Neuropathology and Therapeutic Intervention in Spinal and Bulbar Muscular Atrophy

Abstract
:1. Introduction
2. Clinical and genetic features of SBMA
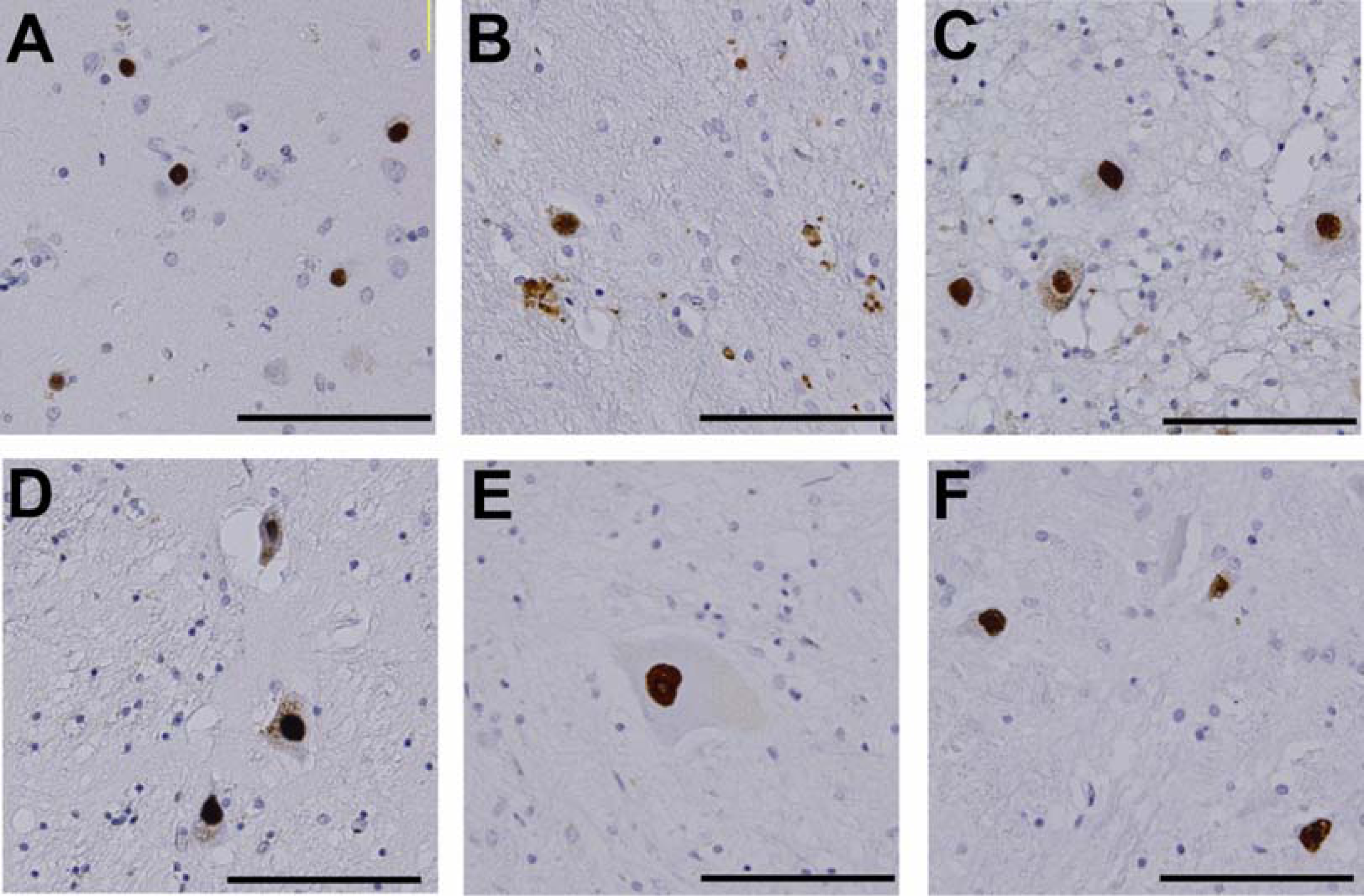
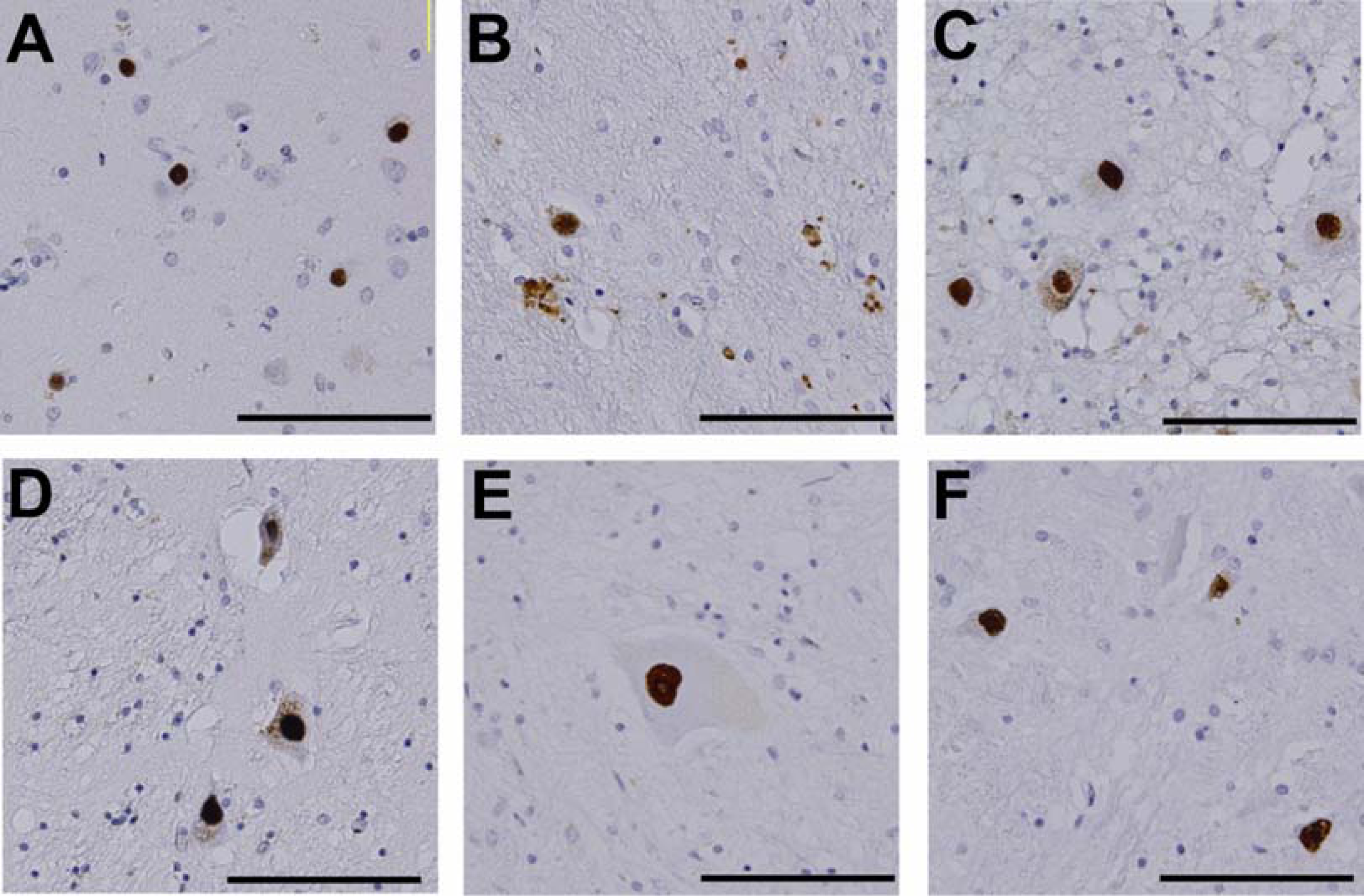
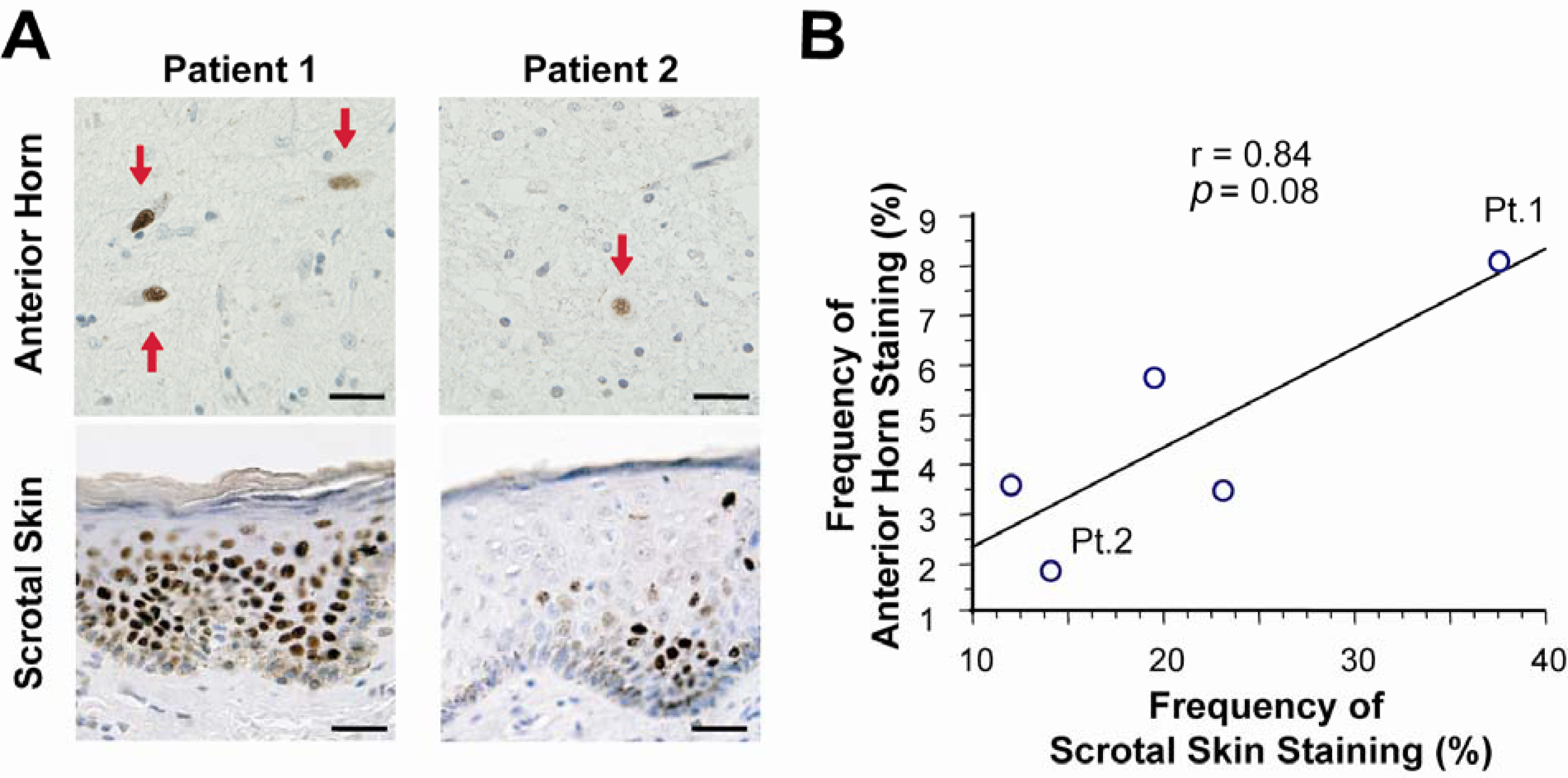
3. Neuropathology and molecular mechanisms of SBMA
4. Protein folding abnormalities in SBMA
5. Therapeutic strategies for SBMA
5. Conclusions
Acknowledgments
References and Notes
- Kennedy, WR; Alter, M; Sung, JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology 1968, 18, 671–680. [Google Scholar]
- Atsuta, N; Watanabe, H; Ito, M; Banno, H; Suzuki, K; Katsuno, M; Tanaka, F; Tamakoshi, A; Sobue, G. Natural history of spinal and bulbar muscular atrophy (SBMA): A study of 223 Japanese patients. Brain 2006, 129, 1446–1455. [Google Scholar]
- La Spada, AR; Wilson, EM; Lubahn, DB; Harding, AE; Fischbeck, KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar]
- Tanaka, F; Doyu, M; Ito, Y; Matsumoto, M; Mitsuma, T; Abe, K; Aoki, M; Itoyama, Y; Fischbeck, KH; Sobue, G. Founder effect in spinal and bulbar muscular atrophy (SBMA). Hum. Mol. Genet 1996, 5, 1253–1257. [Google Scholar]
- Fischbeck, KH. Kennedy disease. J. Inherit. Metab. Dis 1997, 20, 152–158. [Google Scholar]
- Doyu, M; Sobue, G; Mukai, E; Kachi, T; Yasuda, T; Mitsuma, T; Takahashi, A. Severity of X-linked recessive bulbospinal neuronopathy correlates with size of the tandem CAG repeat in androgen receptor gene. Ann. Neurol 1992, 32, 707–710. [Google Scholar]
- La Spada, AR; Roling, DB; Harding, AE; Warner, CL; Spiegel, R; Hausmanowa-Petrusewicz, I; Yee, WC; Fischbeck, KH. Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat. Genet 1992, 2, 301–304. [Google Scholar]
- Gatchel, JR; Zoghbi, HY. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet 2005, 6, 743–755. [Google Scholar]
- Suzuki, K; Katsuno, M; Banno, H; Takeuchi, Y; Atsuta, N; Ito, M; Watanabe, H; Yamashita, F; Hori, N; Nakamura, T; Hirayama, M; Tanaka, F; Sobue, G. CAG repeat size correlates to electrophysiological motor and sensory phenotypes in SBMA. Brain 2008, 131, 229–239. [Google Scholar]
- Sobue, G; Hashizume, Y; Mukai, E; Hirayama, M; Mitsuma, T; Takahashi, A. X-linked recessive bulbospinal neuronopathy - A clinicopathological study. Brain 1989, 112, 209–232. [Google Scholar]
- Bates, G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet 2003, 361, 1642–1644. [Google Scholar]
- Martindale, D; Hackam, A; Wieczorek, A; Ellerby, L; Wellington, C; McCutcheon, K; Singaraja, R; Kazemi-Esfarjani, P; Devon, R; Kim, SU; Bredesen, DE; Tufaro, F; Hayden, MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet 1998, 18, 150–154. [Google Scholar]
- Podolsky, S; Sax, DS; Leopold, NA. Increased frequency of diabetes mellitus in patients with Huntington’s chorea. Lancet 1972, 1, 1356–1358. [Google Scholar]
- Sinnreich, M; Klein, CJ. Bulbospinal muscular atrophy - Kennedy’s disease. Arch. Neurol 2004, 61, 1324–1326. [Google Scholar]
- Li, M; Miwa, S; Kobayashi, Y; Merry, DE; Yamamoto, M; Tanaka, F; Doyu, M; Hashizume, Y; Fischbeck, KH; Sobue, G. Nuclear inclusions of the androgen receptor protein in spinal and bulb muscular atrophy. Ann. Neurol 1998, 44, 249–254. [Google Scholar]
- Kobayashi, Y; Miwa, S; Merry, DE; Kume, A; Mei, L; Doyu, M; Sobue, G. Caspase-3 cleaves the expanded androgen receptor protein of spinal and bulbar muscular atrophy in a polyglutamine repeat length-dependent manner. Biochem. Biophys. Res. Commun 1998, 252, 145–150. [Google Scholar]
- McCampbell, A; Taylor, JP; Taye, AA; Robitschek, J; Li, M; Walcott, J; Merry, D; Chai, Y; Paulson, H; Sobue, G; Fischbeck, KH. CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet 2002, 9, 2197–2202. [Google Scholar]
- Minamiyama, M; Katsuno, M; Adachi, H; Waza, M; Sang, C; Kobayashi, Y; Tanaka, F; Doyu, M; Inukai, A; Sobue, G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet 2004, 13, 1183–1192. [Google Scholar]
- Ranganathan, S; Harmison, GG; Meyertholen, K; Pennuto, M; Burnett, BG; Fischbeck, KH. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum. Mol. Genet 2009, 18, 27–42. [Google Scholar]
- Morfini, G; Pigino, G; Szebenyi, G; You, Y; Pollema, S; Brady, ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat. Neurosci 2006, 9, 907–916. [Google Scholar]
- Katsuno, M; Adachi, H; Minamiyama, M; Waza, M; Tokui, K; Banno, H; Suzuki, K; Onoda, Y; Tanaka, F; Doyu, M; Sobue, G. Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration. J. Neurosci 2006, 26, 12106–12117. [Google Scholar]
- Puls, I; Jonnakuty, C; LaMonte, BH; Holzbaur, EL; Tokito, M; Mann, E; Floeter, MK; Bidus, K; Drayna, D; Oh, SJ; Brown, RH, Jr; Ludlow, CL; Fischbeck, KH. Mutant dynactin in motor neuron disease. Nat. Genet 2003, 33, 455–456. [Google Scholar]
- Arrasate, M; Mitra, S; Schweitzer, ES; Segal, MR; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar]
- Truant, R; Atwal, RS; Desmond, C; Munsie, L; Tran, T. Huntington’s disease: Revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases. FEBS J 2008, 275, 4252–4262. [Google Scholar]
- Nagai, Y; Inui, T; Popiel, HA; Fujikake, N; Hasegawa, K; Urade, Y; Goto, Y; Naiki, H; Toda, T. A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol 2007, 14, 332–340. [Google Scholar]
- Takahashi, T; Kikuchi, S; Katada, S; Nagai, Y; Nishizawa, M; Onodera, O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet 2008, 17, 345–356. [Google Scholar]
- Katsuno, M; Banno, H; Suzuki, K; Takeuchi, Y; Kawashima, M; Tanaka, F; Adachi, H; Sobue, G. Molecular genetics and biomarkers of polyglutamine diseases. Curr. Mol. Med 2008, 8, 221–234. [Google Scholar]
- Adachi, H; Katsuno, M; Minamiyama, M; Waza, M; Sang, C; Nakagomi, Y; Kobayashi, Y; Tanaka, F; Doyu, M; Inukai, A; Yoshida, M; Hashizume, Y; Sobue, G. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain 2005, 128, 659–670. [Google Scholar]
- Yamada, M; Sato, T; Tsuji, S; Takahashi, H. Oligodendrocytic polyglutamine pathology in dentatorubral-pallidoluysian atrophy. Ann. Neurol 2002, 52, 670–674. [Google Scholar]
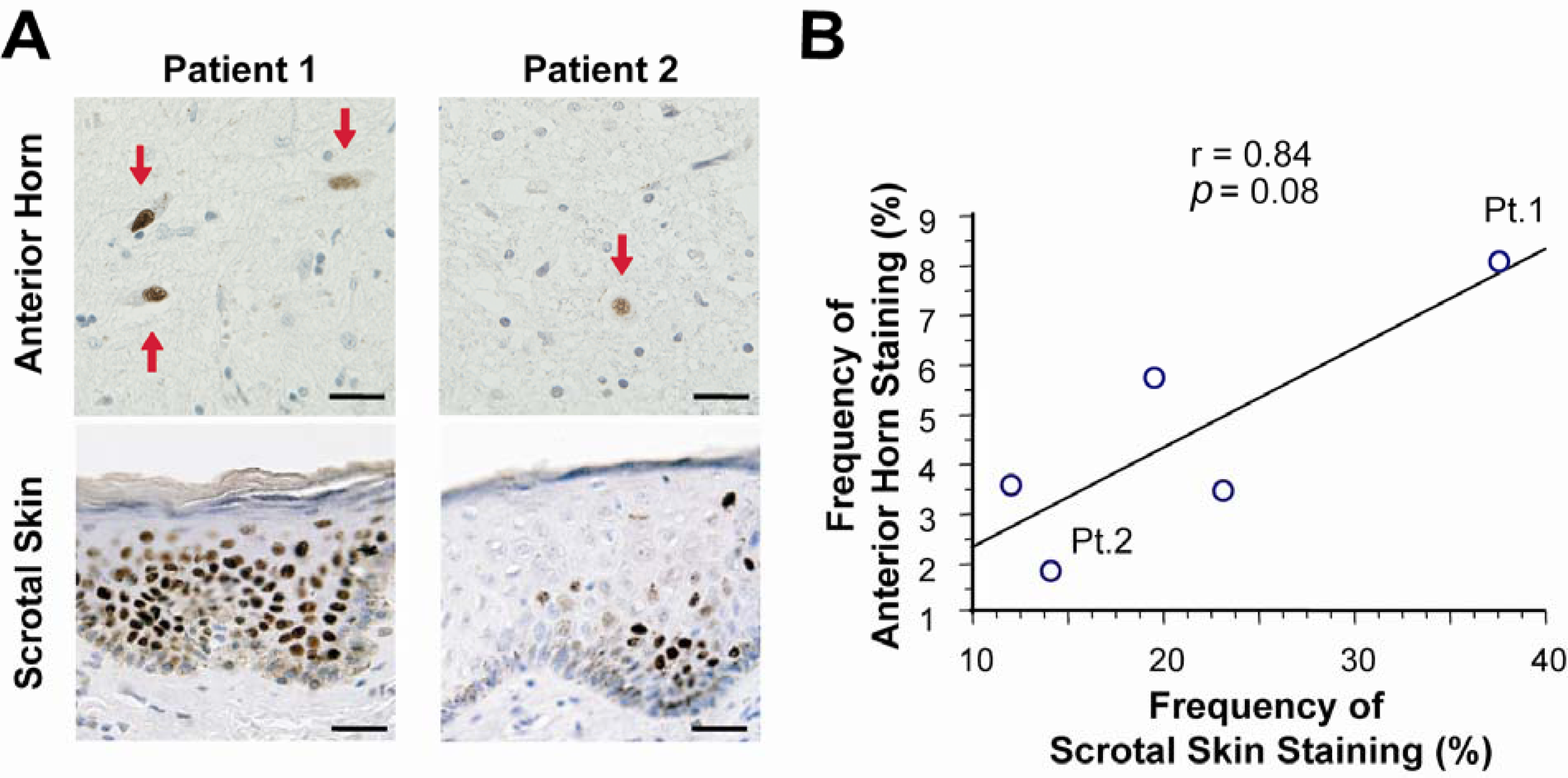
- Banno, H; Adachi, H; Katsuno, M; Suzuki, K; Atsuta, N; Watanabe, H; Tanaka, F; Doyu, M; Sobue, G. Mutant androgen receptor accumulation in spinal and bulbar muscular atrophy scrotal skin: A pathogenic marker. Ann. Neurol 2006, 59, 520–526. [Google Scholar]
- Williams, AJ; Paulson, HL. Polyglutamine neurodegeneration: Protein misfolding revisited. Trends Neurosci 2008, 31, 521–528. [Google Scholar]
- Katsuno, M; Adachi, H; Kume, A; Li, M; Nakagomi, Y; Niwa, H; Sang, C; Kobayashi, Y; Doyu, M; Sobue, G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 2002, 35, 843–854. [Google Scholar]
- Takeyama, K; Ito, S; Yamamoto, A; Tanimoto, H; Furutani, T; Kanuka, H; Miura, M; Tabata, T; Kato, S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron 2002, 35, 855–864. [Google Scholar]
- Chevalier-Larsen, ES; O’Brien, CJ; Wang, HY; Jenkins, SC; Holder, L; Lieberman, AP; Merry, DE. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J. Neurosci 2004, 24, 4778–4786. [Google Scholar]
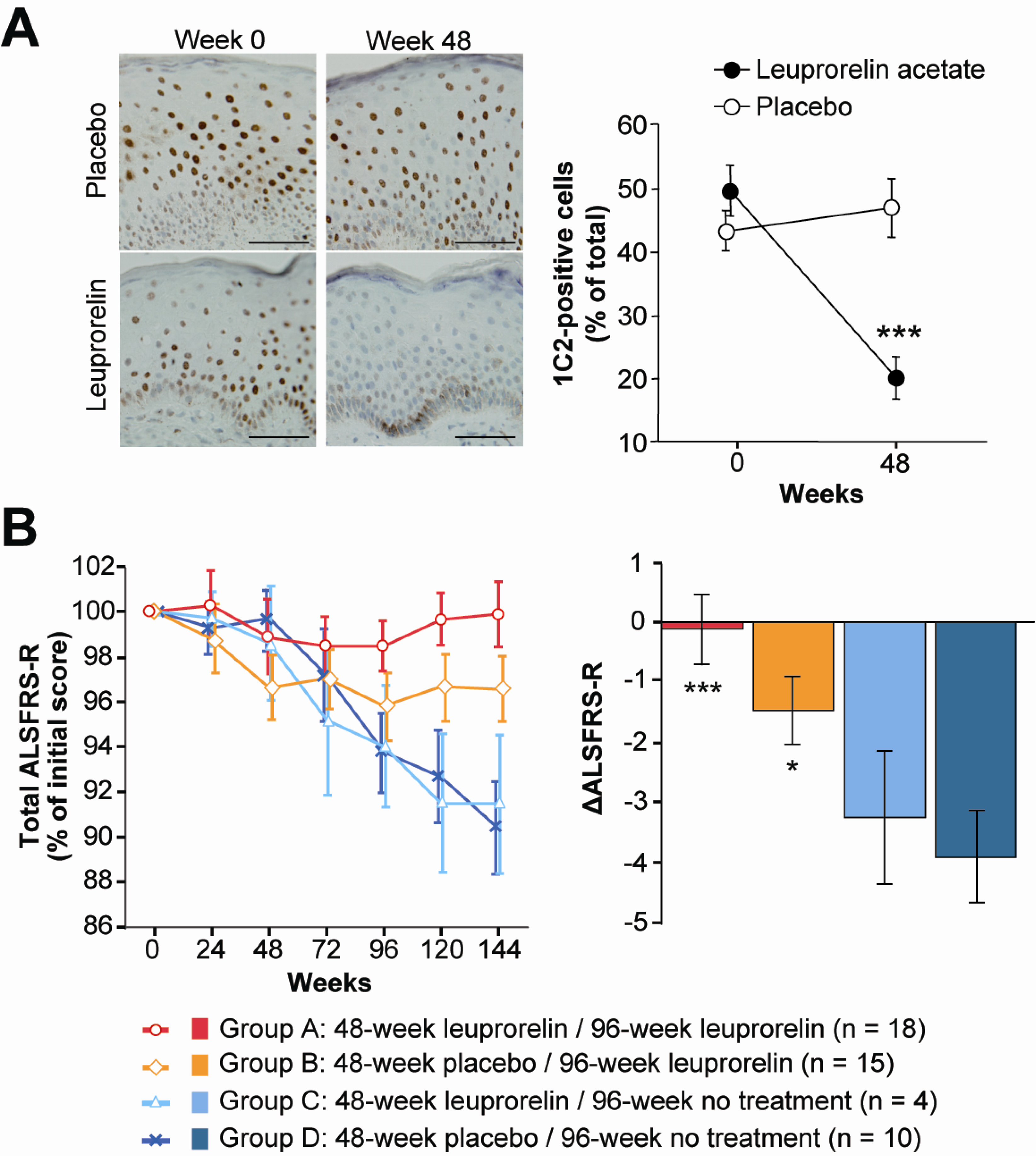
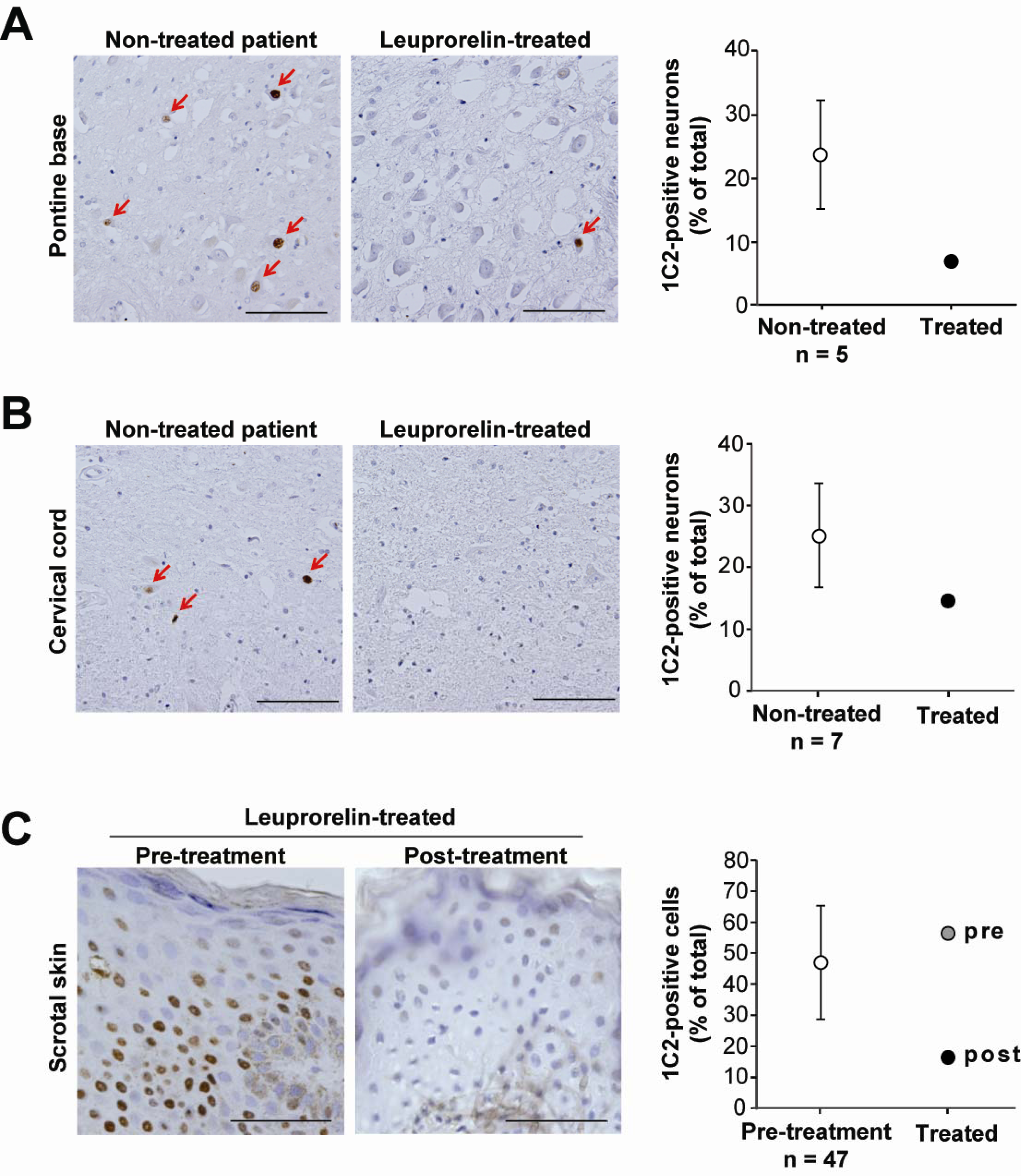
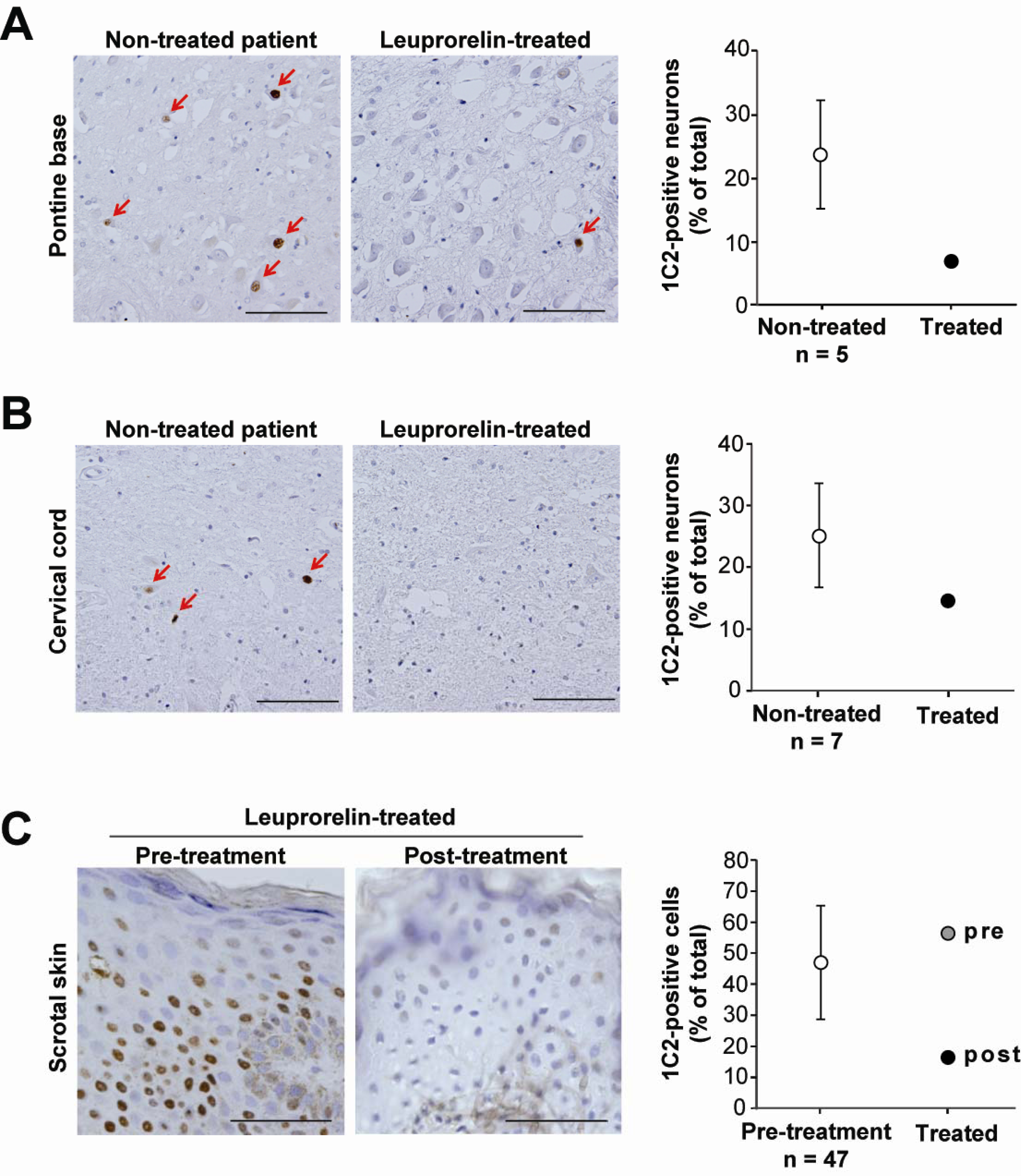
- Katsuno, M; Adachi, H; Doyu, M; Minamiyama, M; Sang, C; Kobayashi, Y; Inukai, A; Sobue, G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat. Med 2003, 9, 768–773. [Google Scholar]
- Banno, H; Katsuno, M; Suzuki, K; Takeuchi, Y; Kawashima, M; Suga, N; Takamori, M; Ito, M; Nakamura, T; Matsuo, K; Yamada, S; Oki, Y; Adachi, H; Minamiyama, M; Waza, M; Atsuta, N; Watanabe, H; Fujimoto, Y; Nakashima, T; Tanaka, F; Doyu, M; Sobue, G. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann. Neurol 2009, 65, 140–150. [Google Scholar]
- Adachi, H; Katsuno, M; Minamiyama, M; Sang, C; Pagoulatos, G; Angelidis, C; Kusakabe, M; Yoshiki, A; Kobayashi, Y; Doyu, M; Sobue, G. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J. Neurosci 2003, 23, 2203–2211. [Google Scholar]
- Katsuno, M; Sang, C; Adachi, H; Minamiyama, M; Waza, M; Tanaka, F; Doyu, M; Sobue, G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc. Natl. Acad. Sci. USA 2005, 102, 16801–16806. [Google Scholar]
- Waza, M; Adachi, H; Katsuno, M; Minamiyama, M; Sang, C; Tanaka, F; Inukai, A; Doyu, M; Sobue, G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med 2005, 11, 1088–1095. [Google Scholar]
- Thomas, M; Harrell, JM; Morishima, Y; Peng, HM; Pratt, WB; Lieberman, AP. Pharmacologic and genetic inhibition of hsp90-dependent trafficking reduces aggregation and promotes degradation of the expanded glutamine androgen receptor without stress protein induction. Hum. Mol. Genet 2006, 15, 1876–1883. [Google Scholar]
- Tokui, K; Adachi, H; Waza, M; Katsuno, M; Minamiyama, M; Doi, H; Tanaka, K; Hamazaki, J; Murata, S; Tanaka, F; Sobue, G. 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in a SBMA model mouse. Hum. Mol. Genet 2009, 18, 898–910. [Google Scholar]
- Steffan, JS; Kazantsev, A; Spasic-Boskovic, O; Greenwald, M; Zhu, YZ; Gohler, H; Wanker, EE; Bates, GP; Housman, DE; Thompson, LM. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar]
- Ying, MY; Xu, R; Wu, XH; Zhu, HX; Zhuang, Y; Han, M; Xu, T. Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J. Biol. Chem 2006, 281, 12580–12586. [Google Scholar]
- Ferrante, RJ; Kubilus, JK; Lee, J; Ryu, H; Beesen, A; Zucker, B; Smith, K; Kowall, NW; Ratan, RR; Luthi-Carter, R; Hersch, SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci 2003, 23, 9418–9427. [Google Scholar]
- Hockly, E; Richon, VM; Woodman, B; Smith, DL; Zhou, XB; Rosa, E; Sathasivam, K; Ghazi-Noori, S; Mahal, A; Lowden, PAS; Steffan, JS; Marsh, JL; Thompson, LM; Lewis, CM; Marks, PA; Bates, GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar]
- Gardian, G; Browne, SE; Choi, DK; Klivenyi, P; Gregorio, J; Kubilus, JK; Ryu, H; Langley, B; Ratan, RR; Ferrante, RJ; Beal, MF. Neuroprotective effects of phenylbutyrate in the N171–82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem 2005, 280, 556–563. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Major clinical features | Affected regions | Causative protein | Gene (locus) |
|---|---|---|---|---|
| Huntington’s disease (HD) | Chorea, cognitive deficits, psychiatric disturbances | Striatum, cerebral cortex | Huntingtin | IT15 (4p16.3) |
| Spinal and bulbar muscular atrophy (SBMA) | Weakness, muscular atrophy, bulbar palsy | Spinal cord, brainstem | Androgen receptor | AR (Xq13-q12) |
| Spinocerebellar ataxia type 1 (SCA1) | Ataxia, bulbar palsy, pyramidal signs, muscular atrophy | Cerebellum, brainstem | Ataxin 1 | SCA1 (6p23) |
| Spinocerebellar ataxia type 2 (SCA2) | Ataxia, slow eye movement, neuropathy | Cerebellum, brainstem | Ataxin 2 | SCA2 (12q24.1) |
| Spinocerebellar ataxia type 3 (SCA3, Machado-Joseph disease) | Ataxia, bulging eye, parkinsonism, spasticity, fasciculations | Cerebellum, basal ganglia, brainstem, spinal cord | Ataxin 3 | SCA3/MJD (14q32.1) |
| Spinocerebellar ataxia type 6 (SCA6) | Ataxia | Cerebellum | α1A-voltage-dependent calcium channel subunit | CACNA1A (19p13) |
| Spinocerebellar ataxia type 7 (SCA7) | Ataxia, retinal degeneration | Cerebellum, retina, brainstem, visual cortex | Ataxin 7 | SCA7 (3p12-p13) |
| Spinocerebellar ataxia type 17 (SCA17) | Ataxia, cognitive deficits, dystonia, parkinsonism | Cerebellum, striatum | TATA box binding protein | TBP (6q27) |
| Dentatorubral-pallidoluysian atrophy (DRPLA) | Ataxia, myoclonic epilepsy, choreoathetosis, cognitive deficits | Cerebellum, cerebral cortex, globus pallidus, red nuclei, subthalamic nuclei | Atrophin 1 | DRPLA (12p13.31) |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Banno, H.; Katsuno, M.; Suzuki, K.; Tanaka, F.; Sobue, G. Neuropathology and Therapeutic Intervention in Spinal and Bulbar Muscular Atrophy. Int. J. Mol. Sci. 2009, 10, 1000-1012. https://doi.org/10.3390/ijms10031000
Banno H, Katsuno M, Suzuki K, Tanaka F, Sobue G. Neuropathology and Therapeutic Intervention in Spinal and Bulbar Muscular Atrophy. International Journal of Molecular Sciences. 2009; 10(3):1000-1012. https://doi.org/10.3390/ijms10031000
Chicago/Turabian StyleBanno, Haruhiko, Masahisa Katsuno, Keisuke Suzuki, Fumiaki Tanaka, and Gen Sobue. 2009. "Neuropathology and Therapeutic Intervention in Spinal and Bulbar Muscular Atrophy" International Journal of Molecular Sciences 10, no. 3: 1000-1012. https://doi.org/10.3390/ijms10031000