Introduction
The monocyclic (poly)enes cyclopropene (
1), cyclopentadiene (
2) and cycloheptatriene (
3) may be regarded as building blocks from which both the fulvene and fulvalene families of molecules can be constructed [
1].
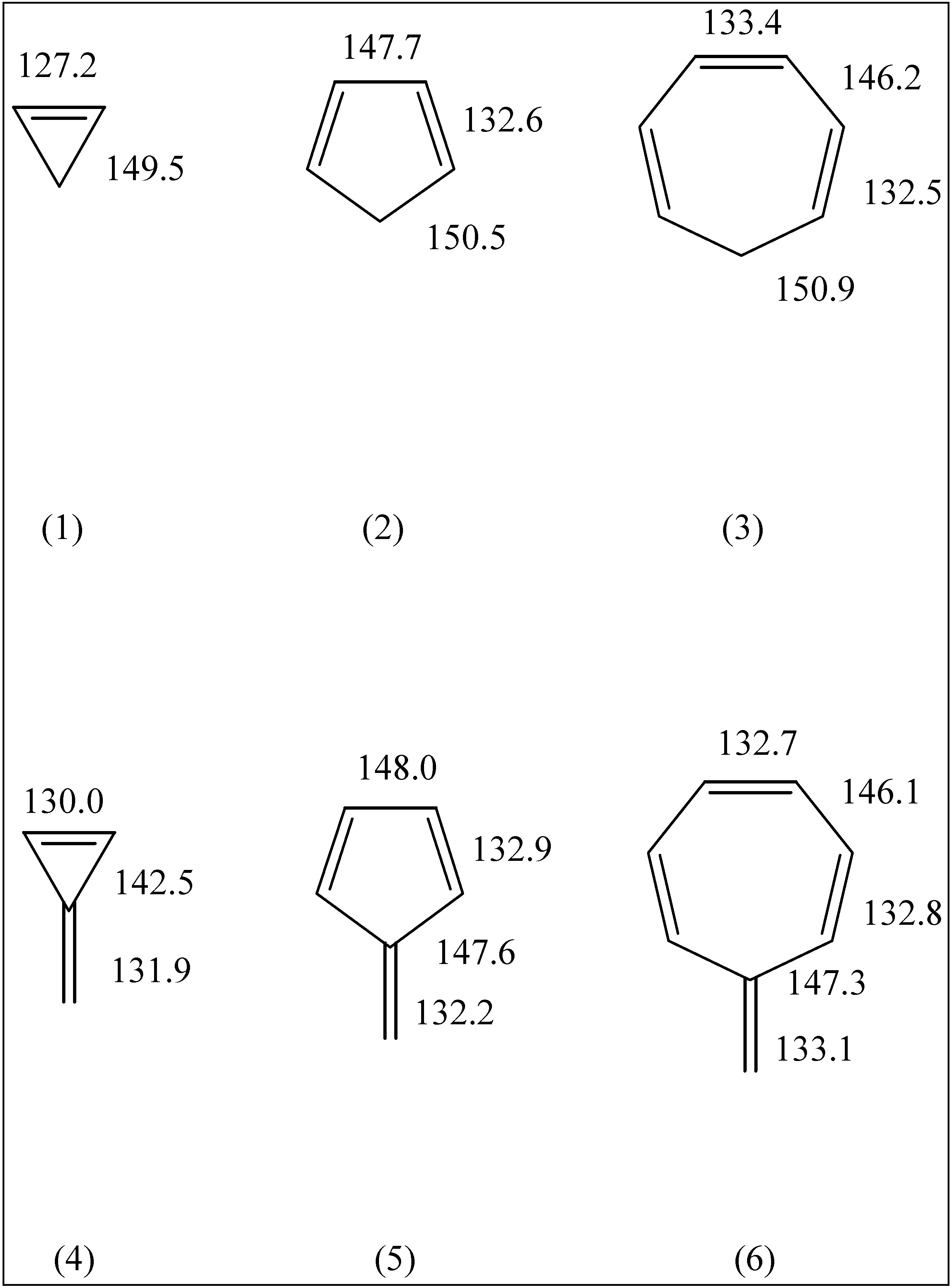
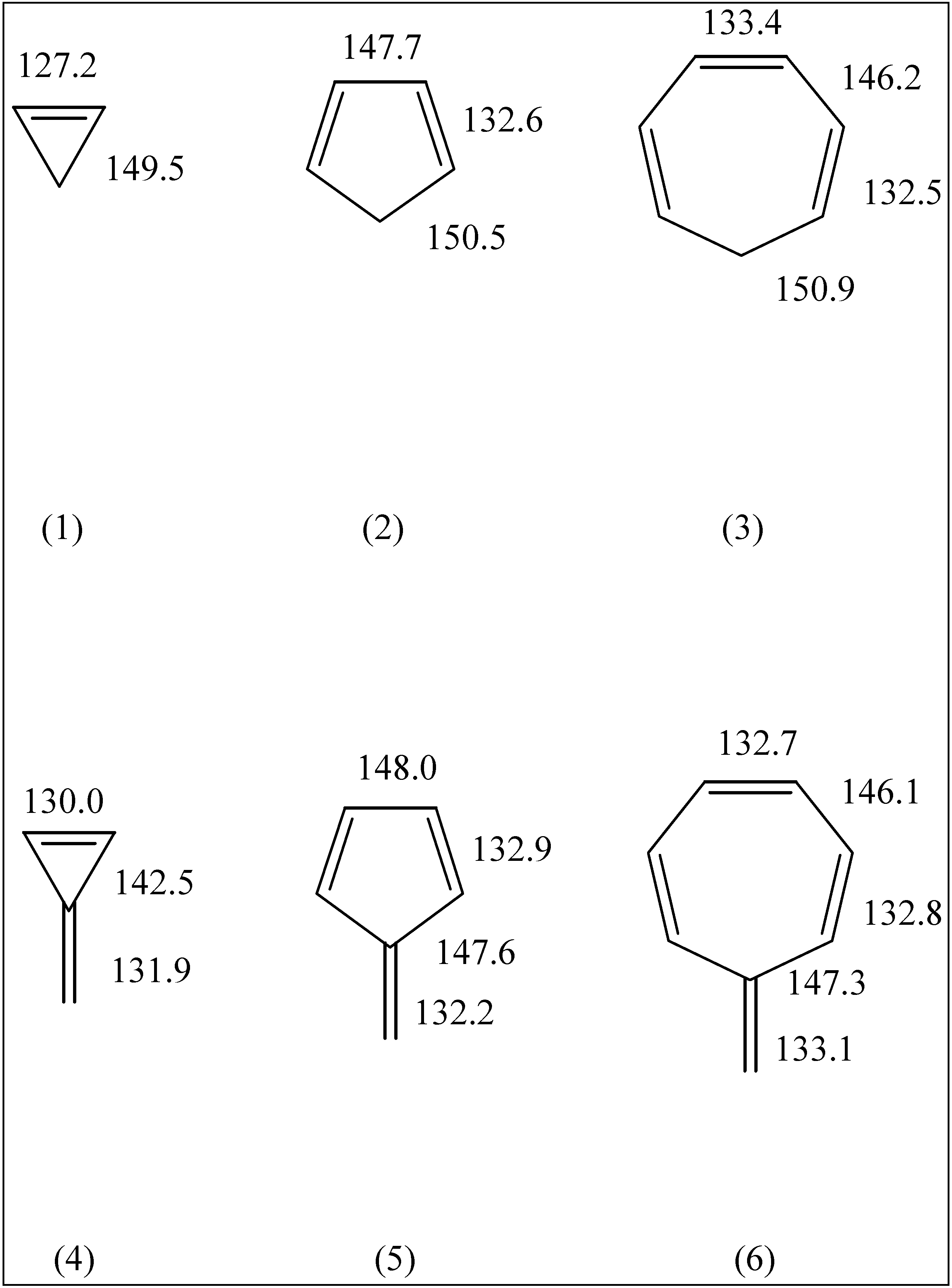
The fulvenes triafulvene (4), pentafulvene (5) and heptafulvene (6) have attracted much interest due to their cross-conjugated structures and questions regarding their aromatic/ antiaromatic character.
Figure 1.
HF geometries of molecules 1 through 6.
Figure 1.
HF geometries of molecules 1 through 6.
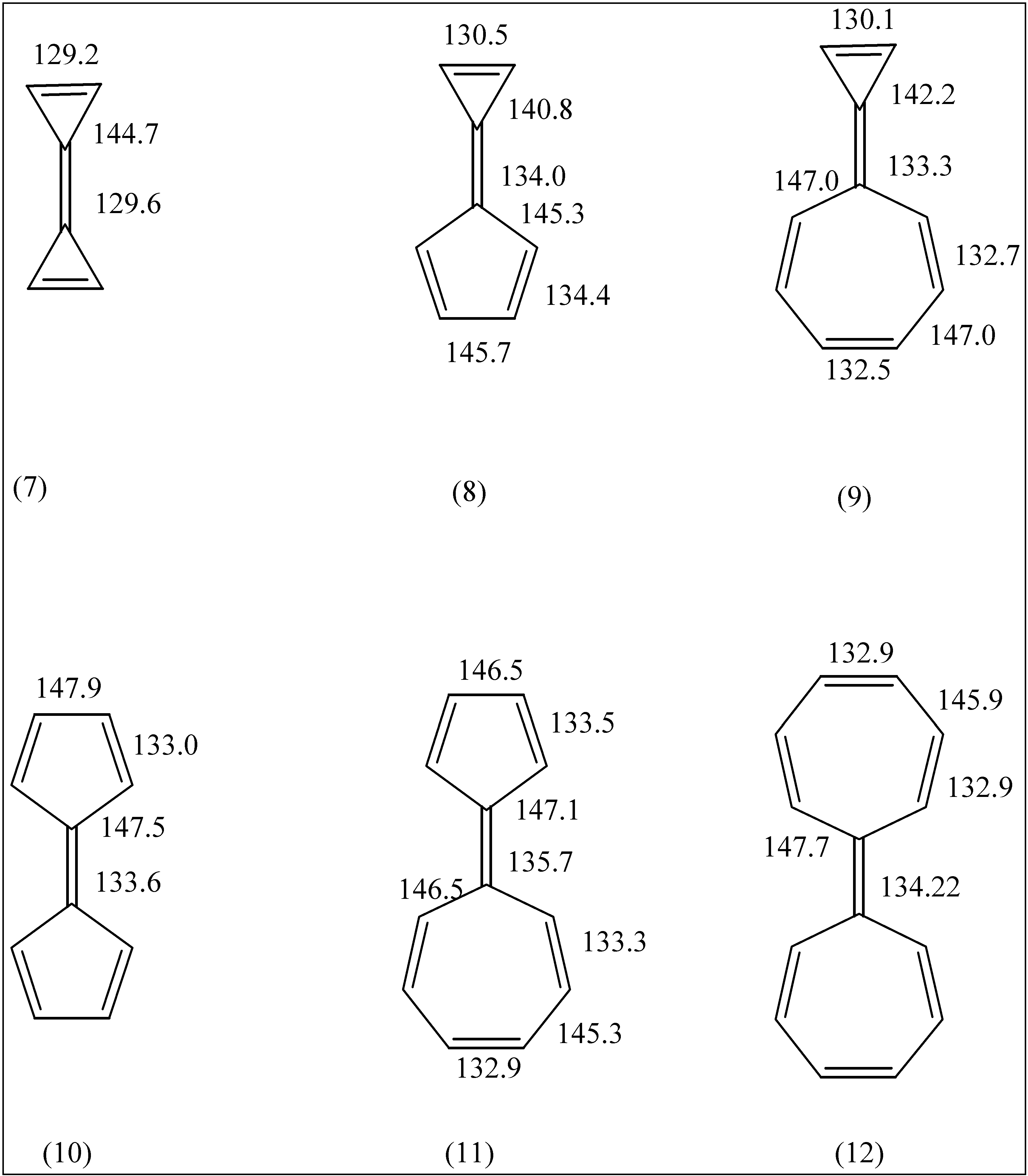
The fulvalenes triafulvalene (7), triapentafulvalene (8), triaheptafulvalene (9) and pentafulvalene (10) have also received a great deal of attention in the literature. The small fulvalenes (7-9) have not been synthesized, but a great deal is known about the chemistry of 12; it is non-planar with a marked alternation of long and short bonds in the seven membered rings and it has a relatively long central C=C bond.
Figure 2.
HF geometries of molecules 7 through 12.
Figure 2.
HF geometries of molecules 7 through 12.
The electric moments of a molecule are quantities of fundamental importance in structural chemistry. When a molecule with permanent electric dipole moment
pe is subject to an external constant electrostatic field
E, the change in the dipole moment can be written in tensor notation as [
2]
Here pe(0) is the dipole in the absence of a field and pe(E) is the dipole moment in the presence of the field. The six independent quantities αij ( j ≥ i) define the dipole polarizability tensor, the ten independent quantities βijk define the first dipole hyperpolarizability and so on.
Hyperpolarizabilities are important when the applied electric field is large. There has recently been an intense search for molecules with large non-zero hyperpolarizabilities [
3], since these substances have potential as the constituents of non-linear optical materials.
The energy U of the molecular charge distribution also changes when an electrostatic field is applied. This change can be written as
Equations (1) and (2) are the key equations for the calculation of molecular polarizabilities and hyperpolarizabilities by gradient techniques [
3]. The dipole polarizability is obtained as the first derivative of the induced dipole with respect to the applied field or the second derivative of the energy. The dipole hyperpolarizability is obtained as the second derivative of the induced dipole with respect to the applied field, and so on. Analytical gradient expressions are available at many levels of theory, otherwise they have to be found by numerical techniques.
The experimental determination of a molecular polarizability is far from straightforward, especially if the molecule has little or no symmetry. For a molecule with symmetry, the principal axes of the polarizability tensor correspond to the symmetry axes. Otherwise the principal axes have to be determined by diagonalization of the polarizability matrix. The principal axes are usually referred to as ‘a’, ‘b’ and ‘c’. We will adopt the convention
where the α
ii are the eigenvalues of the polarizability matrix.
The mean value
can be determined from the refractive index n of a gas according to the equation
where p is the pressure, kB the Boltzmann constant, T the thermodynamic temperature and ꇊ
0 the permittivity of free space. A key assumption in the derivation of equation (5) is that the individual molecules are sufficiently far apart on average that they do not interact with each other.
In a condensed phase, the problem is more complicated because the separation between molecules is of the order of molecular dimensions and their interactions can no longer be ignored. As a result both the external field and the field due to the surrounding molecules polarize each molecule.
The Lorenz-Lorentz equation
applies to non-polar molecules in condensed phases and it can be derived from a detailed consideration of these ideas [
1]. Here, N is the number of molecules in volume V. Rewriting equation (6) in terms of molar quantities defines the molar refractivity
Here M is the molar mass, N
A the Avogadro constant and ρ the density. It appears to be an experimental fact that molar refractivities are additive properties at the molecular level, and a view has long prevailed that the molar refractivity of a molecule is a sum of the molar refractivities of the constituent parts (atoms/ groups) [
5]. Extensive tables of additive atom and group molar refractivities are available [
5,
6]. These tables have been extended to molecular polarizabilities with the compilations of Denbigh [
6] and others.
In the case of molecules with a permanent dipole moment, it is necessary to take account of the orientation polarization. The Debye equation
permits polarizabilities and dipole moments to be determined from measurements of the relative permittivity ∈
r and the density ρ as a function of temperature. Reliable results can only be obtained from dilute solutions [
1].
There is a large literature concerning semi-empirical calculations of dipole polarizabilities and hyperpolarizabilities of organic molecules. It is usually found that the in-plane components of α are well represented at (for example) the AM1 level of theory, but that the perpendicular component is very much smaller than the experimental value.
A few authors have used
Ab Initio techniques to study molecular polarizabilities. It is common knowledge that polarizabilities can only be calculated accurately by employing extended basis sets. In particular, these basis sets have to include diffuse functions that can accurately describe the response of a molecular charge distribution to an external electric field. Such diffuse (s and p) functions are needed in addition to the normal polarization functions; they are denoted by + and ++ in packages such as GAUSSIAN98 [
7].
Once near the Hartree Fock limit, it is necessary to concern oneself with the correlation contribution to such properties. In recent years, density functional techniques have received a great deal of attention in the literature. In Density functional theory (DFT) we write the electronic energy expression [
4,
8] as
where ε
X is the exchange functional and ε
C the correlation functional. In order to calculate ε
X and ε
C it is necessary to give some functional form to the two potentials and then calculate the contribution to the electronic energy as an integral over the electron density (and occasionally the gradient of the electron density). These calculations are performed numerically. There are many variants on the form of the exchange and the correlation functional, most of which are based on the free-electron gas model [
4,
8].
In two earlier notes in this Series [
9,
10], we reported polarizability studies for pentafulvene (
5) and pentafulvalene (
10). The aim of this paper is to collect results for the full series of molecules
1-
12.
Calculations
A. Geometries and dipole moments
All
Ab Initio calculations were made using Gaussian98 [
7]. Geometries were optimized at the HF/6-311G(3d,2p) level of theory.
For the record, the
Ab Initio total energies and electric dipole moments are given in
Table 1.
Table 1.
Total energies ε and dipole moments pe for molecules 1-12.
Table 1.
Total energies ε and dipole moments pe for molecules 1-12.
| Molecule | Key | - ε / Eh* | pe / D* |
| cyclopropene | 1 | 115.858663 | 0.5267 |
| cyclopentadiene | 2 | 192.847328 | 0.3399 |
| cycloheptatriene | 3 | 269.757718 | 0.3343 |
| triafulvene | 4 | 153.715377 | 2.3963 |
| pentafulvene | 5 | 230.709509 | 0.4060 |
| heptafulvene | 6 | 307.621691 | 0.4039 |
| triafulvalene | 7 | 229.345393 | 0 |
| triapentafulvalene | 8 | 306.374585 | 4.9764 |
| triaheptafulvalene | 9 | 383.274605 | 3.7578 |
| pentafulvalene | 10 | 383.359632 | 0 |
| Pentaheptafulvalene | 11 | 460.270703 | 2.2248 |
| heptafulvalene | 12 | 537.177307 | 0 |
A full study of the geometries and electric dipole moments of these molecules at the HF/6-31G* and MP2/6-31G* levels of theory has been given by Scott et. al. [
1]. Since our HF results are very similar to theirs, we simply note the salient C-C bond lengths in
Figure 1 and
Figure 2. All the fulvenes (
4-
6) and the smaller fulvalenes (
7,
9 and
10) are found to be planar. Pentaheptafulvalene (
11) is slightly non-planar whilst heptafulvalene (
12) has a folded C
2h structure. The calculated C-C bond lengths are consistently smaller than the experimental values. Such behaviour is common for HF calculations on molecules with multiple bonds.
B. Polarizabilities
Polarizabilities were calculated at the HF/6-311G(3d,2p) // HF/6-311++G(3d,2p) and HF/6311G(3d,2p) // BLYP/6-311++G(3d,2p) levels of theory. That is, the geometries discussed above were used unchanged, but two sets of extra diffuse s and p functions were added to the basis sets for the purpose of polarizability calculations.
Dipole polarizabilities calculated at the HF level of theory are shown in
Table 2.
Table 2.
Principal dipole polarizability tensor components (HF).
Table 2.
Principal dipole polarizability tensor components (HF).
| Molecule | αaa / au* | αbb | αcc | <α> |
| 1 | 26.863 | 35.555 | 36.031 | 32.816 |
| 2 | 41.076 | 58.844 | 64.517 | 54.812 |
| 3 | 57.287 | 86.666 | 92.616 | 78.856 |
| 4 | 32.943 | 41.913 | 65.566 | 46.807 |
| 5 | 45.628 | 65.052 | 104.717 | 71.799 |
| 6 | 57.889 | 95.501 | 146.247 | 99.879 |
| 7 | 42.299 | 62.725 | 104.999 | 70.008 |
| 8 | 56.058 | 85.029 | 151.931 | 97.673 |
| 9 | 67.858 | 113.537 | 193.133 | 124.843 |
| 10 | 67.812 | 101.817 | 213.058 | 127.562 |
| 11 | 80.122 | 132.732 | 285.781 | 166.212 |
| 12 | 101.169 | 159.462 | 287.267 | 182.633 |
The corresponding results at BLYP level are shown in
Table 3.
Table 3.
Principal dipole polarizability tensor components (BLYP).
Table 3.
Principal dipole polarizability tensor components (BLYP).
| Molecule | αaa / au | αbb | αcc | <α> |
| 1 | 28.153 | 36.908 | 38.820 | 34.627 |
| 2 | 42.523 | 64.667 | 67.458 | 58.216 |
| 3 | 60.273 | 94.617 | 98.119 | 84.336 |
| 4 | 33.809 | 44.115 | 67.630 | 48.518 |
| 5 | 46.266 | 71.108 | 103.363 | 73.579 |
| 6 | 58.484 | 103.872 | 149.783 | 104.046 |
| 7 | 43.342 | 65.394 | 110.142 | 72.959 |
| 8 | 56.904 | 91.458 | 154.105 | 100.822 |
| 9 | 68.827 | 122.486 | 202.319 | 131.211 |
| 10 | 68.581 | 111.201 | 211.555 | 130.446 |
| 11 | 80.706 | 144.867 | 291.086 | 172.22 |
| 12 | 104.681 | 174.461 | 311.392 | 196.845 |
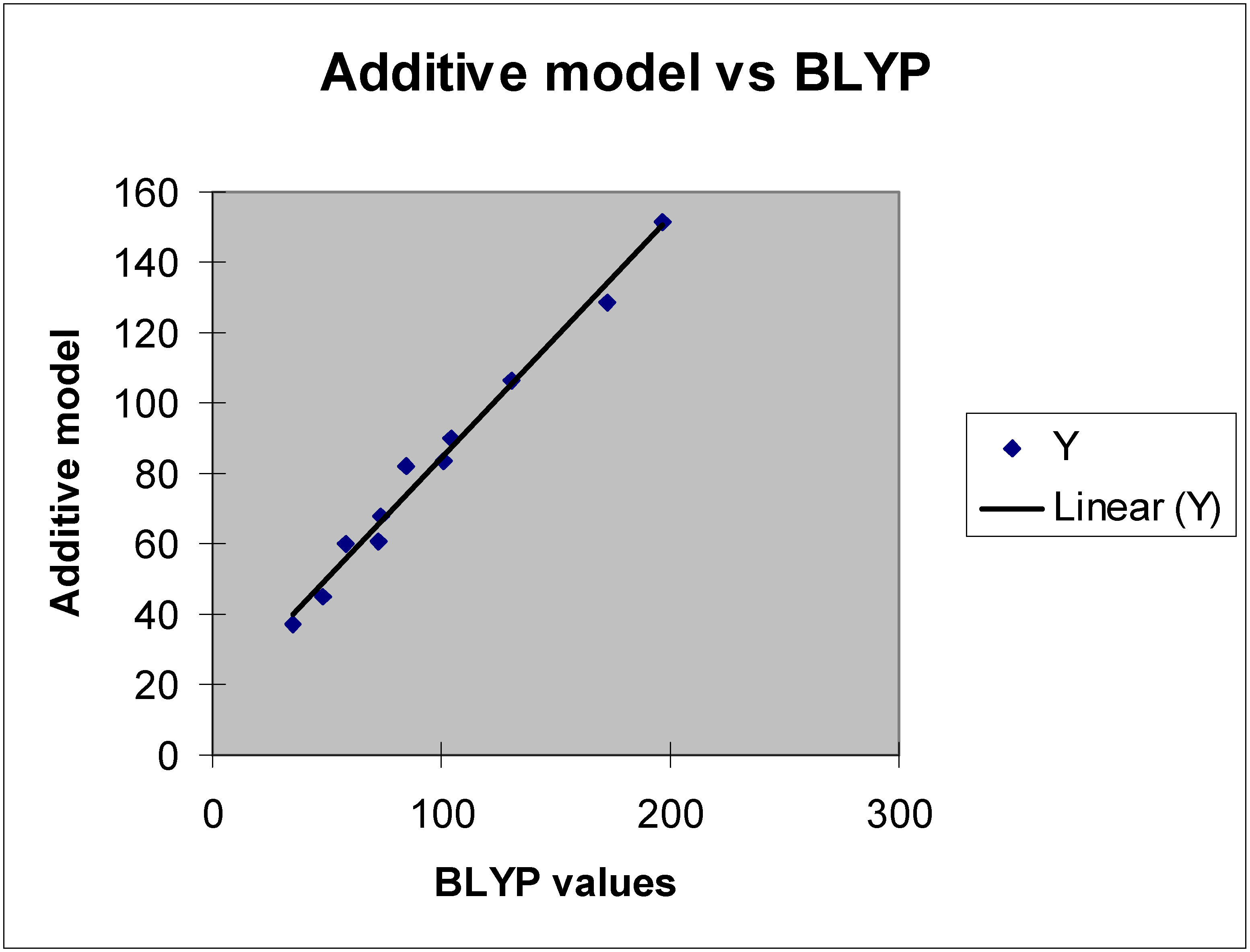
The BLYP polarizabilities are generally a few percent higher than the corresponding values calculated at HF level.
We discussed above the possibility that molecular polarizabilities could be decomposed into contributions from the constituent atoms and/ or groups. In an earlier paper [
11], we suggested the following atom contributions (
Table 4) based on our analysis of a number of conjugated hydrocarbons.
Table 4.
Atom Contributions to <α>.
Table 4.
Atom Contributions to <α>.
| Level of theory | αC / au. | αH/ au |
| HF/6-311++G(3d,2p) | 8.3020 | 2.3606 |
| BLYP/6-311++G(3d,2p) | 7.9110 | 3.3772 |
These values can be used to calculate the mean polarizabilities for molecules
1 through
12. Analysis shows a good straight-line relationship, but poor overall absolute agreement between the BLYP values and those predicted on the basis of simple additivity. Re-analysis of the atom polarizability values needed to give good absolute agreement with experiment along the lines discussed in [
11] suggests that the BLYP values in
Table 4 should be adjusted to 15.68 and –2.75 au.
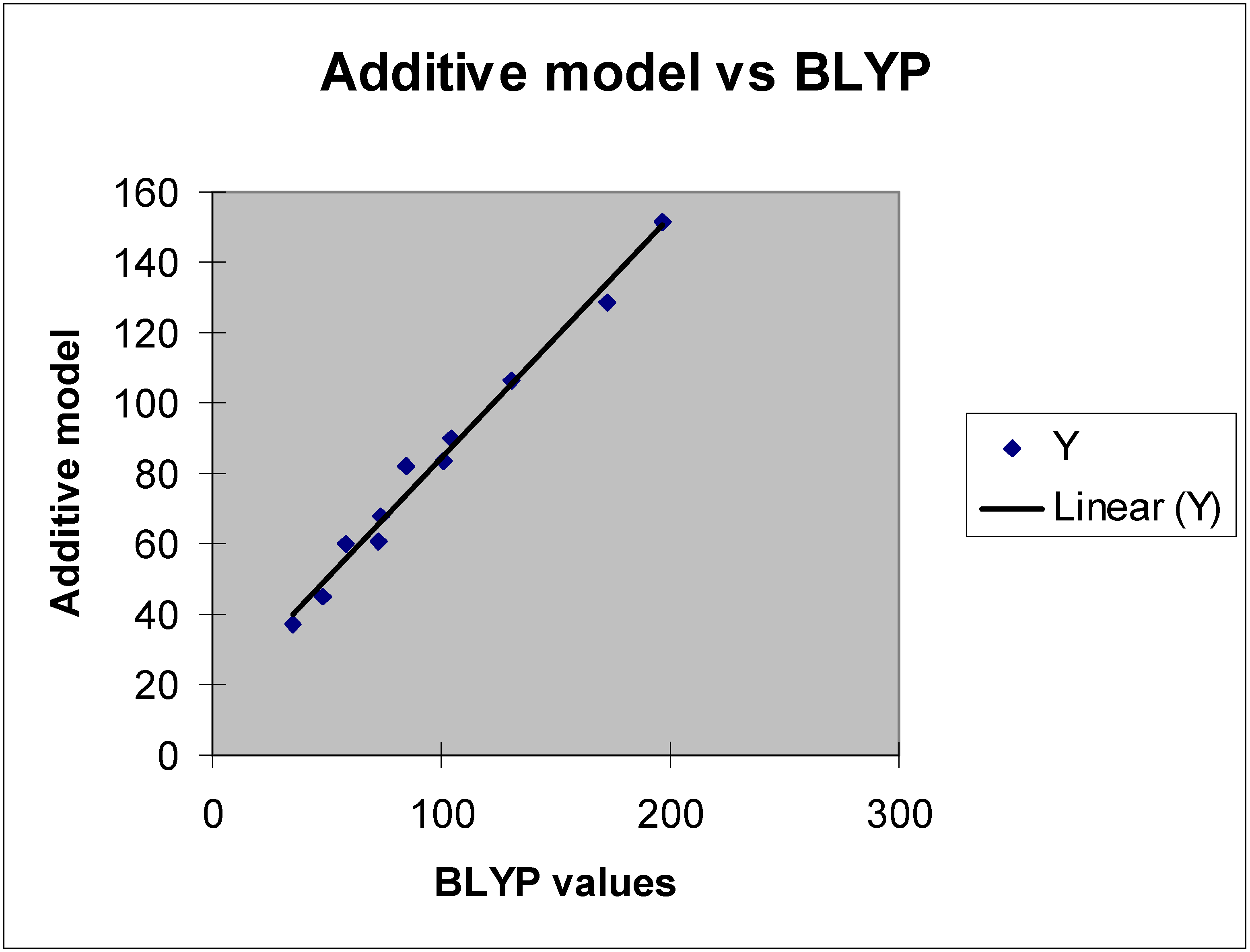
Figure 3.
Regression analysis.
Figure 3.
Regression analysis.
The “additive group” model has been criticised on the grounds that it does not allow for the interaction between atoms and groups in a molecule [12]. Nevertheless, it can sometimes give useful chemical insight. Thus the BLYP results for ethane, ethene and cyclopropane are given in
Table 5, for comparison against cyclopropene (
1).
Table 5.
BLYP mean polarizabilities.
Table 5.
BLYP mean polarizabilities.
| Molecule | <α> / au |
| Ethane | 29.007 |
| Ethene | 27.454 |
| Cyclopropane | 36.900 |
| Cyclopropene (1) | 34.627 |
Cyclopropene has a smaller <α> than cyclopropane. If we regard cyclopropene as ethene plus a carbon atom, then the additive model gives an <α> of 35.365, in modest agreement with the full BLYP calculation. Likewise, ethane plus a carbon atom gives a predicted <α> = 36.918 au for cyclopropane, again in modest agreement with the calculated value.
Cyclopentane has <α> = 63.592 au, which is to be compared with a value for cyclopentadiene (2) plus two hydrogen atoms of 64.970 au.
The Ab Initio dipole polarizabilities for 9 and 10 are almost identical.
There is no experimental data in the literature.
C. Hyperpolarizabilities
Hyperpolarizability data is also hard to come by, both experimentally and theoretically. There is no experimental data in the literature for any of the molecules
1 through
12. The tensor components for molecules
1 through
6 are shown in
Table 6, and those for the remaining molecules in
Table 7.
Table 6.
Hyperpolarizability tensor components (HF/6-311++G(3d,2p)) for (1) through (6)*.
Table 6.
Hyperpolarizability tensor components (HF/6-311++G(3d,2p)) for (1) through (6)*.
| Component | 1 | 2 | 3 | 4 | 5 | 6 |
| aaa | 0 | 0 | -13.99 | .02 | 0 | .02 |
| aab | .01 | 0 | 0 | 0 | 0 | 0 |
| aac | -10.02 | 10.73 | -4.78 | 61.38 | -4.85 | 25.70 |
| abb | 0 | -.02 | 17.99 | .01 | 0 | 0 |
| abc | 0 | 0 | 0 | 0 | 0 | 0 |
| acc | 0 | .01 | -8.92 | .04 | 0 | -.04 |
| bbb | .12 | 0 | 0 | 0 | 0 | 0 |
| bbc | -57.80 | -16.63 | 6.56 | -59.05 | -26.26 | -3.66 |
| bcc | -.10 | 0 | 0 | 0 | 0 | 0 |
| ccc | 19.87 | 35.05 | 0.91 | -10.24 | -2.76 | 210.14 |
Table 7.
Hyperpolarizability tensor components (HF/6-311++G(3d,2p)) for (7) through (12).
Table 7.
Hyperpolarizability tensor components (HF/6-311++G(3d,2p)) for (7) through (12).
| Component | 7 | 8 | 9 | 10 | 11 | 12 |
| aaa | 0 | 0 | -.016 | 0 | -.026 | 0 |
| aab | 0 | 0 | -.730 | 0 | .339 | 0 |
| aac | 0 | 63.617 | 24.409 | 0 | 45.080 | 0 |
| abb | 0 | .168 | -.013 | 0 | .146 | 0 |
| abc | 0 | .003 | 0 | 0 | .015 | 0 |
| acc | 0 | -.189 | .015 | 0 | -.157 | 0 |
| bbb | 0 | -.002 | 0 | 0 | .220 | 0 |
| bbc | 0 | 6.606 | 30.038 | 0 | 140.918 | 0 |
| bcc | 0 | .002 | .008 | 0 | -.187 | 0 |
| ccc | 0 | -329.424 | 676.918 | 0 | 732.070 | 0 |
Molecules (
7), (
10) and (
12) have zero dipole hyperpolarizabilities, on account of their geometries. The dipole hyperpolarizabilities of a conjugated molecule can be greatly enhanced by the presence of a donor and acceptor group located at either end of the conjugation path. Strong donors and acceptors exert the largest effect [
13], and the nature of the conjugation path between the donor and acceptor group is also of great importance. It is interesting to note the large dipole hyperpolarizabilities of molecules
8,
9 and
11, which is consistent with this charge transfer mechanism.
All our calculations refer to static fields. The interaction of molecules with oscillating electric fields leads to different non-linear optical processes typified by the electro-optic Pockels effect (EOPE), second harmonic generation (SHG), optical rectification (OR), DC-electric field induced (EFI)SHG, third harmonic generation (THG), Optical Kerr Effect (OKE), DC-electric field induced (FEI)OR etc. These optical processes have to be handled theoretically using so-called time dependent techniques.



{kind=link}
{kind=link}
{kind=link}