
Large Subunit of the Human Herpes Simplex Virus Terminase as a Promising Target in Design of Anti-Herpesvirus Agents
 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results
2.1. Preparation of Compound 1-Resistant HSV-1 Population
2.2. High-Throughput Sequencing of Resistant Clones
2.3. Molecular Modeling Study
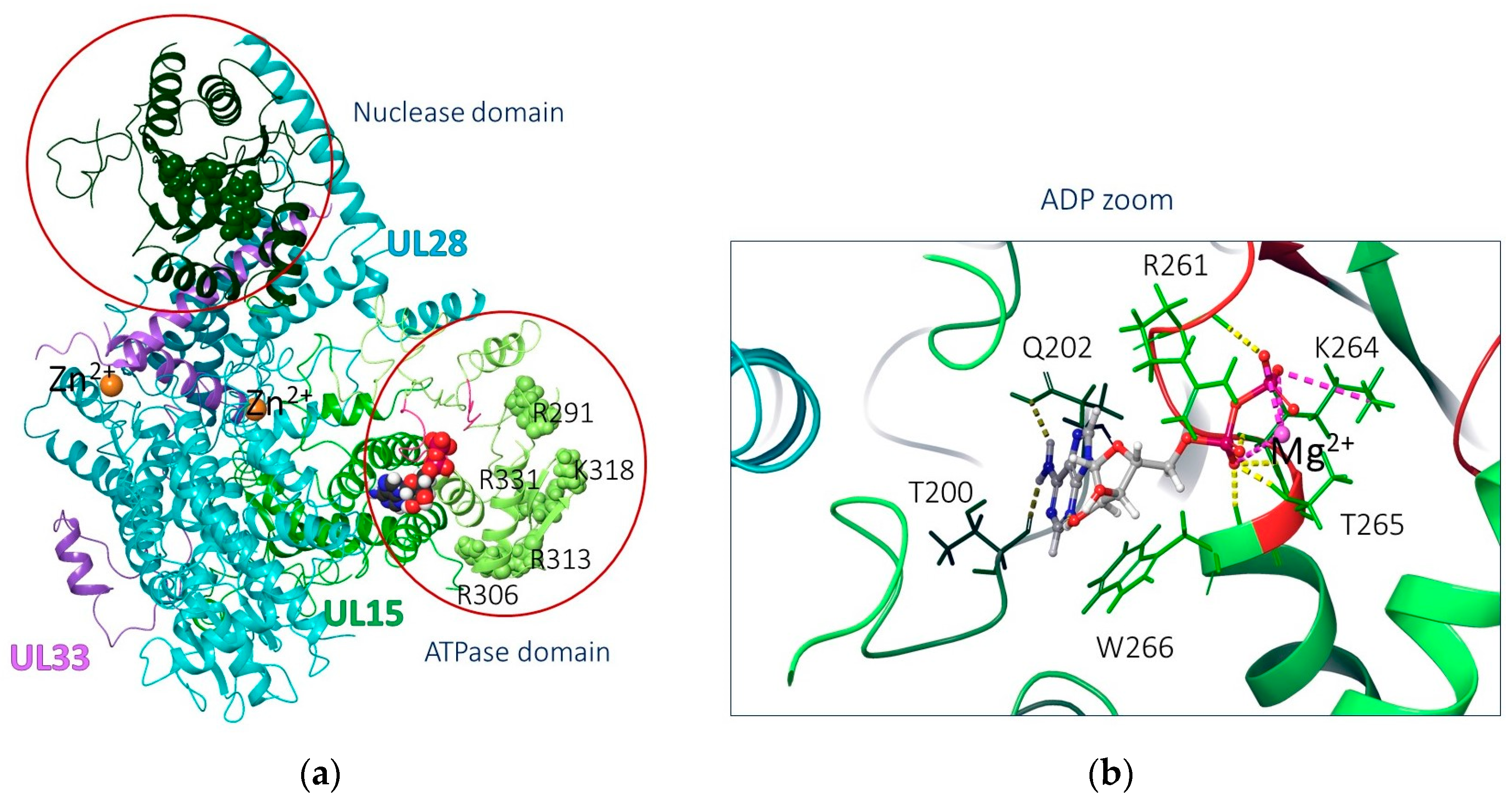
2.3.1. Biological Target Analysis
2.3.2. Molecular Docking Results
Docking to the ATP Domain
Docking to Alternative Binding Sites
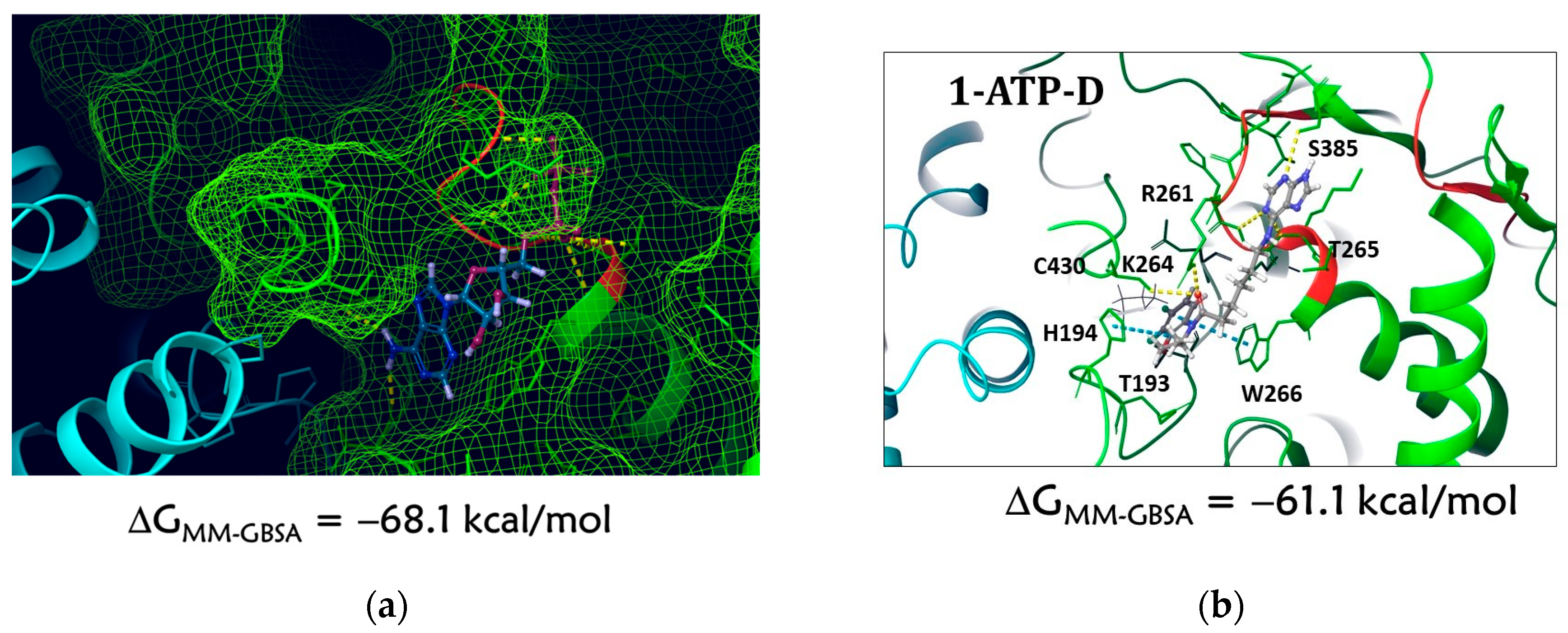
2.3.3. Molecular Dynamic Results
Molecular Dynamics of the 1–ATP-D Complex
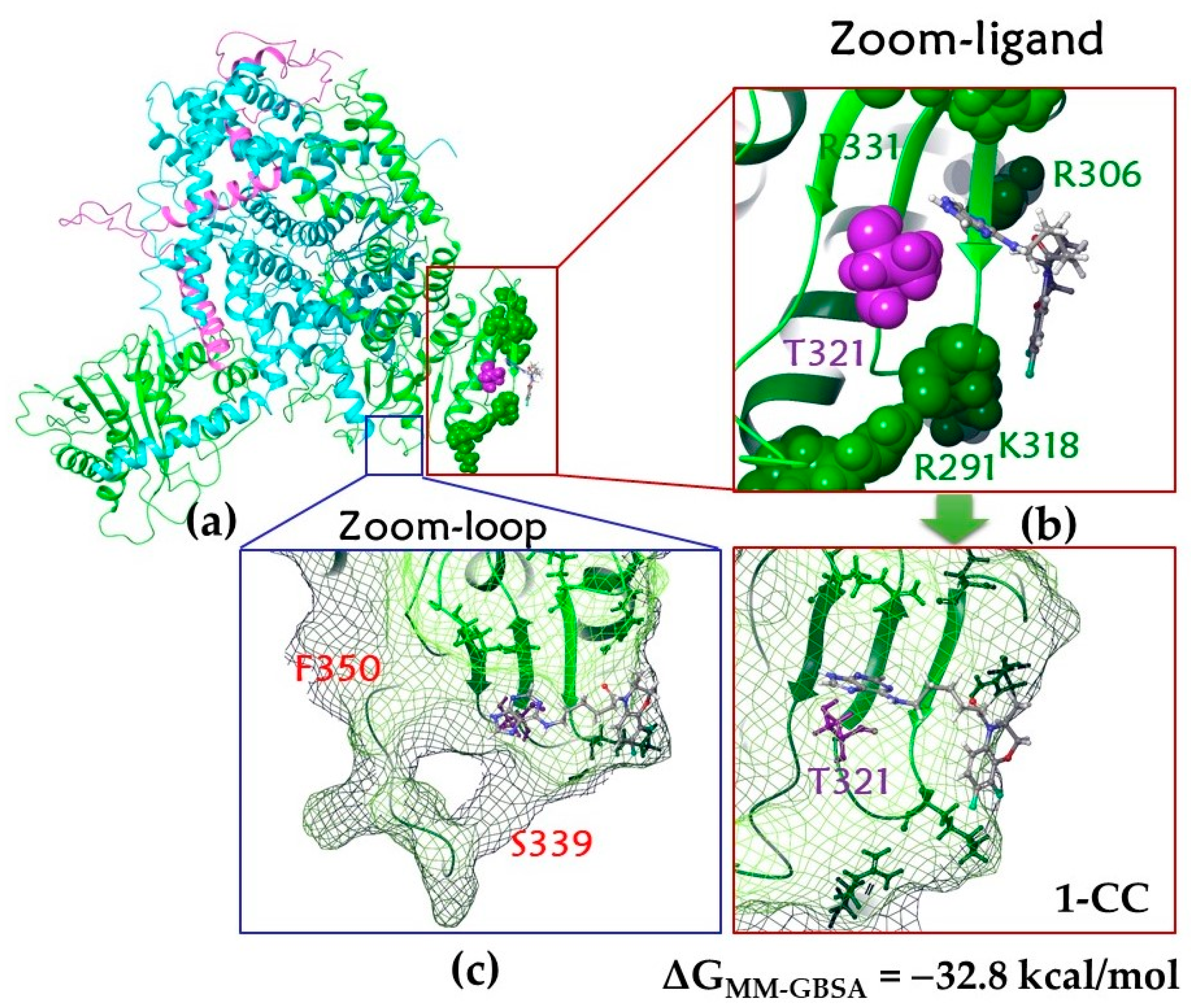
Molecular Dynamics of the 1–CC Complex
Molecular Dynamics of 1–loop1 and 1–loop2 Complexes
3. Materials and Methods
3.1. Compounds
3.2. Viruses
3.3. Cells
3.4. Antiviral Tests
3.5. Cloning
3.6. Isolation of DNA from Herpesvirus-Infected Cells
3.7. High-Throughput Sequencing of Herpesvirus Genomes
3.8. Calculation Technique
3.8.1. Preparation of Protein and Ligand
3.8.2. Binding Site Analysis
3.8.3. Molecular Docking
3.8.4. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.R.; Schofield, J.K.; Tatnall, F.M.; Leigh, I.M. Natural history, management and complications of herpes labialis. J. Med. Virol. 1993, 41 (Suppl. S1), 22–26. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.; Baines, J. Clinical management of herpes simplex virus infections: Past, present, and future. F1000Research 2018, 7, 1726. [Google Scholar] [CrossRef] [PubMed]
- Looker, K.J.; Johnston, C.; Welton, N.J.; James, C.; Vickerman, P.; Turner, K.M.E.; Boily, M.C.; Gottlieb, S.L. The global and regional burden of genital ulcer disease due to herpes simplex virus: A natural history modelling study. BMJ Glob. Health 2020, 5, e001875. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, H.H.; Chemaitelly, H.; Abu-Raddad, L.J. Characterizing the transitioning epidemiology of herpes simplex virus type 1 in the USA: Model-based predictions. BMC Med. 2019, 17, 57. [Google Scholar] [CrossRef]
- Khadr, L.; Harfouche, M.; Omori, R.; Schwarzer, G.; Chemaitelly, H.; Abu-Raddad, L.J. The Epidemiology of Herpes Simplex Virus Type 1 in Asia: Systematic Review, Meta-Analyses, and Meta-Regressions. Clin. Infect. Dis. 2019, 68, 757–772. [Google Scholar] [CrossRef]
- Xu, F.; Sternberg, M.R.; Kottiri, B.J.; McQuillan, G.M.; Lee, F.K.; Nahmias, A.J.; Berman, S.M.; Markowitz, L.E. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 2006, 296, 964–973. [Google Scholar] [CrossRef]
- Kawana, T. Sexually transmitted diseases of alpha herpes virus in women. Nihon Rinsho 2000, 58, 883–889. (In Japanese) [Google Scholar]
- Frangoul, H.; Wills, M.; Crossno, C.; Engel, M.; Domm, J. Acyclovir-resistant herpes simplex virus pneumonia post-unrelated stem cell transplantation: A word of caution. Pediatr. Transplant. 2007, 11, 942–944. [Google Scholar] [CrossRef]
- Généreau, T.; Lortholary, O.; Bouchaud, O.; Lacassin, F.; Vinceneux, P.; De Truchis, P.; Jaccard, A.; Meynard, J.-L.; Verdon, R.; Sereni, D. Herpes Simplex Esophagitis in Patients with AIDS: Report of 34 Cases. Clin. Infect. Dis. 1996, 22, 926–931. [Google Scholar] [CrossRef]
- Ljungman, P.; Ellis, M.N.; Hackman, R.C.; Shepp, D.H.; Meyers, J.D. Acyclovir-Resistant Herpes Simplex Virus Causing Pneumonia after Marrow Transplantation. J. Infect. Dis. 1990, 162, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Sacks, S.L.; Wanklin, R.J.; Reece, D.E.; Hicks, K.A.; Tyler, K.L.; Coen, D.M. Progressive Esophagitis from Acyclovir-Resistant Herpes Simplex. Clinical Roles for DNA Polymerase Mutants and Viral Heterogeneity? Ann. Intern. Med. 1989, 111, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Bacon, T.H.; Levin, M.J.; Leary, J.J.; Sarisky, R.T.; Sutton, D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin. Microbiol. Rev. 2003, 16, 114–128. [Google Scholar] [CrossRef]
- Das, D.; Hong, J. Chapter 12–Herpesvirus Polymerase Inhibitors. In Viral Polymerases: Structures, Functions and Roles as Antiviral Drug Targets; Gupta, S.P., Ed.; Academic Press: London, UK, 2019; pp. 333–356. [Google Scholar] [CrossRef]
- Fyfe, J.A.; Keller, P.M.; Furman, P.A.; Miller, R.L.; Elion, G.B. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl)guanine. J. Biol. Chem. 1978, 253, 8721–8727. [Google Scholar] [CrossRef] [PubMed]
- Balfour, H.H., Jr. Antiviral drugs. N. Engl. J. Med. 1999, 340, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Luczkowiak, J.; Álvarez, M.; Sebastián-Martín, A.; Menéndez-Arias, L. Chapter 4-DNA-Dependent DNA Polymerases as Drug Targets in Herpesviruses and Poxviruses. In Viral Polymerases: Structures, Functions and Roles as Antiviral Drug Targets; Gupta, S.P., Ed.; Academic Press: London, UK, 2019; pp. 95–134. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Resistance of herpes simplex viruses to nucleoside analogues: Mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 2011, 55, 459–472. [Google Scholar] [CrossRef]
- Schmidt, S.; Bohn-Wippert, K.; Schlattmann, P.; Zell, R.; Sauerbrei, A. Sequence analysis of herpes simplex virus 1 thymidine kinase and DNA polymerase genes from over 300 clinical isolates from 1973 to 2014 finds novel mutations that may be relevant for development of antiviral resistance. Antimicrob. Agents Chemother. 2015, 59, 4938–4945. [Google Scholar] [CrossRef]
- Jiang, Y.-C.; Feng, H.; Lin, Y.-C.; Guo, X.-R. New strategies against drug resistance to herpes simplex virus. Int. J. Oral Sci. 2016, 8, 1–6. [Google Scholar] [CrossRef]
- Reusser, P. Herpesvirus resistance to antiviral drugs: A review of the mechanisms, clinical importance and therapeutic options. J. Hosp. Infect. 1996, 33, 235–248. [Google Scholar] [CrossRef]
- Poole, C.L.; James, S.H. Antiviral Therapies for Herpesviruses: Current Agents and New Directions. Clin. Ther. 2018, 40, 1282–1298. [Google Scholar] [CrossRef]
- Ho, H.T.; Woods, K.; Bronson, J.J.; de Boeck, H.; Martin, J.C.; Hitchcock, M.J. Intracellular metabolism of the antiherpes agent (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine. Mol. Pharmacol. 1992, 41, 197–202. [Google Scholar] [PubMed]
- Bestman-Smith, J.; Boivin, G. Drug Resistance Patterns of Recombinant Herpes Simplex Virus DNA Polymerase Mutants Generated with a Set of Overlapping Cosmids and Plasmids. J. Virol. 2003, 77, 7820–7829. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.L.; Patel, A.; Madinger, N.; Bessesen, M.; Krause, P.R.; Weinberg, A. Development of herpes simplex virus disease in patients who are receiving cidofovir. Clin. Infect. Dis. 2005, 41, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, K.; Zhu, X.; Pham, V.D.; Goyette, N.; Piret, J.; Shi, R.; Boivin, G. Impact of Amino Acid Substitutions in Region II and Helix K of Herpes Simplex Virus 1 and Human Cytomegalovirus DNA Polymerases on Resistance to Foscarnet. Antimicrob. Agents Chemother. 2021, 65, e00390-21. [Google Scholar] [CrossRef]
- Khurana, R.N.; Charonis, A.; Samuel, M.A.; Gupta, A.; Tawansy, K.A. Intravenous foscarnet in the management of acyclovir-resistant herpes simplex virus type 2 in acute retinal necrosis in children. Med. Sci. Monit. 2005, 11, CS75–CS78. [Google Scholar]
- Birkmann, A.; Zimmermann, H. HSV antivirals—Current and future treatment options. Curr. Opin. Virol. 2016, 18, 9–13. [Google Scholar] [CrossRef]
- Biron, K.K.; Harvey, R.J.; Chamberlain, S.C.; Good, S.S.; Smith, A.A., III; Davis, M.G.; Talarico, C.L.; Miller, W.H.; Ferris, R.; Dornsife, R.E. Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action. Antimicrob. Agents Chemother. 2002, 46, 2365–2372. [Google Scholar] [CrossRef]
- Imlay, H.N.; Kaul, D.R. Letermovir and Maribavir for the Treatment and Prevention of Cytomegalovirus Infection in Solid Organ and Stem Cell Transplant Recipients. Clin. Infect. Dis. 2021, 73, 156–160. [Google Scholar] [CrossRef]
- Shiraki, K.; Yasumoto, S.; Toyama, N.; Fukuda, H. Amenamevir, a Helicase-Primase Inhibitor, for the Optimal Treatment of Herpes Zoster. Viruses 2021, 13, 1547. [Google Scholar] [CrossRef]
- Maruho Receives Manufacturing and Marketing Approval for a Partial Change of the Indication and Dosage/Administration for Anti-Herpes Virus Agent “Amenalief Tab. 200mg” for the Treatment of Recurrent Herpes Simplex in Japan. Available online: https://www.maruho.co.jp/english/information/20230224.html (accessed on 21 July 2023).
- Williams-Aziz, S.L.; Hartline, C.B.; Harden, E.A.; Daily, S.L.; Prichard, M.N.; Kushner, N.L.; Beadle, J.R.; Wan, W.B.; Hostetler, K.Y.; Kern, E.R. Comparative Activities of Lipid Esters of Cidofovir and Cyclic Cidofovir against Replication of Herpesviruses In Vitro. Antimicrob. Agents Chemother. 2005, 49, 3724–3733. [Google Scholar] [CrossRef]
- Hostetler, K.Y. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antiviral Res. 2009, 82, A84–A98. [Google Scholar] [CrossRef] [PubMed]
- Quenelle, D.C.; Lampert, B.; Collins, D.J.; Rice, T.L.; Painter, G.R.; Kern, E.R. Efficacy of CMX001 against Herpes Simplex Virus Infections in Mice and Correlations with Drug Distribution Studies. J. Infect. Dis. 2010, 202, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, A.E.; Spencer, J.F.; Ying, B.; Buller, R.M.L.; Wold, W.S.; Toth, K. Cidofovir and brincidofovir reduce the pathology caused by systemic infection with human type 5 adenovirus in immunosuppressed Syrian hamsters, while ribavirin is largely ineffective in this model. Antiviral Res. 2014, 112, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Grossi, I.M.; Foster, S.A.; Gainey, M.R.; Krile, R.T.; Dunn, J.A.; Brundage, T.; Khouri, J.M. Efficacy of delayed brincidofovir treatment against a lethal rabbitpox virus challenge in New Zealand White rabbits. Antiviral Res. 2017, 143, 278–286. [Google Scholar] [CrossRef]
- Gosert, R.; Rinaldo, C.H.; Wernli, M.; Major, E.O.; Hirsch, H.H. CMX001 (1-O-hexadecyloxypropyl-cidofovir) Inhibits Polyomavirus JC Replication in Human Brain Progenitor-Derived Astrocytes. Antimicrob. Agents Chemother. 2011, 55, 2129–2136. [Google Scholar] [CrossRef]
- Lowe, D.M.; Alderton, W.K.; Ellis, M.R.; Parmar, V.; Miller, W.H.; Roberts, G.B.; Fyfe, J.A.; Gaillard, R.; Ertl, P.; Snowden, W. Mode of action of (R)-9-[4-hydroxy-2-(hydroxymethyl)butyl]guanine against herpesviruses. Antimicrob. Agents Chemother. 1995, 39, 1802–1808. [Google Scholar] [CrossRef]
- A Phase 2b Trial of EPB-348 for the Treatment of Herpes Zoster. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT00831103 (accessed on 24 July 2023).
- Tyring, S.K.; Plunkett, S.; Scribner, A.R.; Broker, R.E.; Herrod, J.N.; Handke, L.T.; Wise, J.M.; Martin, P.A.; Valomaciclovir Zoster Study Group. Valomaciclovir versus valacyclovir for the treatment of acute herpes zoster in immunocompetent adults: A randomized, double-blind, active-controlled trial. J. Med. Virol. 2012, 84, 1224–1232. [Google Scholar] [CrossRef]
- Ng, T.I.; Shi, Y.; Huffaker, H.J.; Kati, W.; Liu, Y.; Chen, C.M.; Lin, Z.; Maring, C.; Kohlbrenner, W.E.; Molla, A. Selection and Characterization of Varicella-Zoster Virus Variants Resistant to (R)-9-[4-Hydroxy-2-(hydroxymethy)butyl]guanine. Antimicrob. Agents Chemother. 2001, 45, 1629–1636. [Google Scholar] [CrossRef]
- Zhu, W.; Burnette, A.; Dorjsuren, D.; Roberts, P.E.; Huleihel, M.; Shoemaker, R.H.; Marquez, V.E.; Agbaria, R.; Sei, S. Potent Antiviral Activity of North-Methanocarbathymidine against Kaposi’s Sarcoma-Associated Herpesvirus. Antimicrob. Agents Chemother. 2005, 49, 4965–4973. [Google Scholar] [CrossRef]
- Prichard, M.N.; Keith, K.A.; Quenelle, D.C.; Kern, E.R. Activity and Mechanism of Action of N-Methanocarbathymidine against the Herpesvirus and Orthopoxvirus Infections. Antimicrob. Agents Chemother. 2006, 50, 1336–1341. [Google Scholar] [CrossRef]
- Zalah, L.; Huleihel, M.; Manor, E.; Konson, A.; Ford, H., Jr.; Marquez, V.E.; Johns, D.G.; Agbaria, R. Metabolic pathways of N-methanocarbathymidine, a novel antiviral agent, in native and herpes simplex virus type 1 infected Vero cells. Antiviral Res. 2002, 55, 63–75. [Google Scholar] [CrossRef] [PubMed]
- N&N Pharmaceuticals Inc. Commences Clinical Trials on Its Novel Antiviral Drug. Available online: http://www.prnewswire.com/news-releases/nn-pharmaceuticals-inc-commences-clinical-trials-on-its-novel-antiviral-drug-300254173.html (accessed on 24 July 2023).
- N-Methanocarbathymidine Granted Orphan Drug Designation in the US. Available online: http://www.prnewswire.com/news-releases/n-methanocarbathymidine-granted-orphan-drug-designation-in-the-us-300334437.html (accessed on 24 July 2023).
- Kleymann, G.; Fischer, R.; Betz, U.A.K.; Hendrix, M.; Bender, W.; Schneider, U.; Handke, G.; Eckenberg, P.; Hewlett, G.; Pevzner, V. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat. Med. 2002, 8, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Field, H.J.; Huang, M.L.; Lay, E.M.; Mickleburgh, I.; Zimmermann, H.; Birkmann, A. Baseline sensitivity of HSV-1 and HSV-2 clinical isolates and defined acyclovir-resistant strains to the helicase-primase inhibitor pritelivir. Antiviral Res. 2013, 100, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Quenelle, D.C.; Birkmann, A.; Goldner, T.; Pfaff, T.; Zimmermann, H.; Bonsmann, S.; Collins, D.J.; Rice, T.L.; Prichard, M.N. Efficacy of pritelivir and acyclovir in the treatment of herpes simplex virus infections in a mouse model of herpes simplex encephalitis. Antiviral Res. 2018, 149, 1–6. [Google Scholar] [CrossRef]
- Trial on Efficacy and Safety of Pritelivir Tablets for Treatment of Acyclovir-resistant Mucocutaneous HSV (Herpes Simplex Virus) Infections in Immunocompromised Subjects (PRIOH-1). Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03073967 (accessed on 24 July 2023).
- Thorough QT/QTc of Pritelivir in Healthy Subjects. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05671029 (accessed on 24 July 2023).
- Krasnov, V.P.; Musiyak, V.V.; Vozdvizhenskaya, O.A.; Galegov, G.A.; Andronova, V.L.; Gruzdev, D.A.; Chulakov, E.N.; Vigorov, A.Y.; Ezhikova, M.A.; Kodess, M.I.; et al. N-[omega-(Purin-6-yl)aminoalkanoyl] Derivatives of Chiral Heterocyclic Amines as Promising Anti-Herpesvirus Agents. Eur. J. Org. Chem. 2019, 2019, 4811–4821. [Google Scholar] [CrossRef]
- Krasnov, V.P.; Levit, G.L.; Musiyak, V.V.; Gruzdev, D.A.; Charushin, V.N. Fragment-based approach to novel bioactive purine derivatives. Pure Appl. Chem. 2020, 92, 1277–1295. [Google Scholar] [CrossRef]
- Vozdvizhenskaya, O.A.; Andronova, V.L.; Galegov, G.A.; Levit, G.L.; Krasnov, V.P.; Charushin, V.N. Synthesis and antiherpetic activity of novel purine conjugates with 7,8-difluoro-3-methyl-3,4-dihydro-2H-1,4-benzoxazine. Chem. Heterocycl. Comp. 2021, 57, 490–497. [Google Scholar] [CrossRef]
- Krasnov, V.P.; Zarubaev, V.V.; Gruzdev, D.A.; Vozdvizhenskaya, O.A.; Vakarov, S.A.; Musiyak, V.V.; Chulakov, E.N.; Volobueva, A.S.; Sinegubova, E.O.; Ezhikova, M.A.; et al. Novel purine conjugates with N-heterocycles: Synthesis and anti-influenza activity. Chem. Heterocycl. Comp. 2021, 57, 498–504. [Google Scholar] [CrossRef]
- Krasnov, V.P.; Musiyak, V.V.; Levit, G.L.; Gruzdev, D.A.; Andronova, V.L.; Galegov, G.A.; Orshanskaya, I.R.; Sinegubova, E.O.; Zarubaev, V.V.; Charushin, V.N. Synthesis of Pyrimidine Conjugates with 4-(6-Aminohexanoyl)-7,8-difluoro-3,4-dihydro-3-methyl-2H-[1,4]benzoxazine and Evaluation of Their Antiviral Activity. Molecules 2022, 27, 4236. [Google Scholar] [CrossRef]
- Andronova, V.L.; Galegov, G.A.; Musiyak, V.V.; Vozdvizhenskaya, O.A.; Levit, G.L.; Krasnov, V.P. Antiviral effect of novel purine conjugate LAS-131 against Herpes simplex virus type 1 (Herpesviridae: Alphaherpesvirinae: Simplexvirus: Human alphaherpesvirus 1) in vitro. Voprosy Virusologii 2020, 65, 373–380. (In Russian) [Google Scholar] [CrossRef]
- Sarisky, R.T.; Nguyen, T.T.; Duffy, K.E.; Wittrock, R.J.; Leary, J.J. Difference in Incidence of Spontaneous Mutations between Herpes Simplex Virus Types 1 and 2. Antimicrob. Agents Chemother. 2000, 44, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Gus’kova, A.A.; Skoblov, M.Y.; Korovina, A.N.; Yasko, M.V.; Karpenko, I.L.; Kukhanova, M.K.; Andronova, V.L.; Galegov, G.A.; Skoblov, Y.S. Antiherpetic Properties of Acyclovir 5’-Hydrogenphosphonate and the Mutation Analysis of Herpes Virus Resistant Strains. Chem. Biol. Drug Des. 2009, 74, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Bowtie 2. Available online: https://bowtie-bio.sourceforge.net/bowtie2/index.shtml (accessed on 25 August 2023).
- Mullaney, J.; Moss, H.W.; McGeoch, D.J. Gene UL2 of Herpes Simplex Virus Type 1 Encodes a Uracil-DNA Glycosylase. J. Gen. Virol. 1989, 70, 449–454. [Google Scholar] [CrossRef]
- Bogani, F.; Corredeira, I.; Fernandez, V.; Sattler, U.; Rutvisuttinunt, W.; Defais, M.; Boehmer, P.E. Association between the Herpes Simplex Virus-1 DNA Polymerase and Uracil DNA Glycosylase. J. Biol. Chem. 2010, 285, 27664–27672. [Google Scholar] [CrossRef] [PubMed]
- Pyles, R.B.; Thompson, R.L. Evidence that the Herpes Simplex Virus Type 1 Uracil DNA Glycosylase Is Required for Efficient Viral Replication and Latency in the Murine Nervous System. J. Virol. 1994, 68, 4963–4972. [Google Scholar] [CrossRef]
- Weller, S.K.; Coen, D.M. Herpes Simplex Viruses: Mechanisms of DNA Replication. Cold Spring Harbor Perspect. Biol. 2012, 4, a013011. [Google Scholar] [CrossRef]
- Sauerbrei, A.; Bohn-Wippert, K.; Kaspar, M.; Krumbholz, A.; Karrasch, M.; Zell, R. Database on natural polymorphisms and resistance-related non-synonymous mutations in thymidine kinase and DNA polymerase genes of herpes simplex virus types 1 and 2. J. Antimicrob. Chemother. 2016, 71, 6–16. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef]
- Desai, P.; Sexton, G.L.; McCaffery, J.M.; Person, S. A Null Mutation in the Gene Encoding the Herpes Simplex Virus Type 1 UL37 Polypeptide Abrogates Virus Maturation. J. Virol. 2001, 75, 10259–10271. [Google Scholar] [CrossRef]
- Bucks, M.A.; Murphy, M.A.; O’Regan, K.J.; Courtney, R.J. Identification of interaction domains within the UL37 tegument protein of herpes simplex virus type 1. Virology 2011, 416, 42–53. [Google Scholar] [CrossRef]
- Kelly, B.J.; Mijatov, B.; Fraefel, C.; Cunningham, A.L.; Diefenbach, R.J. Identification of a single amino acid residue which is critical for the interaction between HSV-1 inner tegument proteins pUL36 and pUL37. Virology 2012, 422, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.H.; Cunningham, A.L.; Diefenbach, R.J. The Major Determinant for Addition of Tegument Protein pUL48 (VP16) to Capsids in Herpes Simplex Virus Type 1 Is the Presence of the Major Tegument Protein pUL36 (VP1/2). J. Virol. 2010, 84, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Pasdeloup, D.; McElwee, M.; Beilstein, F.; Labetoulle, M.; Rixon, F.J. Herpesvirus Tegument Protein pUL37 Interacts with Dystonin/BPAG1 to Promote Capsid Transport on Microtubules during Egress. J. Virol. 2013, 87, 2857–2867. [Google Scholar] [CrossRef]
- Sari, T.K.; Pritchard, S.M.; Cunha, C.W.; Wudiri, G.A.; Laws, E.I.; Aguilar, H.C.; Taus, N.S.; Nicola, A.V. Contributions of Herpes Simplex Virus 1 Envelope Proteins to Entry by Endocytosis. J. Virol. 2013, 87, 13922–13926. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, J.M.; Lazear, H.M.; Awasthi, S.; Wang, F.; Friedman, H.M. The Herpes Simplex Virus 1 IgG Fc Receptor Blocks Antibody-Mediated Complement Activation and Antibody-Dependent Cellular Cytotoxicity In Vivo. J. Virol. 2011, 85, 3239–3249. [Google Scholar] [CrossRef]
- Kramer, T.; Enquist, L.W. Directional spread of alphaherpesviruses in the nervous system. Viruses 2013, 5, 678–707. [Google Scholar] [CrossRef]
- Johnson, D.C.; Huber, M.T. Directed Egress of Animal Viruses Promotes Cell-to-Cell Spread. J. Virol. 2002, 76, 1–8. [Google Scholar] [CrossRef]
- Krummenacher, C.; Baribaud, I.; Eisenberg, R.J.; Cohen, G.H. Cellular Localization of Nectin-1 and Glycoprotein D during Herpes Simplex Virus Infection. J. Virol. 2003, 77, 8985–8999. [Google Scholar] [CrossRef]
- Smith, G. Herpesvirus Transport to the Nervous System and Back Again. Annu. Rev. Microbiol. 2012, 66, 153–176. [Google Scholar] [CrossRef]
- Krummenacher, C.; Carfí, A.; Eisenberg, R.J.; Cohen, G.H. Entry of Herpesviruses into Cells: The Enigma Variations. Adv. Exp. Med. Biol. 2013, 790, 178–195. [Google Scholar] [CrossRef]
- Dingwell, K.S.; Brunetti, C.R.; Hendricks, R.L.; Tang, Q.; Tang, M.; Rainbow, A.J.; Johnson, D.C. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 1994, 68, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Heming, J.D.; Huffman, J.B.; Jones, L.M.; Homa, F.L. Isolation and Characterization of the Herpes Simplex Virus 1 Terminase Complex. J. Virol. 2014, 88, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, P.; Wang, N.; Chen, Z.; Su, D.; Zhou, Z.H.; Rao, Z.; Wang, X. Architecture of the herpesvirus genome-packaging complex and implications for DNA translocation. Protein Cell 2020, 11, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Scholtes, L.; Baines, J.D. Effects of Major Capsid Proteins, Capsid Assembly, and DNA Cleavage/Packaging on the pUL17/pUL25 Complex of Herpes Simplex Virus 1. J. Virol. 2009, 83, 12725–12737. [Google Scholar] [CrossRef]
- Albright, B.S.; Kosinski, A.; Szczepaniak, R.; Cook, E.; Stow, N.D.; Conway, J.F.; Weller, S.K. The Putative Herpes Simplex Virus 1 Chaperone Protein UL32 Modulates Disulfide Bond Formation during Infection. J. Virol. 2015, 89, 443–453. [Google Scholar] [CrossRef]
- Yang, K.; Wills, E.G.; Baines, J.D. A Mutation in UL15 of Herpes Simplex Virus 1 That Reduces Packaging of Cleaved Genomes. J. Virol. 2011, 85, 11972–11980. [Google Scholar] [CrossRef]
- Guo, P. High resolution structure of hexameric herpesvirus DNA-packaging motor elucidates revolving mechanism and ends 20-year fervent debate. Protein Cell 2020, 11, 311–315. [Google Scholar] [CrossRef]
- DelToro, D.; Ortiz, D.; Ordyan, M.; Sippy, J.; Oh, C.S.; Keller, N.; Feiss, M.; Catalano, C.E.; Smith, D.E. Walker-A Motif Acts to Coordinate ATP Hydrolysis with Motor Output in Viral DNA Packaging. J. Mol. Biol. 2016, 428, 2709–2729. [Google Scholar] [CrossRef]
- Yu, D.; Weller, S.K. Genetic Analysis of the UL 15 Gene Locus for the Putative Terminase of Herpes Simplex Virus Type 1. Virology 1998, 243, 32–44. [Google Scholar] [CrossRef]
- Chen, W.; Xiao, H.; Wang, X.; Song, S.; Han, Z.; Li, X.; Yang, F.; Wang, L.; Song, J.; Liu, H.; et al. Structural changes of a bacteriophage upon DNA packaging and maturation. Protein Cell 2020, 11, 374–379. [Google Scholar] [CrossRef]
- Wang, N.; Chen, W.; Zhu, L.; Zhu, D.; Feng, R.; Wang, J.; Zhu, B.; Zhang, X.; Chen, X.; Liu, X.; et al. Structures of the portal vertex reveal essential protein-protein interactions for Herpesvirus assembly and maturation. Protein Cell 2020, 11, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Grainge, I.; Zhao, Z.; Vieweger, M. Two classes of nucleic acid translocation motors: Rotation and revolution without rotation. Cell Biosci. 2014, 4, 54. [Google Scholar] [CrossRef]
- Pi, F.; Zhao, Z.; Chelikani, V.; Yoder, K.; Kvaratskhelia, M.; Guo, P. Development of Potent Antiviral Drugs Inspired by Viral Hexameric DNA-Packaging Motors with Revolving Mechanism. J. Virol. 2016, 90, 8036–8046. [Google Scholar] [CrossRef]
- De Clerck, E.; Descamps, J.; Verheist, G.; Walker, R.T.; Jones, A.S.; Torrence, P.F.; Shugar, D. Comparative Efficacy of Antiherpes Drugs against Different Strains of Herpes Simplex Virus. J. Infect. Dis. 1980, 141, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Andronova, V.L.; Galegov, G.A.; Yasko, M.V.; Kukhanova, M.K.; Skoblov, Y.S. Comparative study of resistance to acycloguanosine and acycloguanosine H-phosphonate in herpes simplex virus. Voprosy Virusologii 2010, 55, 31–34. (In Russian) [Google Scholar] [PubMed]
- Schrödinger. Available online: https://www.schrodinger.com/releases/release-2021-4 (accessed on 10 December 2021).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | IC50 (µg/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound 1 | ACV | PCV | GCV | BVDU | IDU | CDV | Ara-A | FOS | |
| HSV-1/L2 | 1.95 | 0.39 | 0.78 | 0.56 | 0.098 | 3.9 | 3.9 | 15.6 | 31.25 |
| HSV-1/L2/R1 | 9.75 | 0.39 | 0.78 | 1.12 | 0.114 ± 0.08 | 3.9 | 3.9 | 15.6 | 31.25 |
| Clone 1 | 20.0 | 0.39 | 0.78 | 0.78 | 0.122 | 3.9 | 3.9 | 15.6 | 31.25 |
| Clone 2 | 20.0 | 0.39 | 0.78 | 0.78 | 0.106 ± 0.008 | 3.9 | 3.9 | 15.6 | 31.25 |
| Clone 3 | 20.0 | 0.39 | 0.78 | 0.89 ± 0.11 | 0.098 | 3.9 | 3.9 | 15.6 | 31.25 |
| Sample | Total Reads × 106 | Software Used for Mapping | |
|---|---|---|---|
| bowtie2 | bowtie2 (Local) | ||
| Clone 1 | 5.75 | 15.33% | 15.54% |
| Clone 2 | 5.11 | 23.39% | 23.69% |
| Clone 3 | 5.36 | 23.80% | 24.13% |
| Wild type | 6.41 | 20.41% | 20.67% |
| Gene Array | Product | Position on Reference Genome | Mutation in Genome | Product |
|---|---|---|---|---|
| TRL3-UL3 | UL2 (uracil-DNA glycosylase) | 9940 | G→A | R19H |
| UL13.5-UL15 | UL15 (large subunit of terminase) | 29,982 | C→T | T321I |
| UL27.5-UL30 | UL30 (DNA polymerase) | 65,203 | C→T | E799D |
| UL37-UL39.6 | UL37 (tegument protein) | 81,716 | C→T | R790H |
| US5-US7 | US7 (glycoprotein I) | 140,281 | G→A | A165T |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasnov, V.P.; Andronova, V.L.; Belyavsky, A.V.; Borisevich, S.S.; Galegov, G.A.; Kandarakov, O.F.; Gruzdev, D.A.; Vozdvizhenskaya, O.A.; Levit, G.L. Large Subunit of the Human Herpes Simplex Virus Terminase as a Promising Target in Design of Anti-Herpesvirus Agents. Molecules 2023, 28, 7375. https://doi.org/10.3390/molecules28217375
Krasnov VP, Andronova VL, Belyavsky AV, Borisevich SS, Galegov GA, Kandarakov OF, Gruzdev DA, Vozdvizhenskaya OA, Levit GL. Large Subunit of the Human Herpes Simplex Virus Terminase as a Promising Target in Design of Anti-Herpesvirus Agents. Molecules. 2023; 28(21):7375. https://doi.org/10.3390/molecules28217375
Chicago/Turabian StyleKrasnov, Victor P., Valeriya L. Andronova, Alexander V. Belyavsky, Sophia S. Borisevich, George A. Galegov, Oleg F. Kandarakov, Dmitry A. Gruzdev, Olga A. Vozdvizhenskaya, and Galina L. Levit. 2023. "Large Subunit of the Human Herpes Simplex Virus Terminase as a Promising Target in Design of Anti-Herpesvirus Agents" Molecules 28, no. 21: 7375. https://doi.org/10.3390/molecules28217375