Synthesis and Catalytic Activity of Two New Cyclic Tetraaza Ligands

1
Institut für Organische Chemie, Universität Regensburg, D-93040 Regensburg, Germany
2
Current address: Department of Chemistry, Technical University of Denmark, Denmark
*
Author to whom correspondence should be addressed.
Molecules 2003, 8(5), 453-458; https://doi.org/10.3390/80500453
Submission received: 11 April 2003
/
Revised: 9 May 2003
/
Accepted: 9 May 2003
/
Published: 31 May 2003
{kind=link}
{kind=link}
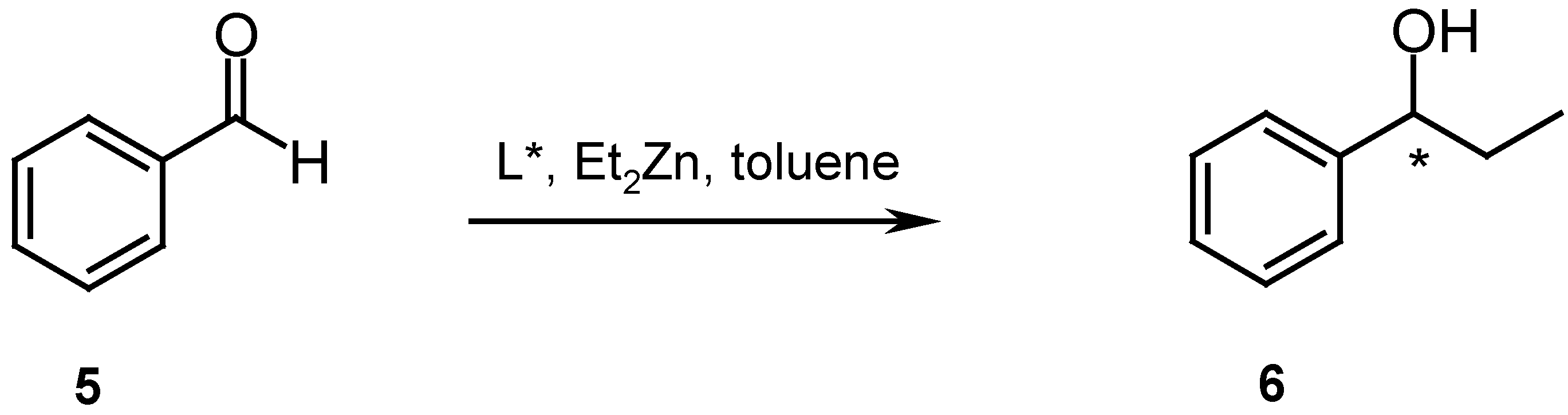{kind=link}
Abstract
:Two new chiral cyclic tetraaza ligands were synthesized and characterized. Their catalytic activity was tested in the asymmetric addition of diethylzinc to benzaldehyde. The expected secondary alcohol was obtained in moderate yields, but with very low enantioselectivity.
Introduction
The importance of nitrogen-containing ligands as catalysts for many asymmetric transformations has grown in the last years [1] because of their high stability, easy preparation and promising results [2]. In 1969 Uhlemann developed the synthesis of a new chiral Schiff base 1 from o-amino-benzaldehyde and 1,2-cyclohexanediamine [3]. Compounds containing this optically active trans-cyclohexane-1,2-diamine moiety have proven to be very useful in both asymmetric synthesis [4] and diastereomeric recognition of peptides [5].
We report in this paper the synthesis and characterization of two new tetraaza ligands containing 1,2-cyclohexanediamine as the chiral unit and their ability to catalyse the addition of diethylzinc to benzaldehyde.
Results and Discussion
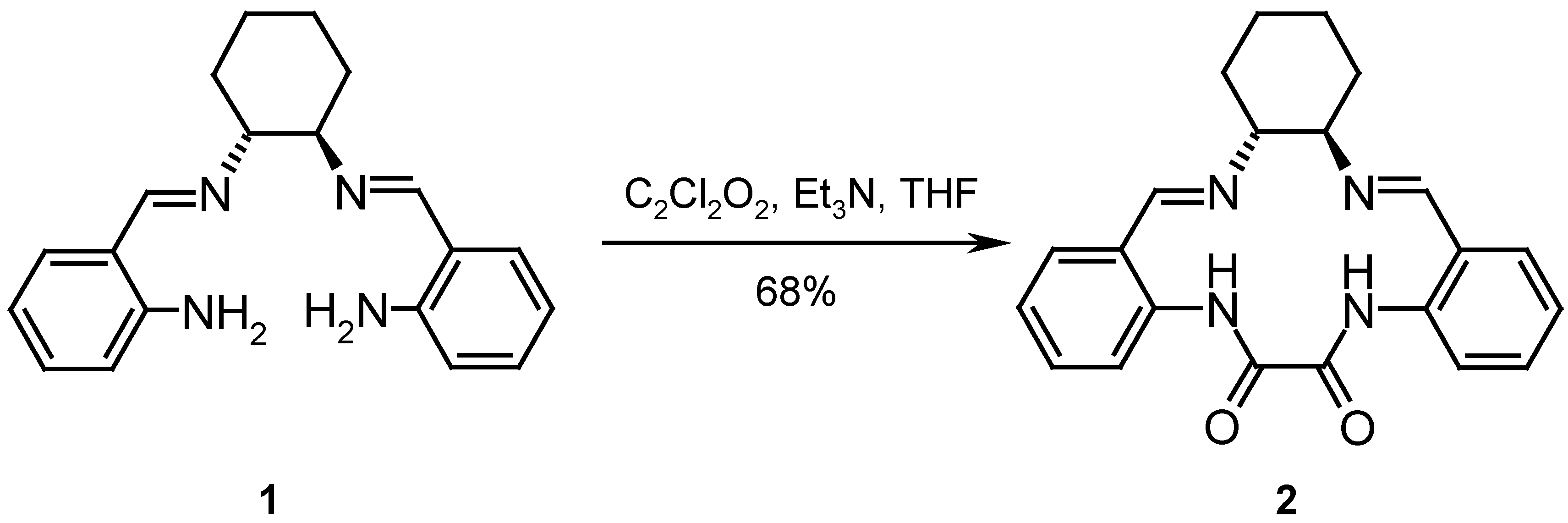
The ligand 2 was synthesized from compound 1 by reaction with oxalyl chloride in THF in the presence of Et3N as catalyst, according to a procedure reported in the literature [6] (Scheme 1). After flash chromatography of the crude product, the cyclic compound 2 was obtained in a 68 % yield as colorless crystals.
Scheme 1.
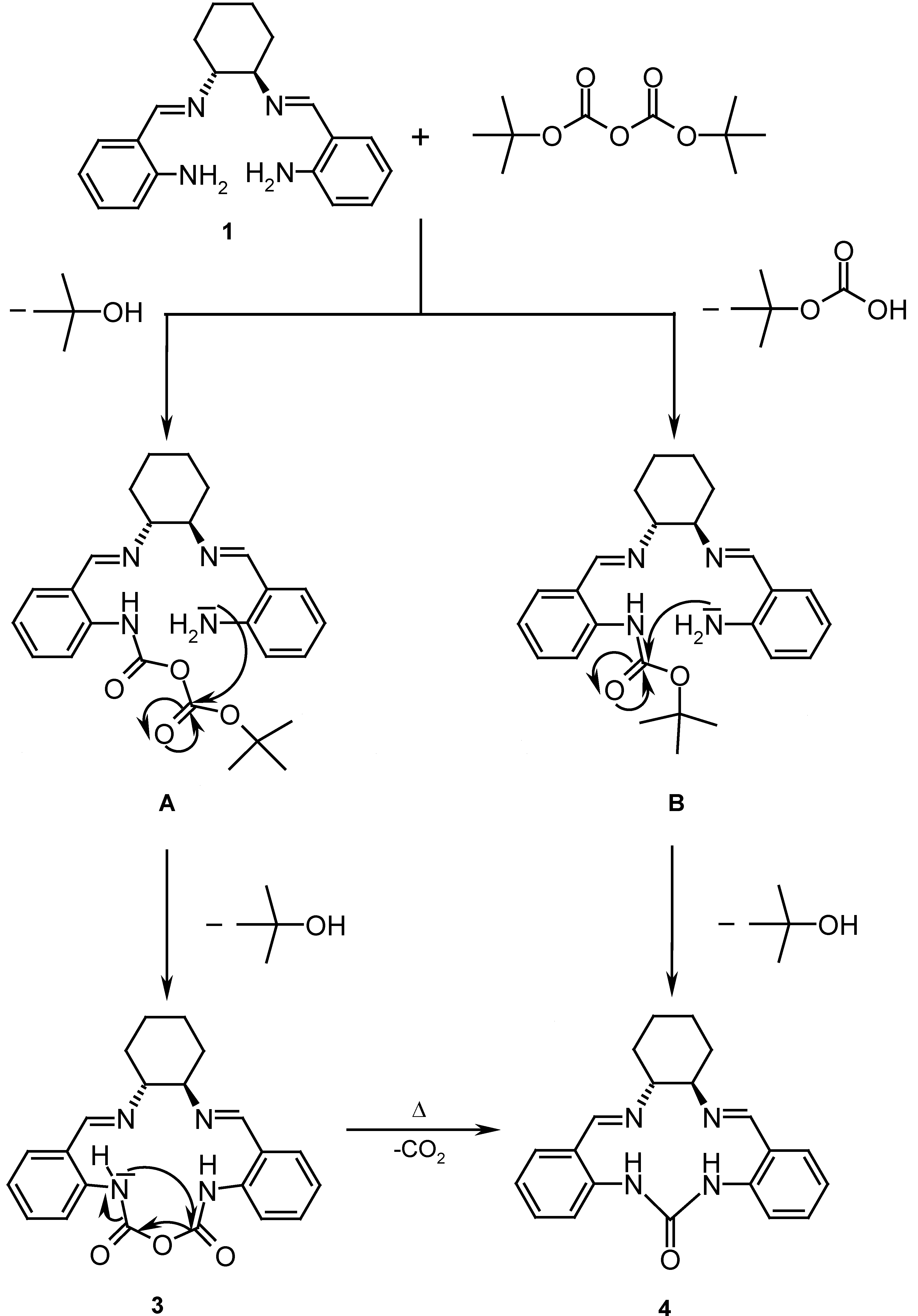
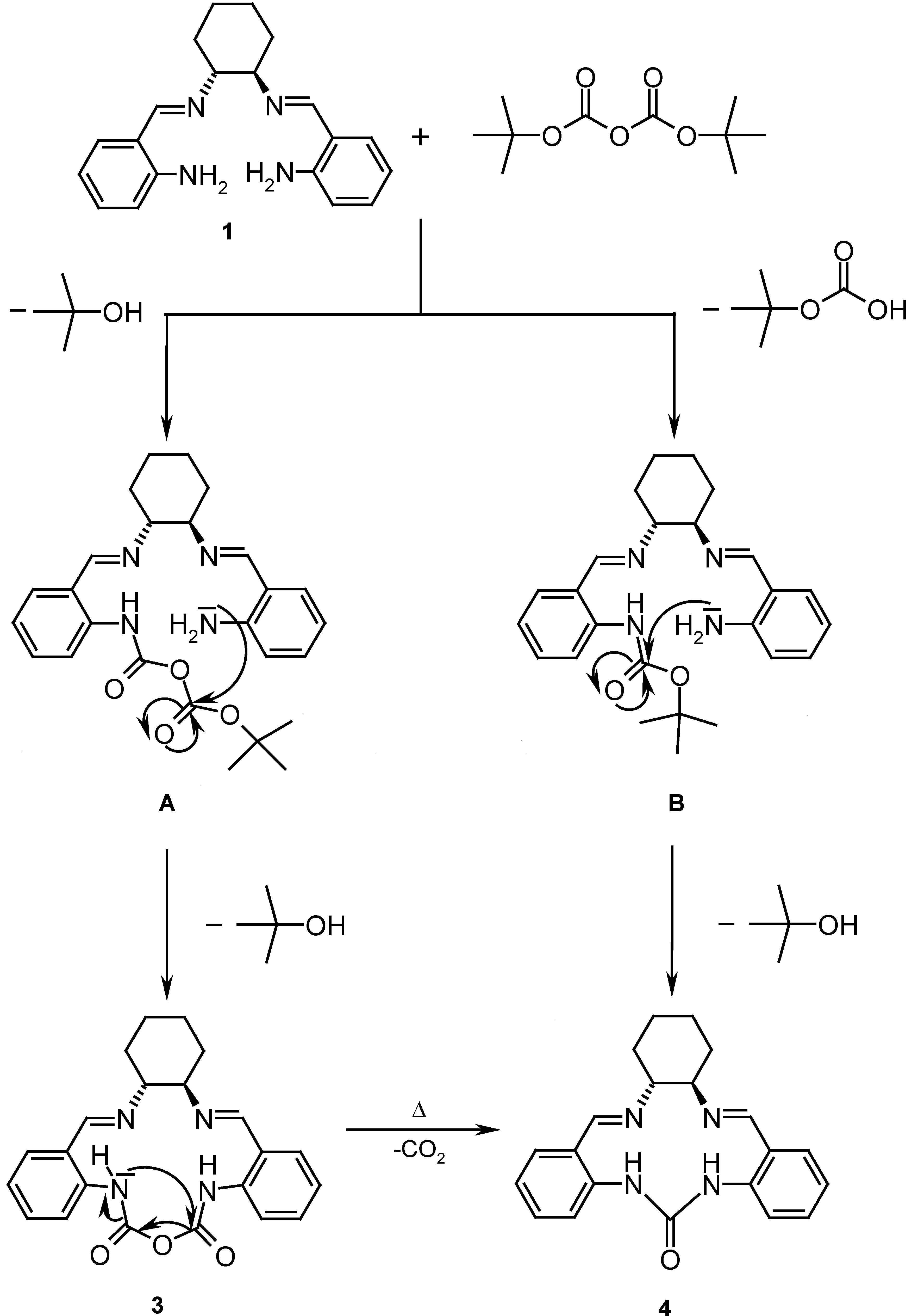
The synthesis of ligand 4 using 1 as starting material was attempted by reaction with di-tert-butyl dicarbonate (Boc2O) in CH2Cl2 using 4-dimethylaminopyridine (DMAP) as catalyst. Such reactions for similar substrates have been reported to give yields of 87-96 % [7]. According to the mechanism shown in Scheme 2, an intermediate urethane B is formed by reaction of the amine nitrogen with the Boc2O. Intramolecular nucleophilic addition of the second arylamine to the urethane or an isocyanate, which may evolve from B by elimination of tert-butanol should yield urea 4.
After performing the reaction and successive chromatographic separations, a colorless product was obtained. This product, however, displays in its 13C-NMR spectrum two carbonyl group signals at δ = 151.3 and δ = 151.7, while only one was expected. 1H-NMR spectra and mass spectrometry (molecular ion m/z = 391) indicated a different product with structure 3. A likely mechanism for its formation is the reaction of 1 with Boc2O and loss of tert-butanol to A followed by intramolecular cyclization to yield 3. Only a few reports of stable carbamic anhydrides exist in the literature [8]. The proton and carbon NMR spectra of 3 indicate a non C2 symmetric conformation of the molecule. The 1H-13C HMBC NMR data show that each N-H group can be correlated to one CO carbon.
It was thought that upon heating compound 3, it would release CO2, thus leading to 4. This was tested by differential scanning calorimetry (DSC) [9], which showed a reaction of 3 at 259 °C, which indeed led to a loss of weight corresponding to CO2 elimination, however, the high temperature at which this reaction occurs makes it of limited synthetic interest [10].
Scheme 2.
Catalysis
The cyclic diimine ligands 2 and 3 were examined as catalysts for the asymmetric addition of diethylzinc to benzaldehyde (Scheme 3).
Scheme 3.
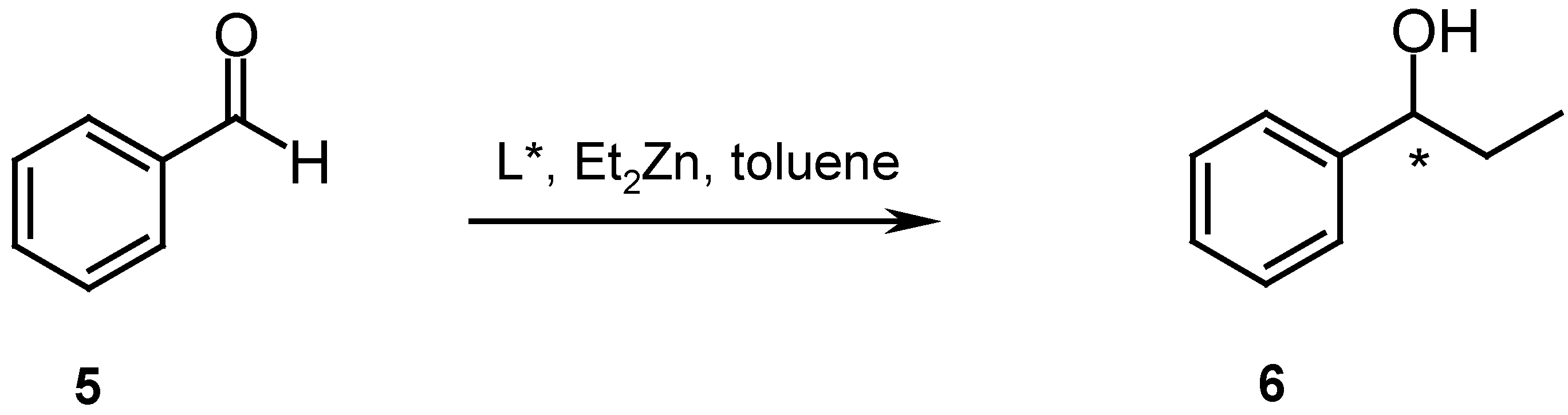
Moderate yields (51 and 88 %, respectively) of 1-phenylpropanol (6) were obtained, but no or very low enantioselectivities were observed [11]. The possible cause of these low selectivities could be the small size of the available cavity, which is not large enough to accommodate the Zn atom to form the intermediate zincate essential for the intramolecular alkyl transfer reaction.
Experimental
General
Melting points were determined with a Büchi SMP 20 and are uncorrected. IR-spectra were recorded with a Bio-Rad FTS 3000 MX FT-IR. 1H-NMR and 13C-NMR were recorded with a Bruker ARX 400 or a Bruker AC 250 instruments at 250 and 62.9 MHz, respectively. The 1H chemical shifts are reported in δ (ppm) relative to CDCl3 (7.26 ppm), DMSO-d6, (2.49 ppm) and TMS (0 ppm), while 13C chemical shifts are reported in δ (ppm) relative to CDCl3 (77 ppm), DMSO-d6 (36.9 ppm) and TMS (0 ppm). MS-spectra were recorded on a Varian CH-5 (EI) and a Finnigan MAT SSQ 7000 (ESI). Solvents were distilled and dried according to standard laboratory methods [12].
Synthesis of the ligands
7,10-Cyclooxalamide-N,N´-bis-(phenyl-2-ylmethylene)-cyclohexane-1R,2R-diamine (2): Schiff base 1 (400 mg, 1.25 mmol) and dry Et3N (0.35 ml, 2.51 mmol) were dissolved in dry THF (20 mL) under nitrogen with stirring. A solution of oxalyl chloride (0.15 ml, 1.77 mmol) in dry THF (25 mL) was added with a syringe pump over a period of 2.5 h. The reaction mixture was then stirred for 19 h at room temperature. The mixture was concentrated to half of its volume and the colourless precipitate of the product was filtered off. Flash chromatography of the raw product in CHCl3 afforded the cyclic compound 2 (318 mg, 0.85 mmol, 68 %), mp = 223-225 °C (dec.); = -17° (c 1.0, CHCl3); IR (KBr): ν = 3440, 2963, 2925, 2854, 2359, 1683, 1635, 1577, 1506, 1440, 1290, 1261, 1159, 1093, 1025, 939, 859, 802, 753 cm-1; 1H-NMR (CDCl3): δ = 1.53-2.13 (m, 8H), 3.79 (s, 2H), 7.00 (dd, 2H, J = 7.6 Hz), 7.18 (d, 2H, J = 7.6 Hz), 7.40 (dd, 2H, J = 7.9, 7.6 Hz), 8.52 (s, 2H, HC=N), 8.87 (d, 2H, J = 7.9 Hz), 14.22 (s, 2H, PhNH); 13C-NMR (CDCl3): δ = 24.38, 33.1, 73.59, 119.95, 122.75, 123.52, 130.75, 132.85, 138.57, 159.74, 163.24; MS (PI-EIMS) m/z (%): 374.2 (8) [M+.], 228.2 (22) [M+. – N=CH-Ph-NHCO], 147.1 (100) [N=CH-Ph-NHCO]
7,10-Cyclodicarbonic-diamide-N,N´-bis-(phenyl-2-ylmethylene)-cyclohexane-1R,2R-diamine (3): Di-tert-butyl dicarbonate (Boc2O, 657 mg, 3.01 mmol), 4-dimethylaminopyridine (DMAP, 41 mg, 0.34 mmol) and Schiff base 1 (500 mg, 1.56 mmol) were dissolved with stirring in dry CH2Cl2 (10 mL) under nitrogen. The reaction mixture was stirred for 30 minutes at room temperature and then at 40 °C for 6 h. Evaporation of the solvent and crystallization of the residue from EtOH (50 mL) afforded crude 3. After two purifications by column chromatography (eluting with AcOEt) a colorless product (230 mg, 0.59 mmol, 38 %) was obtained, mp = 253-255 °C (dec.); = -20° (c 1.5, DMSO); IR (KBr): ν = 3413, 3208, 3062, 2923, 2856, 1679, 1608, 1498, 1463, 1385, 1296, 1270, 1227, 1157, 1139, 938, 794, 754 cm-1; 1H-NMR (DMSO-d6): δ = 1.31-1.45 (m, 3H), 1.73-1.77 (m, 3H), 1.90-1.95 (m, 1H), 2.63-2.67 (m, 1H), 3.62-3.71 (m, 1H), 5.04-5.09 (m, 1H), 5.96 (s, 1H, HC=N), 6.51 (s, 1H, HC=N), 6.79-7.16 (m, 4H), 7.20-7.23 (m, 3H), 7.29-7.32 (m, 1H), 9.68 (s, 1H, PhNH), 9.71 (s, 1H, PhNH); 13C-NMR (DMSO-d6): δ = 24.93, 25.58, 30.72, 31.96, 53.96, 63.76, 81.24, 87.63, 113.29, 113.34, 118.03, 118.21, 120.87, 120.92, 127.28, 127.43, 129.27, 129.46, 136.47, 136.53, 151.33, 151.67; MS (CI-MS) m/z (%): 391.3 (100) [MH+.], 244.2 (73), [MH+. – N=CH-Ph-NHCO]
Catalysis: Preparation of 1-Phenyl-1-propanol (6): The ligand (0.05 mmol, 5 mol %) was dissolved in dry toluene (6 mL) under nitrogen, diethylzinc (1.1 M solution in toluene, 0.1 mL, 0.11 mmol) was added, and the mixture was allowed to stir for 1 h at room temperature, then cooled to 0 °C or maintained at room temperature. The remaining diethylzinc (2.17 ml, 2.39 mmol) was added slowly. After five minutes the aldehyde (1 mmol) was added. The reaction was stirred until no more aldehyde was observed (TLC), then quenched with 2 M HCl (6 mL). The layers were separated and the aqueous phase was extracted with Et2O (3 x 10 mL). The combined organic extracts were dried with Na2SO4, filtered and concentrated under reduced pressure. The product was purified by short path distillation to give the alcohol as colorless oil. Rf = 0.32 (hexane/ethyl acetate 5:1). - = +30.6 (c = 1.11, CHCl3) for the (R)-enantiomer [13]; 1H-NMR (CDCl3): δ = 0.89 (t, 3H, J = 7.5 Hz, CH3), 1.74-1.83 (m, 2H, J = 13.6, 7.5, 7.0 Hz, CH2), 2.46 (s, OH), 4.59 (t, 1H, J = 7.0 Hz, CH), 7.29-7.37 (m, 5H, ArH); GC Analysis: HP 5890 II Chromatograph, FID Detector 300 °C, Injector Temperature 260 °C, Column Restek Rt βDEX cst, 30 m, 0.32 mm, 0.25 μm, Oven Temperature 85 °C, Carrier gas H2, Column Head Pressure 3 bar. Retention times: 24.36 min (R), 26.45 min (S)
References and Notes
- Fache, F.; Schulz, E.; Tommasino, M.; Lemaire, M. Chem. Rev. 2000, 100, 2159–2231.
- Alcón, M. J.; Iglesias, M.; Sánchez, F.; Viani, I. J. Organomet. Chem. 2001, 634, 25–33.
- Uhlemann, E.; Plath, M. Z. Chem. 1969, 9, 234–235.
- Trost, B. J. Am. Chem. Soc. 1997, 119, 7879–7880.
- Skog, K. Tetrahedron Lett. 1992, 33, 1751–1754.
- Lindauer, D.; Atzrodt, J.; Beckert, R.; Görls, H. Liebigs Ann. 1995, 199–201.
- Knölker, H.-J.; Braxmeier, T.; Schlechtingen, G. Synlett 1996, 502–504.
- Christie, C. C.; Kirby, G. W.; McGuigan, H.; Mackinnon, J.W. J. Chem. Soc. Perkin Trans. 1 1985, 2469–2473.Ye, Y.; Aulinger, K.; Arnold, N.; Spahl, W.; Steglich, W. Tetrahedron Lett. 1997, 38, 8013–8016.Wallace, E. G. U.S. Patent 4117153, 1978. 1978-09-26.Nilsson, J. L. G.; Sievertsson, H.; Dahlbom, R. Acta Chem. Scand. 1968, 22, 683–685.Sievertsson, H.; Nilsson, J. L. G. Acta Chem. Scand. 1970, 24, 939–945.Bogentoft, C.; Sievertsson, H. Acta Chem. Scand. 1972, 26, 4172–4174.
- A sample of 7.041 mg of 3 was heated under N2 from 25 to 300 °C at a rate of 10 °C/min. The reaction set in at 259.1 °C and the sample lost 0.818 mg in weight, which corresponds to the loss of CO2 (theoretically 0.794 mg).
- The preparative synthesis of 4 thus never succeeded. Possible alternate ways of obtaining this compound could be to react the tetraaza ligand 1 with urea while heating (Davis, T. L.; Underwood, H. W., Jr. J. Am. Chem. Soc. 1922, 44, 2595–2604.), with phosgene in benzene (Jones, L. W.; Root, F. B. J. Am. Chem. Soc. 1926, 48, 181–195.), with diphosgene in dioxane (Cordier, D.; Coulet, P. R. J. Chem. Soc. Perkin Trans. 2 1994, 4, 891–894.) or with carbon dioxide in the presence of a strong base or with methyl chloroformate and Et3N in CH2Cl2 (Naito, R.; Takeuchi, M.; Morihira, K.; Hayakawa, M.; Ikeda, K.; Shibanuma, T.; Isomura, Y. Chem. Pharm. Bull. 1998, 43, 1286–1294.).
- While with ligand 2 the racemic alcohol was obtained, ligand 3 gave the R-isomer in 6 % ee.
- a)Collective of authors. In Organikum, 18. Edition ed; Deutscher Verlag der Wissenschaften: Berlin, 1990; pp. 638–659.b)Tietze, L. F.; Eicher, T. Reaktionen und Synthesen in organisch-chemischen Grundpraktikum, 2nd Edition ed; Georg Thieme Verlag: Stuttgart, 1991; pp. 547–551. [Google Scholar]
- Oriyama, T. Chem. Lett. 1984, 12, 2071–2074.
- Sample availability: Samples of compounds 1, 2 and 3 are available from MDPI.
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Fonseca, M.H.; Hjelmgaard, T.; König, B. Synthesis and Catalytic Activity of Two New Cyclic Tetraaza Ligands. Molecules 2003, 8, 453-458. https://doi.org/10.3390/80500453
AMA Style
Fonseca MH, Hjelmgaard T, König B. Synthesis and Catalytic Activity of Two New Cyclic Tetraaza Ligands. Molecules. 2003; 8(5):453-458. https://doi.org/10.3390/80500453
Chicago/Turabian StyleFonseca, María Hechavarría, Thomas Hjelmgaard, and Burkhard König. 2003. "Synthesis and Catalytic Activity of Two New Cyclic Tetraaza Ligands" Molecules 8, no. 5: 453-458. https://doi.org/10.3390/80500453