
(1E,3E)-1,4-Dinitro-1,3-butadiene—Synthesis, Spectral Characteristics and Computational Study Based on MEDT, ADME and PASS Simulation



Abstract
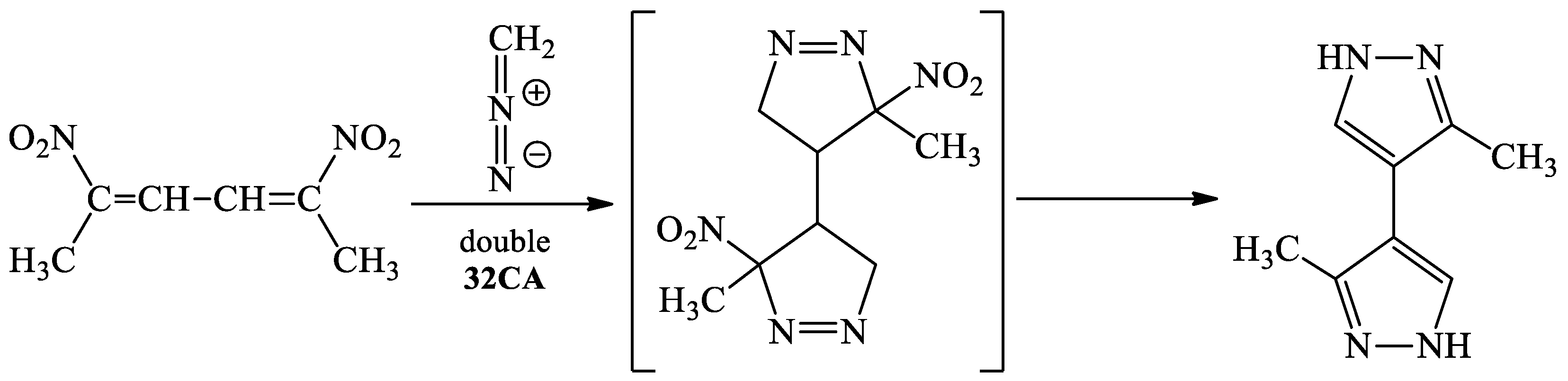
:1. Introduction
2. Results and Discussion
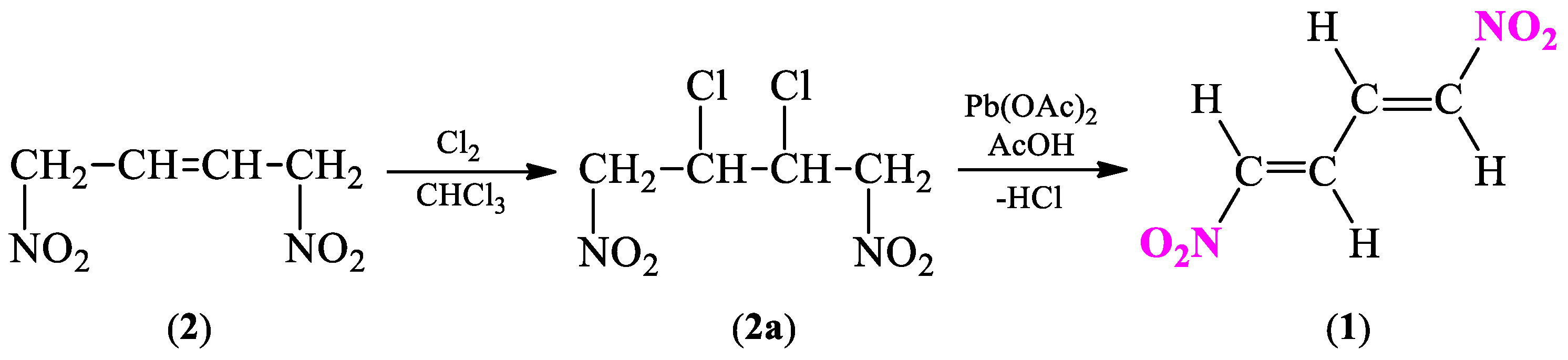
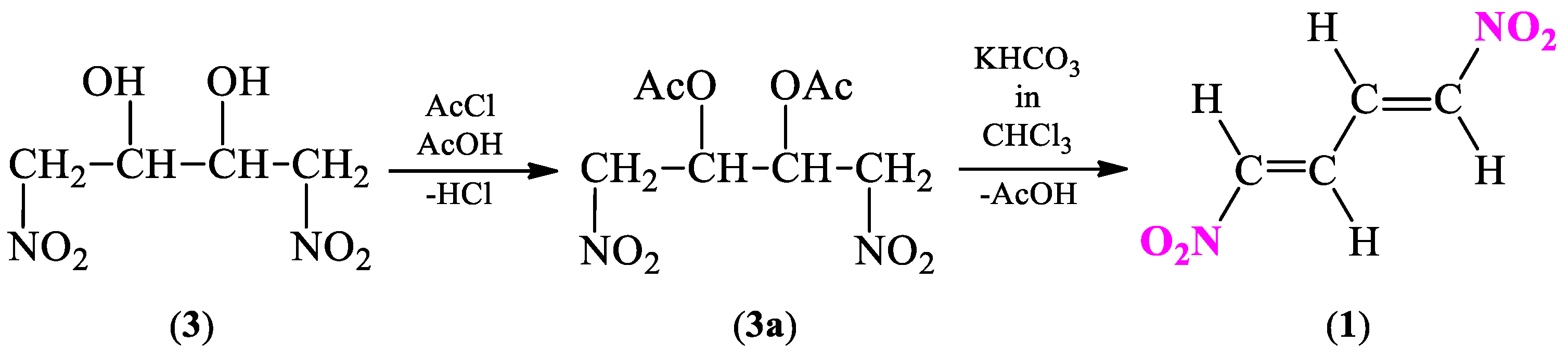
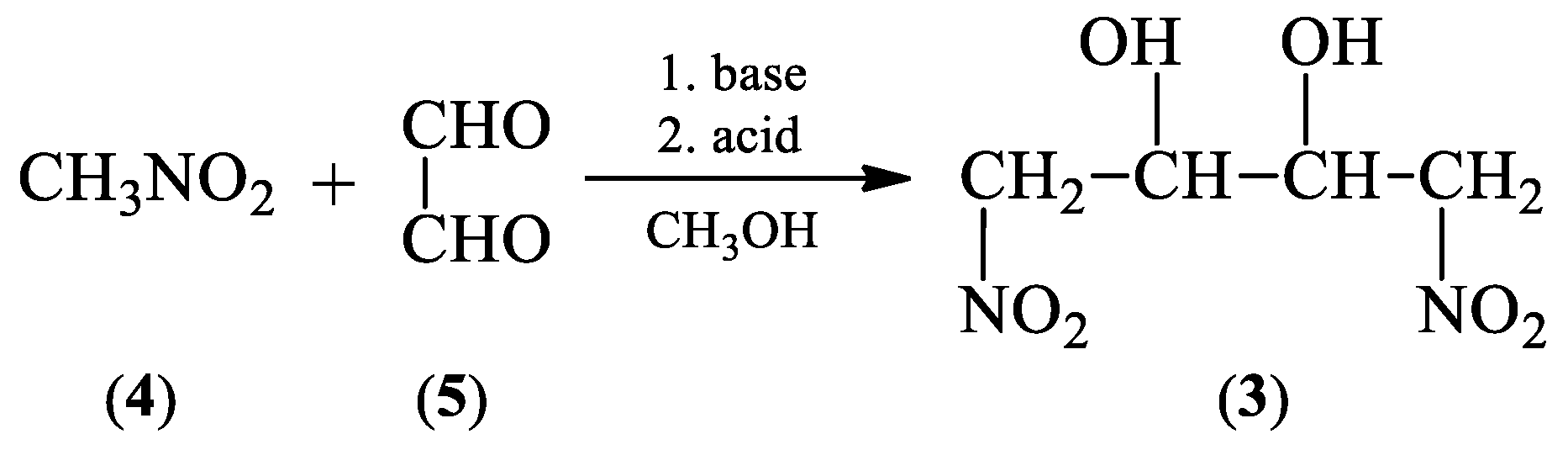
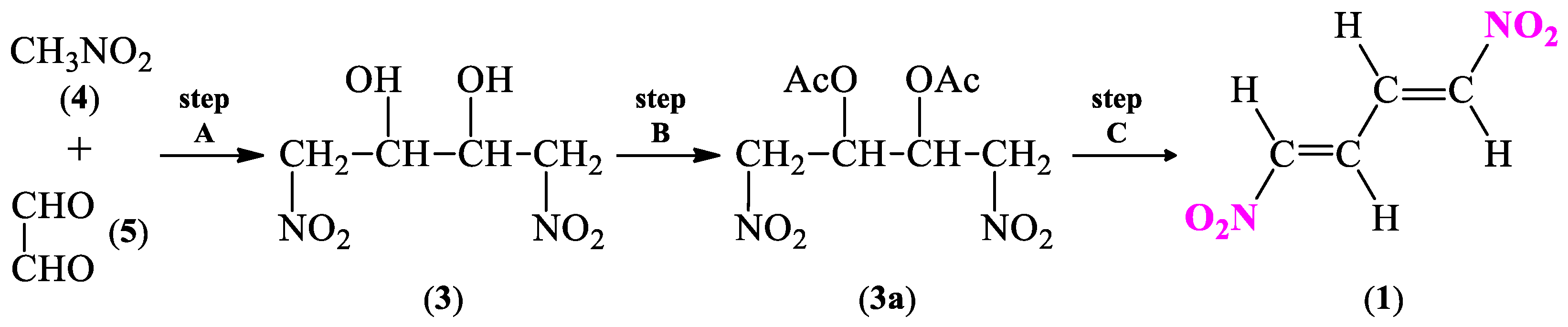
2.1. Synthetic and Computational Aspects of (1E,3E)-1,4-Dinitro-1,3-butadiene Preparation
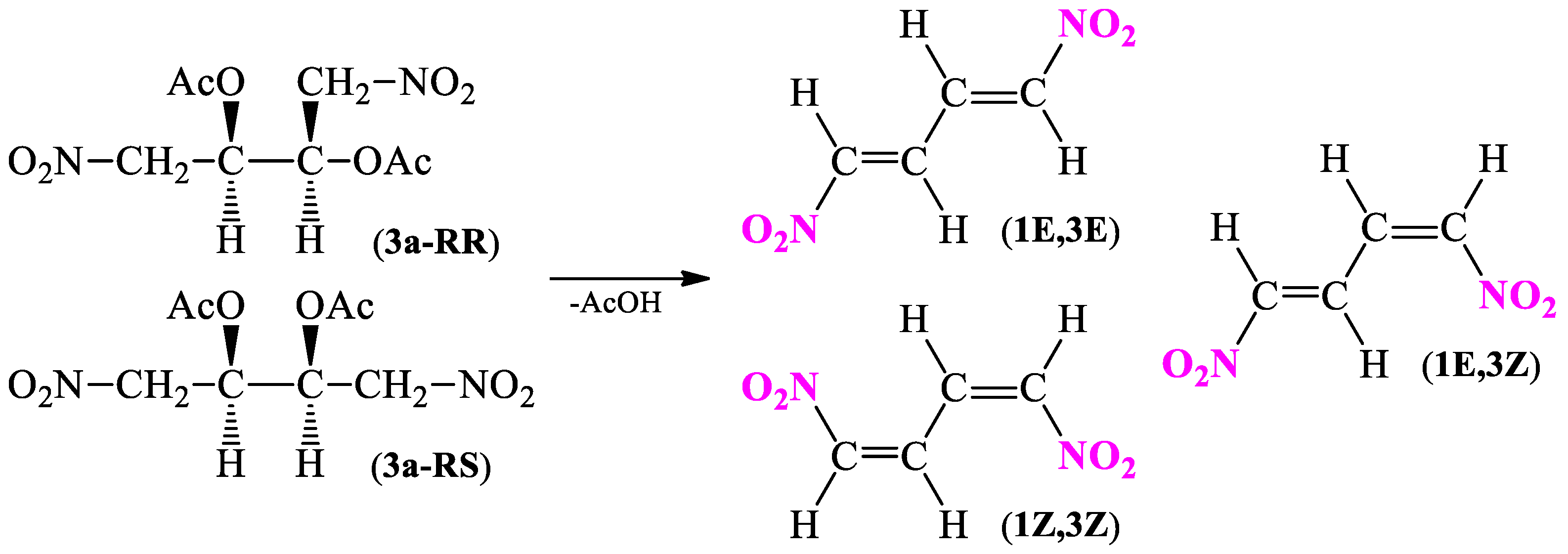
2.1.1. Synthesis Protocol and Its Necessary Modifications
2.1.2. Spectral Characteristics
2.1.3. Understanding the Geometric Isomerism Based on DFT Calculations
2.2. Study of Electron Density Distribution, Bioactivity and Pharmacokinetics Indices Based on MEDT, ADME and PASS
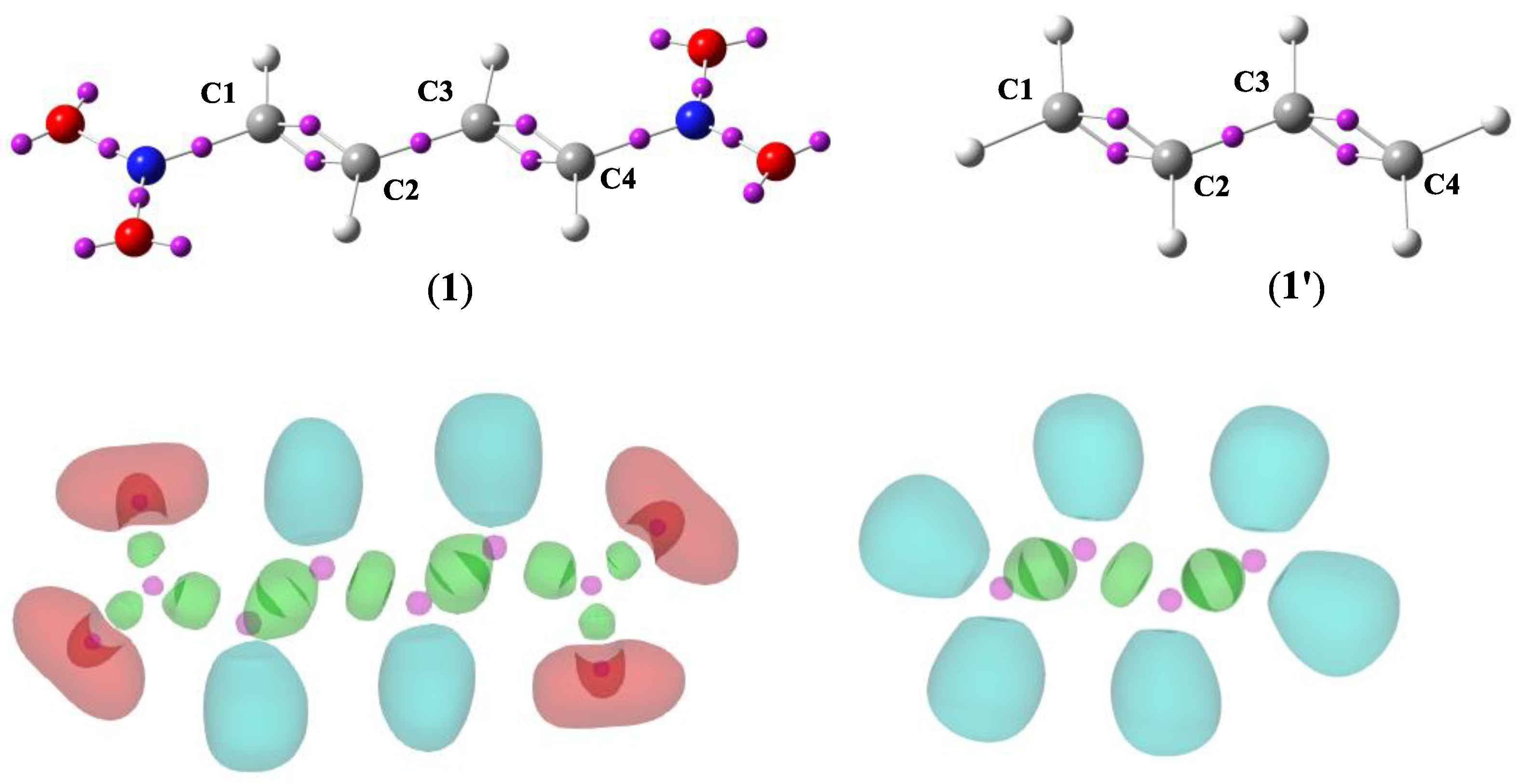
2.2.1. Analysis of the Electronic Structure of the Molecules 1 and 1′ Based on ELF, NPA and MEP
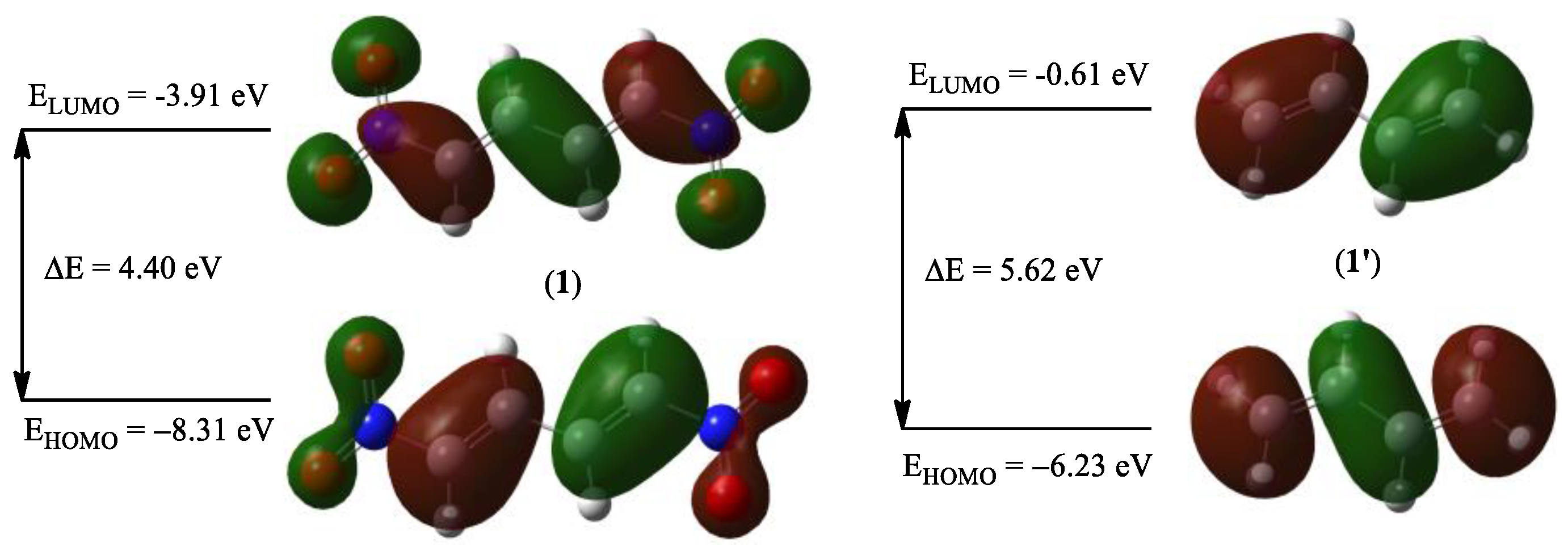
2.2.2. Analysis of the CDFT Reactivity Indices for the Compounds 1 and 1′
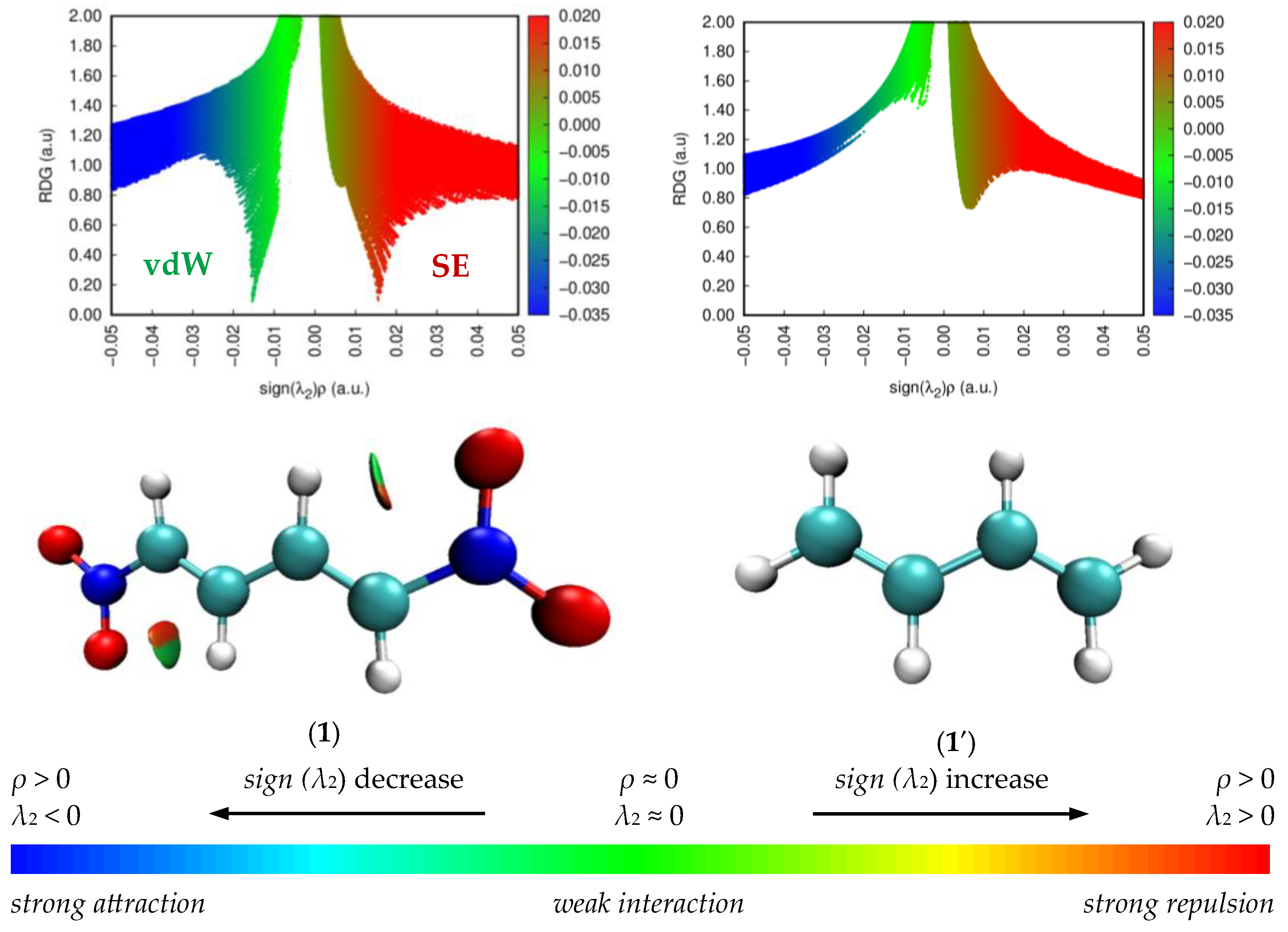
2.2.3. Analysis of the Non-Covalent Interactions in the Molecules 1 and 1′ Based on NCI
2.2.4. Analysis of Drug-Likeness and ADME Studies of the Molecule 1
2.2.5. Assessment of Antimicrobial Activities Based on PASS for the Molecule 1
3. Materials and Methods
3.1. Materials
3.2. General Procedure for 1,4-Dinitro-2,3-butanediol (3) Isomers Synthesis
3.3. General Procedure for 2,3-Diacetoxy-1,4-dinitrobutane (3a) Isomers Synthesis
3.4. General Procedure for (1E,3E)-1,4-Dinitro-1,3-butadiene (1) Synthesis
3.5. Analytical Techniques
3.6. Computational Details
4. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ballini, R.; Petrini, M.; Rosini, G. Nitroalkanes as Central Reagents in the Synthesis of Spiroketals. Molecules 2008, 13, 319–330. [Google Scholar] [CrossRef]
- Nishiwaki, N. A Walk through Recent Nitro Chemistry Advances. Molecules 2020, 25, 3680. [Google Scholar] [CrossRef] [PubMed]
- Ballini, R.; Palmieri, A. Nitroalkanes: Synthesis, Reactivity, and Applications; Wiley-VCH: Weinheim, Germany, 2021. [Google Scholar]
- Zawadzińska, K.; Gaurav, G.K.; Jasiński, R. Preparation of Conjugated Nitroalkenes: Short Review. Sci. Radices 2022, 1, 69–83. [Google Scholar] [CrossRef]
- Boguszewska-Czubara, A.; Łapczuk-Krygier, A.; Rykała, K.; Biernasiuk, A.; Wnorowski, A.; Popiolek, Ł.; Maziarka, A.; Hordyjewska, A.; Jasiński, R. Novel Synthesis Scheme and In Vitro Antimicrobial Evaluation of a Panel of (E)-2-aryl-1-cyano-1-nitroethenes. J. Enzym. Inhib. Med. Chem. 2016, 31, 900–907. [Google Scholar] [CrossRef]
- Boguszewska-Czubara, A.; Kula, K.; Wnorowski, A.; Biernasiuk, A.; Popiolek, Ł.; Miodowski, D.; Demchuk, O.M.; Jasiński, R. Novel Functionalized β-nitrostyrenes: Promising Candidates for New Antibacterial Drugs. Saudi Pharm. J. 2019, 27, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Latif, N.; Girgis, N.S.; Assad, F.M.; Grant, N. (Nitroethenyl) Salicylic Acid Anilides and Related Substances. A New Group of Molluscicidal and Microbicidal Compounds. Liebigs Ann. 1985, 6, 1202–1209. [Google Scholar] [CrossRef]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Recent Advances in Asymmetric Organocatalyzed Conjugate Additions to Nitroalkenes. Molecules 2017, 22, 895. [Google Scholar] [CrossRef] [PubMed]
- Al-Najjar, H.J.; Barakat, A.; Al-Majid, A.M.; Mabkhot, Y.N.; Weber, M.; Ghabbour, H.A.; Fun, H.-K. A Greener, Efficient Approach to Michael Addition of Barbituric Acid to Nitroalkene in Aqueous Diethylamine Medium. Molecules 2014, 19, 1150–1162. [Google Scholar] [CrossRef]
- Kras, J.; Sadowski, M.; Zawadzińska, K.; Nagatsky, R.; Woliński, P.; Kula, K.; Łapczuk, A. Thermal [3+2] Cycloaddition Reactions as Most Universal Way for the Effective Preparation of Five-Membered Nitrogen Containing Heterocycles. Sci. Radices 2023, 2, 247–267. [Google Scholar] [CrossRef]
- Kula, K.; Łapczuk, A.; Sadowski, M.; Kras, J.; Zawadzińska, K.; Demchuk, O.M.; Gaurav, G.K.; Wróblewska, A.; Jasiński, R. On the Question of the Formation of Nitro-Functionalized 2,4-Pyrazole Analogs on the Basis of Nitrylimine Molecular Systems and 3,3,3-Trichloro-1-Nitroprop-1-Ene. Molecules 2022, 27, 8409. [Google Scholar] [CrossRef]
- Szlachcic, P.; Uchacz, T.; Gryl, M.; Danel, A.; Wojtasik, K.; Kolek, P.; Jarosz, B.; Stadnicka, K.M. Combined XRD and DFT studies towards understanding the impact of intramolecular H-bonding on the reductive cyclization process in pyrazole derivatives. J. Mol. Struct. 2019, 1200, 127087. [Google Scholar] [CrossRef]
- Zawadzińska, K.; Ríos-Gutiérrez, M.; Kula, K.; Woliński, P.; Mirosław, B.; Krawczyk, T.; Jasiński, R. The Participation of 3,3,3-Trichloro-1-nitroprop-1-ene in the [3+2] Cycloaddition Reaction with Selected Nitrile N-Oxides in the Light of the Experimental and MEDT Quantum Chemical Study. Molecules 2021, 26, 6774. [Google Scholar] [CrossRef] [PubMed]
- Kras, J.; Woliński, P.; Nagatsky, R.; Demchuk, O.M.; Jasiński, R. Full Regio- and Stereoselective Protocol for the Synthesis of New Nicotinoids via Cycloaddition Processes with the Participation of Trans-Substituted Nitroethenes: Comprehensive Experimental and MEDT Study. Molecules 2023, 28, 3535. [Google Scholar] [CrossRef] [PubMed]
- Jasiński, R.; Żmigrodzka, M.; Dresler, E.; Kula, K. A Full Regio- and Stereoselective Synthesis of 4-Nitroisoxazolidines via Stepwise [3+2] Cycloaddition Reactions between (Z)-C-(9-anthryl)-N-arylnitrones and (E)-3,3,3-trichloro-1-nitroprop-1-ene: Comprehensive Experimental and Theoretical Study. J. Heterocycl. Chem. 2017, 54, 3314–3320. [Google Scholar] [CrossRef]
- Fryźlewicz, A.; Łapczuk-Krygier, A.; Kula, K.; Demchuk, O.M.; Dresler, E.; Jasiński, R. Regio- and Stereoselective Synthesis of Nitrofunctionalized 1,2-Oxazolidine Analogs of Nicotine. Chem. Heterocycl. Comp. 2020, 56, 120–122. [Google Scholar] [CrossRef]
- García-Mingüens, E.; Ferrándiz-Saperas, M.; de Gracia Retamosa, M.; Nájera, C.; Yus, M.; Sansano, J.M. Enantioselective 1,3-Dipolar Cycloaddition Using (Z)-α-Amidonitroalkenes as a Key Step to the Access to Chiral cis-3,4-Diaminopyrrolidines. Molecules 2022, 27, 4579. [Google Scholar] [CrossRef] [PubMed]
- Żmigrodzka, M.; Sadowski, M.; Kras, J.; Dresler, E.; Demchuk, O.M.; Kula, K. Polar [3+2] Cycloaddition between N-Methyl Azomethine Ylide and Trans-3,3,3-trichloro-1-nitroprop-1-ene. Sci. Radices 2022, 1, 26–35. [Google Scholar] [CrossRef]
- Hamada, T.; Iwai, K.; Nishiwaki, N. Synthesis and Characterization of Multiple Functionalized Cyclohexanone Using Diels–Alder Reaction of α-Nitrocinnamate. Reactions 2022, 3, 615–624. [Google Scholar] [CrossRef]
- Woliński, P.; Kącka-Zych, A.; Wróblewska, A.; Wielgus, E.; Dolot, R.; Jasiński, R. Fully Selective Synthesis of Spirocyclic-1,2-oxazine N-Oxides via Non-Catalysed Hetero Diels-Alder Reactions with the Participation of Cyanofunctionalysed Conjugated Nitroalkenes. Molecules 2023, 28, 4586. [Google Scholar] [CrossRef]
- Dresler, E.; Wróblewska, A.; Jasiński, R. Understanding the Molecular Mechanism of Thermal and LA-Catalysed Diels–Alder Reactions between Cyclopentadiene and Isopropyl 3-Nitroprop-2-Enate. Molecules 2023, 28, 5289. [Google Scholar] [CrossRef]
- Ballini, R.; Araújo, N.; Gil, M.V.; Román, E.; Serrano, J.A. Conjugated Nitrodienes. Synthesis and Reactivity. Chem. Rev. 2013, 113, 3493–3515. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Kula, K. Nitro-functionalized Analogues of 1,3-Butadiene: An Overview of Characteristic, Synthesis, Chemical Transformations and Biological Activity. Curr. Chem. Lett. 2024, 13, 15–30. [Google Scholar] [CrossRef]
- Zapol’skii, V.A.; Bilitewski, U.; Kupiec, S.R.; Ramming, I.; Kaufmann, D.E. Polyhalonitrobutadienes as Versatile Building Blocks for the Biotargeted Synthesis of Substituted N-Heterocyclic Compounds. Molecules 2020, 25, 2863. [Google Scholar] [CrossRef]
- Kaberdin, R.V.; Potkin, V.I.; Zapol’skii, V.A. Nitrobutadienes and their halogen derivatives: Synthesis and reactions. Russ. Chem. Rev. 1997, 66, 827–842. [Google Scholar] [CrossRef]
- Petrillo, G.; Benzi, A.; Bianchi, L.; Maccagno, M.; Pagano, A.; Tavani, C.; Spinelli, D. Recent advances in the use of conjugated nitro or dinitro-1,3-butadienes as building-blocks for the synthesis of heterocycles. Tetrahedron Lett. 2020, 61, 152297–152309. [Google Scholar] [CrossRef]
- Al-Jumaili, M.H.A.; Hamad, A.A.; Hashem, H.E.; Hussein, A.D.; Muhaidi, M.J.; Ahmed, M.A.; Albanaa, A.H.A.; Siddique, F.; Bakr, E.A. Comprehensive Review on the Bis-heterocyclic Compounds and Their Anticancer Efficacy. J. Mol. Struct. 2023, 1271, 133970. [Google Scholar] [CrossRef]
- Iftikhar, R.; Khan, F.Z.; Naeem, N. Recent Synthetic Strategies of Small Heterocyclic Organic Molecules with Optoelectronic Applications: A Review. Mol. Divers. 2023, 27, 1–37. [Google Scholar] [CrossRef]
- Ren, F.; Zhang, Y.; Gong, D.; He, X.; Shi, J.; Zhang, Q.; Tu, G. Novel swivel-cruciform 5, 5′-bibenzothiadiazole based small molecule donors for efficient organic solar cells. Org. Electron. 2020, 77, 105521. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, B.; Koh, C.W.; Zhou, X.; Sun, H.; Yu, J.; Yang, K.; Wang, H.; Liao, Q.; Woo, H.Y.; et al. Facile Synthesis of Polycyclic Aromatic Hydrocarbon (PAH)–Based Acceptors with Fine-Tuned Optoelectronic Properties: Toward Efficient Additive-Free Nonfullerene Organic Solar Cells. Adv. Energy Mater. 2019, 9, 1803976. [Google Scholar] [CrossRef]
- Spinelli, D.; Budriesi, R.; Cosimelli, B.; Severi, E.; Micucci, M.; Baroni, M.; Fusi, F.; Ioan, P.; Cross, S.; Frosini, M.; et al. Playing with Opening and Closing of Heterocycles: Using the Cusmano-Ruccia Reaction to Develop a Novel Class of Oxadiazolothiazinones, Active as Calcium Channel Modulators and P-Glycoprotein Inhibitors. Molecules 2014, 19, 16543–16572. [Google Scholar] [CrossRef]
- Synkiewicz-Musialska, B.; Szwagierczak, D.; Kulawik, J.; Pałka, N.; Piasecki, P. Structural, Thermal and Dielectric Properties of Low Dielectric Permittivity Cordierite-Mullite-Glass Substrates at Terahertz Frequencies. Materials 2021, 14, 4030. [Google Scholar] [CrossRef] [PubMed]
- Synkiewicz-Musialska, B. LTCC Glass-Ceramics based on Diopside/Cordierite/Al2O3 for Ultra-High Frequency Applications. Sci. Radices 2022, 2, 190–201. [Google Scholar] [CrossRef]
- Koc, E. Synthesis of Novel Nitroso Acetal Derivatives via Tandem 6 pi-electrocyclization/[3+2]-cycloaddition of 1-nitro-2-methyl-1,3-butadiene. Org. Commun. 2017, 10, 298–303. [Google Scholar] [CrossRef]
- Durden, J.A.; Heywood, D.L.; Sousa, A.A.; Spurr, H.W. Synthesis and Microbial Toxicity of Dinitrobutadienes and Related Compounds. J. Agric. Food Chem. 1970, 18, 50–56. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Rowley, G.L.; Frankel, M.B. Synthesis of Aliphatic Dinitrodienes. J. Org. Chem. 1969, 34, 1512–1513. [Google Scholar] [CrossRef]
- Perekalin, V.V.; Lerner, O.M. Synthesis of Conjugated Dinitrodiene. Dokl. Akad. Nauk SSSR 1959, 129, 1303–1305. [Google Scholar]
- Novikov, S.S.; Korsakova, I.S.; Babievskii, K.K. Synthesis of 1,4-dinitro-1,3-butadiene. Russ. Chem. Bull. 1960, 9, 882–884. [Google Scholar] [CrossRef]
- Carroll, F.I. Structure of the Isomers of 1,4-dinitro-2,3-butanediol. J. Org. Chem. 1966, 31, 366–368. [Google Scholar] [CrossRef]
- Plaut, H. Dinitrodiols and Their Alkali and Alkaline Earth Metal Salts, and Method of Preparation Thereof. U.S. Patent Publication No. US2616923A, 4 November 1952. [Google Scholar]
- Carroll, F.I.; Kerbow, S.C.; Wall, M.E. The Synthesis of 1,4-dichloro-1,4-dinitro-1,3-butadiene. Can. J. Chem. 1966, 44, 2115–2117. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A Simple Measure of Electron Localization in Atomic and Molecular Systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Leboeuf, M.; Koster, A.M.; Jug, K. Topological analysis of the molecular electrostatic potential. J. Chem. Phys. 1999, 111, 4893–4905. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Parr, R.G.; Gadre, S.R.; Bartolotti, L.J. Local Density Functional Theory of Atoms and Molecules. Proc. Natl. Acad. Sci. USA 1979, 76, 2522–2526. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Utnicka, J.; Wójtowicz, A.; Kula, K. The Global and Local Reactivity of C,N-diarylnitryle Imines in [3+2] Cycloaddition Processes with Trans-β-nitrostyrene according to Molecular Electron Density Theory: A computational study. Curr. Chem. Lett. 2023, 12, 421–430. [Google Scholar] [CrossRef]
- Aurell, M.J.; Domingo, L.R.; Pérez, P.; Contreras, R. A Theoretical Study on the Regioselectivity Of 1,3-Dipolar Cycloadditions Using DFT-based Reactivity Indexes. Tetrahedron 2004, 60, 11503–11509. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Bębenek, E.; Sokal, A.; Książek, M.; Chrobak, E. Crystal Structure and Spectroscopic Analysis of 3-Diethoxyphosphoryl-28-[1-(1-deoxy-β-D-glucopyranosyl)-1H-1,2,3-triazol-4-yl]carbonylbetulin. Crystals 2023, 13, 1488. [Google Scholar] [CrossRef]
- Benmerabet, A.; Bouhadiba, A.; Belhocine, Y.; Rahali, S.; Sbei, N.; Seydou, M.; Boucheriha, I.; Omeiri, I.; Assaba, I.M. DFT Investigation on the Complexation of β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin as Recognition Hosts with Trichloroethylene. Atoms 2023, 11, 153. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. A Useful Classification of Organic Reactions Based on the Flux of the Electron Density. Sci. Radices 2023, 2, 1–24. [Google Scholar] [CrossRef]
- Parr, R.G.; von Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The Nucleophilicity N Index in Organic Chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Non-covalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- SwissADME. Swiss Institute of Bioinformatics. Available online: http://www.swissadme.ch/ (accessed on 5 December 2023).
- Di, L.; Kerns, E. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Wishart, D.S. Improving early drug discovery through ADME modelling: An overview. Drugs R D 2007, 8, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.; Doerksen, R. Topological Polar Surface Area: A Useful Descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Darvas, F.; Keseru, G.; Papp, A.; Dormán, G.; Urge, L.; Krajcsi, P. In Silico and Ex silico ADME approaches for drug discovery. Curr. Top. Med. Chem. 2002, 2, 1287–1304. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed]
- Way2Drug, PASS Online. Available online: http://www.way2drug.com/passonline/ (accessed on 6 December 2023).
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Comp. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Poroikov, V.; Filimonov, D. Computer-Aided Prediction of Biological Activity Spectra. Application for Finding and Optimization of New Leads. In Rational Approaches to Drug Design, 1st ed.; Holtje, H.-D., Sippl, W., Eds.; Prous Science: Barcelona, Spain, 2001; pp. 403–407. [Google Scholar]
- Jasiński, R.; Mirosław, B.; Demchuk, O.M.; Babyuk, D.; Łapczuk-Krygier, A. In the search for experimental and quantumchemical evidence for zwitterionic nature of (2E)-3-[4-(dimethylamino)phenyl]-2-nitroprop-2-enenitrile—An extreme example of donor-π-acceptor push-pull molecule. J. Mol. Struct. 2016, 1108, 689–697. [Google Scholar] [CrossRef]
- Kula, K.; Dresler, E.; Demchuk, O.M.; Jasiński, R. New aldimine N-oxides as precursors for preparation of heterocycles with potential biological activity. Przem. Chem. 2015, 94, 1385–1387. [Google Scholar] [CrossRef]
- Zawadzińska, K.; Gadocha, Z.; Pabian, K.; Wróblewska, A.; Wielgus, E.; Jasiński, R. The First Examples of [3+2] Cycloadditions with the Participation of (E)-3,3,3-Tribromo-1-Nitroprop-1-Ene. Materials 2022, 15, 7584. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B. GAUSSIAN 09, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A Complete Basis Set Model Chemistry. The Total Energies of Closed-Shell Atoms and Hydrides of the First-Row Atoms. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Dresler, E.; Allnajar, R.; Jasiński, R. Sterical index:A novel, simple tool for the interpretation of organic reaction mechanisms. Sci. Radices 2023, 2, 69–74. [Google Scholar] [CrossRef]
- Kula, K.; Łapczuk-Krygier, A. A DFT computational study on the [3+2] cycloaddition between parent thionitrone and nitroethene. Curr. Chem. Lett. 2018, 7, 27–34. [Google Scholar] [CrossRef]
- Kula, K.; Sadowski, M. Regio- and stereoselectivity of [3+2] cycloaddition reactions between (Z)-C-(9-anthryl)-N-methylnitrone and analogues of trans- -nitrostyrene in the light of MEDT computational study. Chem. Heterocycl. Compd. 2023, 59, 138–144. [Google Scholar] [CrossRef]
- Zawadzińska, K.; Kula, K. Application of β-phosphorylated nitroethenes in [3+2] cycloaddition reactions involving benzonitrile N-oxide in the light of DFT computational study. Organics 2021, 2, 26–37. [Google Scholar] [CrossRef]
- Jasiński, R.; Magdalena Kubik, M.; Łapczuk-Krygier, A.; Kącka, A.; Dresler, E.; Boguszewska-Czubara, A. An experimental and theoretical study of the hetero Diels-Alder reactions between (E)-2-aryl-1-cyano-1-nitroethenes and ethyl vinyl ether: One-step or zwitterionic, two-step mechanism? React. Kinet. Mech. Catal. 2014, 113, 333–345. [Google Scholar] [CrossRef]
- Jasiński, R.; Mróz, K.; Kącka, A. Experimental and Theoretical DFT Study on Synthesis of Sterically Crowded 2,3,3,(4)5-Tetrasubstituted-4-Nitroisoxazolidines via 1,3-Dipolar Cycloaddition Reactions Between Ketonitrones and Conjugated Nitroalkenes. J. Heterocycl. Chem. 2016, 53, 1424–1429. [Google Scholar] [CrossRef]
- Jasiński, R.; Mróz, K. Kinetic Aspects of [3+2] Cycloaddition Reactions between (E)-3,3,3-Trichloro-1-Nitroprop-1-Ene and Ketonitrones. React. Kinet. Mech. Catal. 2015, 116, 35–41. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization, Visualization Handbook; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware Inc.: New York, NY, USA, 2015. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- World Health Organization. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans “Some Industrial Chemicals”; International Agency for Research on Cancer: Lyon, France, 2000; pp. 487–501.
- World Health Organization. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans “Cadmium, Nickel, Some Epoxides, Miscellaneous Industrial Chemicals and General Considerations on Volatile Anaesthetics”; International Agency for Research on Cancer: Lyon, France, 1976; pp. 487–501.
- Barson, M. NIOSH-Pocket Guide to Chemical Hazards; U.S Department of Health & Human Services: Pittsburgh, PA, USA, 2007.

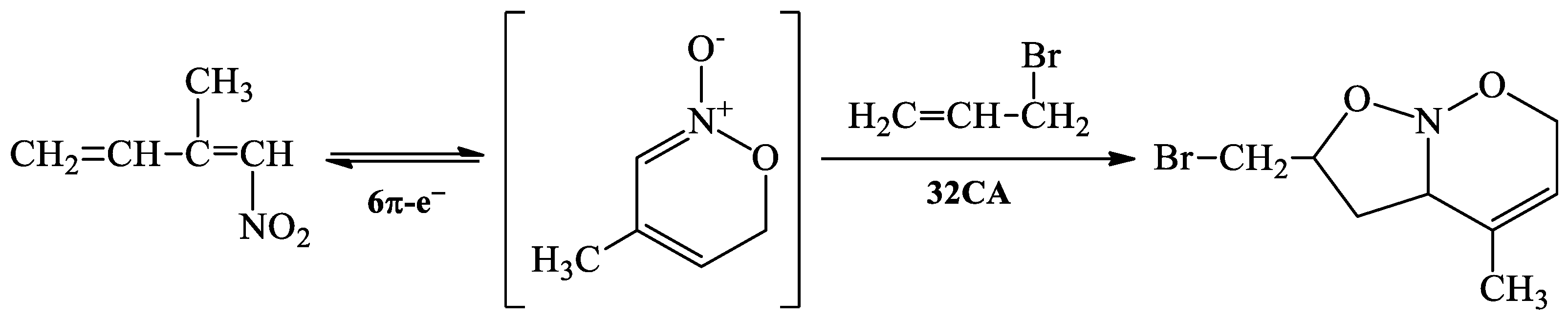

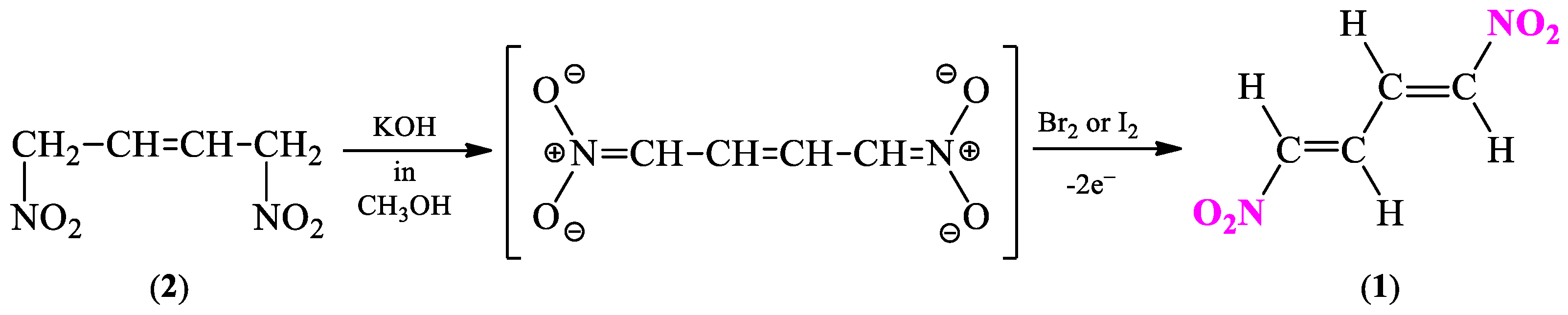




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acidic Agent | Basic Agent | Yield | Reference |
|---|---|---|---|
| SO2 | NaOH | 80.5 | Novikov et al. [39] |
| SO2 | NaOH | 61.0 | Durden et al. [35] |
| SO2 | KOH | 36.4 | Carroll [40] |
| H2SO4 or H3PO4 or AcOH or (COOH)2 | KOH | 24.0 | Plaut [41] |
| Fraction | Solvent | |||||
|---|---|---|---|---|---|---|
| DMFA | Nitromethane | Methanol | Acetone | Chloroform | Diethyl Ether | |
| (ε = 37.781) | (ε = 36.562) | (ε = 32.613) | (ε = 20.493) | (ε = 4.7113) | (ε = 4.240) | |
| 1,4-dinitrobutane-2,3-diol (3) | ✓✓✓ | ✓✓✓ | ✓✓✓ | ✓✓✓ | x | ✓✓ |
| Colored substances | ✓✓✓ | ✓✓✓ | ✓✓✓ | ✓✓✓ | ✓✓✓ | ✓ |
| Potassium acetate | ✓✓✓ | x | ✓✓✓ | ✓✓✓ | x | x |
| Transformation | ΔH | ΔG | ΔS |
|---|---|---|---|
| 3a-RR → 1E,3E + 2AcOH | 33.64 | 11.98 | 72.61 |
| 3a-RR → 1E,3Z + 2AcOH | 36.09 | 14.27 | 73.17 |
| 3a-RR → 1Z,3Z + 2AcOH | 37.31 | 15.11 | 74.45 |
| 3a-RS → 1E,3E + 2AcOH | 30.31 | 7.65 | 75.99 |
| 3a-RS → 1E,3Z + 2AcOH | 32.76 | 9.94 | 76.54 |
| 3a-RS → 1Z,3Z + 2AcOH | 33.98 | 10.78 | 77.83 |
| 1 | 1′ | |
|---|---|---|
| ELF Basins | N [e] | N [e] |
| V(C1,C2) | 1.73 | 1.72 |
| V’(C1,C2) | 1.73 | 1.72 |
| V(C2,C3) | 2.23 | 2.20 |
| V(C1,C2) | 1.73 | 1.72 |
| V’(C1,C2) | 1.73 | 1.72 |
| [eV] | 1 | 1′ |
|---|---|---|
| HOMO energy | −8.31 | −6.23 |
| LUMO energy | −3.91 | −0.61 |
| Energy gap, ΔE | 4.40 | 5.62 |
| Ionization potential, l | 8.31 | 6.23 |
| Electron affinity, A | 3.91 | 0.61 |
| Electronic chemical potential, μ | −6.11 | −3.42 |
| Mulliken electronegativity, X | −4.40 | −5.62 |
| Chemical hardness, η | 4.40 | 5.62 |
| Chemical softness, S | 0.23 | 0.18 |
| Global electrophilicity, ω | 4.24 | 1.04 |
| Global nucleophilicity, N | 0.81 | 2.89 |
| Physicochemical Properties | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Formula | C4H4N2O4 | |||||||||||||
| Molecular weight MW | 144.09 g/mol | |||||||||||||
| #heavy atoms | 10 | |||||||||||||
| #aromatic heavy atoms | 0 | |||||||||||||
| #rotatable bonds | 3 | |||||||||||||
| #H-bond acceptors | 4 | |||||||||||||
| #H-bond donors | 0 | |||||||||||||
| Molar refractivity MR | 36.6 | |||||||||||||
| Topological polar surface area TPSA | 91.64 Å2 | |||||||||||||
| Lipophilicity Log Po/w | ||||||||||||||
| iLOGP | XLOGP | WLOGP | MLOGP | SILICOS-IT | Consensus | |||||||||
| −1.9 | 0.81 | 1.61 | −0.78 | −2.34 | −0.52 | |||||||||
| Water Solubility Log S | ||||||||||||||
| Log S (ESOL) | Solubility | Class | Log S (Ali) | Solubility | Class | |||||||||
| −1.05 | 1.300 mg/mL | very soluble | −2.32 | 0.696 mg/mL | soluble | |||||||||
| Pharmacokinetics | ||||||||||||||
| IG absorption | BBB permeant | CYP1A2 INH | CYP2C19 INH | CYP2C9 INH | CYP2D6 INH | CYP3A4 INH | Log Kp Skin permeation | |||||||
| High | No | No | No | No | No | No | −6.60 cm/s | |||||||
| Medicinal Chemistry Friendliness | ||||||||||||||
| PAINS | Brenk | Synthetic accessibility | ||||||||||||
| 0 alert | 2 alerts | 29.5% | ||||||||||||
| Lipinski et al. [62] (Pfizer) | Ghose et al. [63] (Amgen) | Veber et al. [64] (GSK) | Egan et al. [65] (Pharmacia) | Muegge et al. [66] (Bayer) |
|---|---|---|---|---|
| MW ≤ 500 Da MLOGP ≤ 4.15 #H-bond donors ≤ 5 #H-bond acceptors ≤ 10 | 160 Da ≤ MW ≤ 480 Da −0.4 ≤ WLOGP ≤ 5.6 40 ≤ MR ≤ 130 20 ≤ #atoms ≤ 70 | #rotatable bonds ≤ 10 TPSA ≤ 140 Å2 | WLOGP ≤ 5.88 TPSA ≤ 131.6 Å2 | 200 Da ≤ MW ≤ 600 Da −0.4 ≤ XLOGP ≤ 5.6 TPSA ≤ 150 Å2 #rings ≤ 7 #carbons > 4 #heteroatoms > 1 #rotatable bonds ≤ 15 #H-bond donors ≤ 5 #H-bond acceptors ≤ 10 |
| Antimicrobial Activity | Pa | Pi |
|---|---|---|
| Antiviral (Picornavirus) | 0.581 | 0.024 |
| Antifungal | 0.416 | 0.047 |
| Antibacterial | 0.396 | 0.031 |
| Antiparasitic | 0.274 | 0.065 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadowski, M.; Synkiewicz-Musialska, B.; Kula, K. (1E,3E)-1,4-Dinitro-1,3-butadiene—Synthesis, Spectral Characteristics and Computational Study Based on MEDT, ADME and PASS Simulation. Molecules 2024, 29, 542. https://doi.org/10.3390/molecules29020542
Sadowski M, Synkiewicz-Musialska B, Kula K. (1E,3E)-1,4-Dinitro-1,3-butadiene—Synthesis, Spectral Characteristics and Computational Study Based on MEDT, ADME and PASS Simulation. Molecules. 2024; 29(2):542. https://doi.org/10.3390/molecules29020542
Chicago/Turabian StyleSadowski, Mikołaj, Beata Synkiewicz-Musialska, and Karolina Kula. 2024. "(1E,3E)-1,4-Dinitro-1,3-butadiene—Synthesis, Spectral Characteristics and Computational Study Based on MEDT, ADME and PASS Simulation" Molecules 29, no. 2: 542. https://doi.org/10.3390/molecules29020542