-dependent protein folding is linked to operation of the vitamin K cycle in the endoplasmic reticulum. A protein disulfide isomerase-VKORC1 redox enzyme complex appears to be responsible for vitamin K1 2,3-epoxide reduction. J. Biol. Chem 2007, 282, 2626–2635. [Google Scholar]
- Csala, M; Szarka, A; Margittai, E; Mile, V; Kardon, T; Braun, L; Mandl, J; Bánhegyi, G. Role of vitamin E in ascorbate-dependent protein thiol oxidation in rat liver endoplasmic reticulum. Arch. Biochem. Biophys 2001, 388, 55–59. [Google Scholar]
- Ito, K; Inaba, K. The disulfide bond formation (Dsb) system. Curr. Opin. Struct. Biol 2008, 18, 450–458. [Google Scholar]
- Bader, M; Muse, W; Ballou, DP; Gassner, C; Bardwell, JC. Oxidative protein folding is driven by the electron transport system. Cell 1999, 98, 217–227. [Google Scholar]
- Tu, BP; Weissman, JS. The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 2002, 10, 983–994. [Google Scholar]
- Gross, E; Sevier, CS; Heldman, N; Vitu, E; Bentzur, M; Kaiser, CA; Thorpe, C; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar]
- Sevier, CS; Cuozzo, JW; Vala, A; Aslund, F; Kaiser, CA. A flavoprotein oxidase defines a new endoplasmic reticulum pathway for biosynthetic disulphide bond formation. Nat. Cell. Biol 2001, 3, 874–882. [Google Scholar]
- Tu, BP; Ho-Schleyer, SC; Travers, KJ; Weissman, JS. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 2000, 290, 1571–1574. [Google Scholar]
- Appenzeller-Herzog, C; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta 2008, 1783, 535–548. [Google Scholar]
- Tian, G; Xiang, S; Noiva, R; Lennarz, WJ; Schindelin, H. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell (Cambridge, MA, USA) 2006, 124, 61–73. [Google Scholar]
- Lundstroem, J; Holmgren, A. Determination of the reduction–oxidation potential of the thioredoxin-like domains of protein disulfide-isomerase from the equilibrium with glutathione and thioredoxin. Biochemistry 1993, 32, 6649–6655. [Google Scholar]
- Hawkins, HC; Freedman, RB. The reactivities and ionization properties of the active-site dithiol groups of mammalian protein disulfide-isomerase. Biochem. J 1991, 275, 335–339. [Google Scholar]
- Walker, KW; Gilbert, HF. Scanning and escape during protein disulfide isomerase-assisted protein folding. J. Biol. Chem 1997, 272, 8845–8848. [Google Scholar]
- Sahdev, S; Khattar, SK; Saini, KS. Production of active eukaryotic proteins through bacterial expression systems: A review of the existing biotechnology strategies. Mol. Cell. Biochem 2008, 307, 249–264. [Google Scholar]
- Moroder, L; Besse, D; Musiol, H-J; Rudolph-Boehner, S; Sideler, F. Oxidative folding of cystine-rich peptides versus regioselective cysteine pairing strategies. Biopolymers 1996, 40, 207–234. [Google Scholar]
- Cabrele, C; Fiori, S; Pegoraro, S; Moroder, L. Redox-active cyclic bis(cysteinyl)peptides as catalysts for in vitro oxidative protein folding. Chem. Biol 2002, 9, 731–740. [Google Scholar]
- Cabrele, C; Cattani-Scholz, A; Renner, C; Behrendt, R; Oesterhelt, D; Moroder, L. Photomodulation of the redox and folding adjuvant properties of bis(cysteinyl) peptides. Eur. J. Org. Chem 2002, 2002, 2144–2150. [Google Scholar]
- Woycechowsky, KJ; Raines, RT. The CXC motif: A functional mimic of protein disulfide isomerase. Biochemistry 2003, 42, 5387–5394. [Google Scholar]
- Woycechowsky, KJ; Wittrup, KD; Raines, RT. A small-molecule catalyst of protein folding in vitro and in vivo. Chem. Biol 1999, 6, 871–879. [Google Scholar]
- DeCollo, TV; Lees, WJ. Effects of aromatic thiols on thiol–disulfide interchange reactions that occur during protein folding. J. Org. Chem 2001, 66, 4244–4249. [Google Scholar]
- Beld, J; Woycechowsky, KJ; Hilvert, D. Selenoglutathione: Efficient oxidative protein folding by a diselenide. Biochemistry 2007, 46, 5382–5390. [Google Scholar]
- Spallholz, JE. On the nature of selenium toxicity and carcinostatic activity. Free Radic. Biol. Med 1994, 17, 45–64. [Google Scholar]
- López-Mirabal, HR; Winther, JR. The thiol oxidant dipyridyl disulfide can supply the PDI-Ero1p pathway with additional oxidative equivalents. Antonie Van Leeuwenhoek 2007, 92, 463–472. [Google Scholar]
- Fink, M; Nieves, P; Chang, S; Narayan, M. Non-redox-active small-molecules can accelerate oxidative protein folding by novel mechanisms. Biophys. Chem 2008, 132, 104–109. [Google Scholar]
- Papp, E; Csermely, P. Chemical chaperones: mechanisms of action and potential use. Handb. Exp. Pharmacol 2006, 172, 405–416. [Google Scholar]
- Lees, WJ. Small-molecule catalysts of oxidative protein folding. Curr. Opin. Chem. Biol 2008, 12, 740–745. [Google Scholar]
| List of abbreviations: | |
|---|---|
| BMC | (±)-trans-1,2-bis(mercaptoacetamido)cyclohexane; |
| DMSO | dimethyl sulfoxide; |
| DPS | dipyridyl disulfide; |
| Dsb | disulfide bond formation proteins; |
| ER | endoplasmic reticulum; |
| Ero1 | endoplasmic reticulum oxidoreductase 1; |
| ERP72 | endoplasmic reticulum protein 72; |
| FAD | flavin adeninde dinucleotide; |
| FMN | flavin-mononucleotide; |
| GLO | gulonolactone oxidase; |
| GRP78 | glucose regulated protein 78; |
| GRP94 | glucose regulated protein; |
| GSeSeG | diselenide analogue of oxidized glutathione; |
| GSH | reduced glutathione; |
| GSSG | oxidized glutathione; |
| H2O2 | hydrogen peroxide; |
| PDI | protein disulfide isomerase; |
| ROS | reactive oxygen species; |
| UPR | unfolded protein response; |
| VKORC1 | vitamin K1 2,3-epoxide reductase subunit 1. |

{kind=link}
{kind=link}
| Compounds | E°' (mV) |
|---|---|
| Linear CXXC (active-site sequence of Trx) | −190 |
| Cyclic CXXC (active-site sequence of PDI) | −130 |
| Photoactive CXXC cis/trans (active-site sequence of Trx-reductase) | −147 (cis) / −201(trans) |
| CXC-containing peptides (CGC) | −167 |
| Aromatic thiols: (1.) R = CH2COOH; (2.) R = SO3H | −170 (1.) / −220 (2.) |
| GSeH | −407 |
| Selenocystamine | −348 |
| BMC | −240 |
| GSH | −250 |
| ascorbate / dehydroascorbic acid | 80 |
| tocopherol / tocopheryl radical | 480 |
| vitamin K / vitamin K epoxide | 303 |
| dipyridyl-disulfide / dipyridyl-dithiol | 147 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Margittai, É.; Csala, M.; Mandl, J.; Bánhegyi, G. Participation of Low Molecular Weight Electron Carriers in Oxidative Protein Folding. Int. J. Mol. Sci. 2009, 10, 1346-1359. https://doi.org/10.3390/ijms10031346
Margittai É, Csala M, Mandl J, Bánhegyi G. Participation of Low Molecular Weight Electron Carriers in Oxidative Protein Folding. International Journal of Molecular Sciences. 2009; 10(3):1346-1359. https://doi.org/10.3390/ijms10031346
Chicago/Turabian StyleMargittai, Éva, Miklós Csala, József Mandl, and Gábor Bánhegyi. 2009. "Participation of Low Molecular Weight Electron Carriers in Oxidative Protein Folding" International Journal of Molecular Sciences 10, no. 3: 1346-1359. https://doi.org/10.3390/ijms10031346
APA StyleMargittai, É., Csala, M., Mandl, J., & Bánhegyi, G. (2009). Participation of Low Molecular Weight Electron Carriers in Oxidative Protein Folding. International Journal of Molecular Sciences, 10(3), 1346-1359. https://doi.org/10.3390/ijms10031346