
Hydrogen Bonding in the Dimer and Monohydrate of 2-Adamantanol: A Test Case for Dispersion-Corrected Density Functional Methods

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
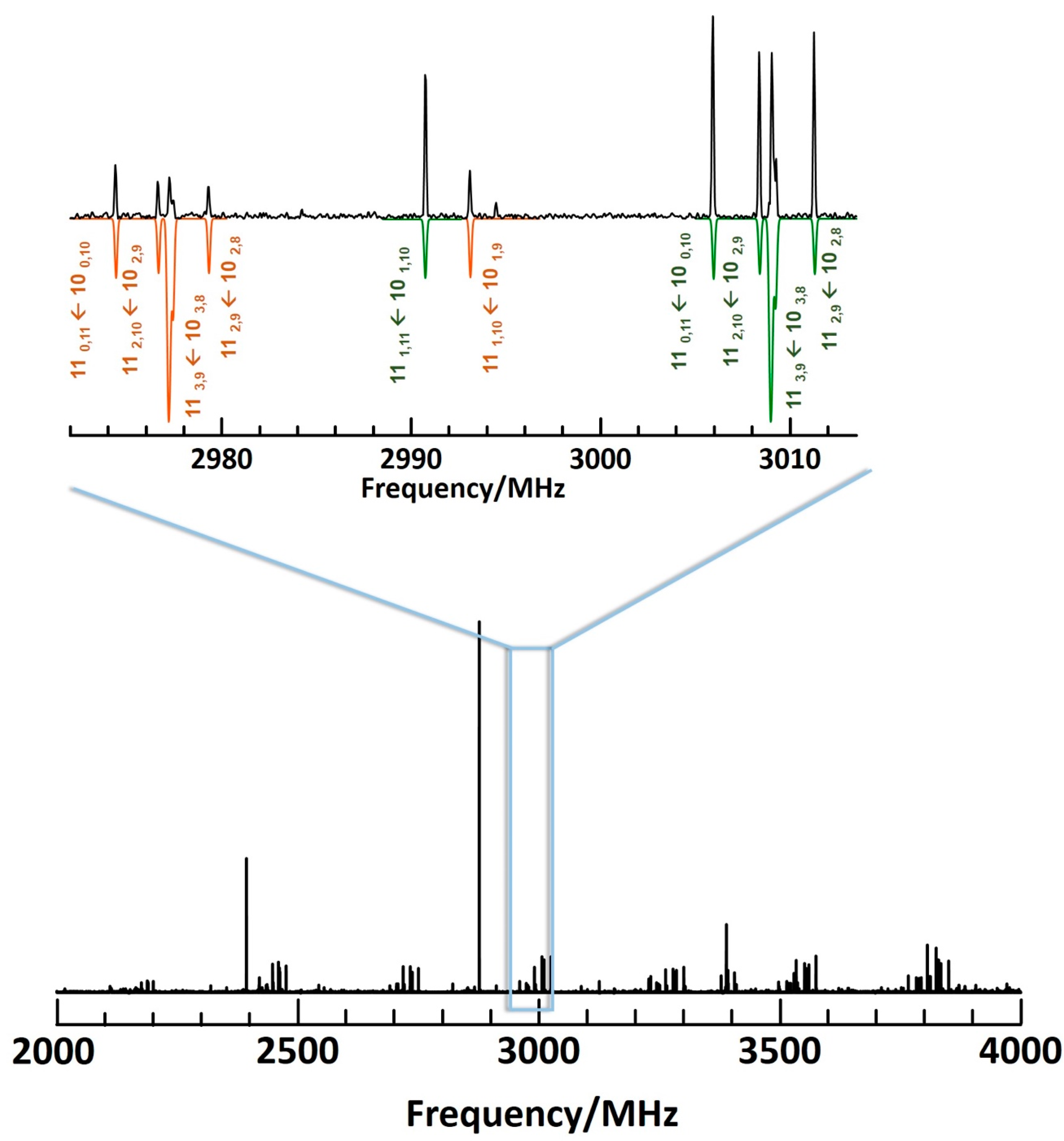
3. Results
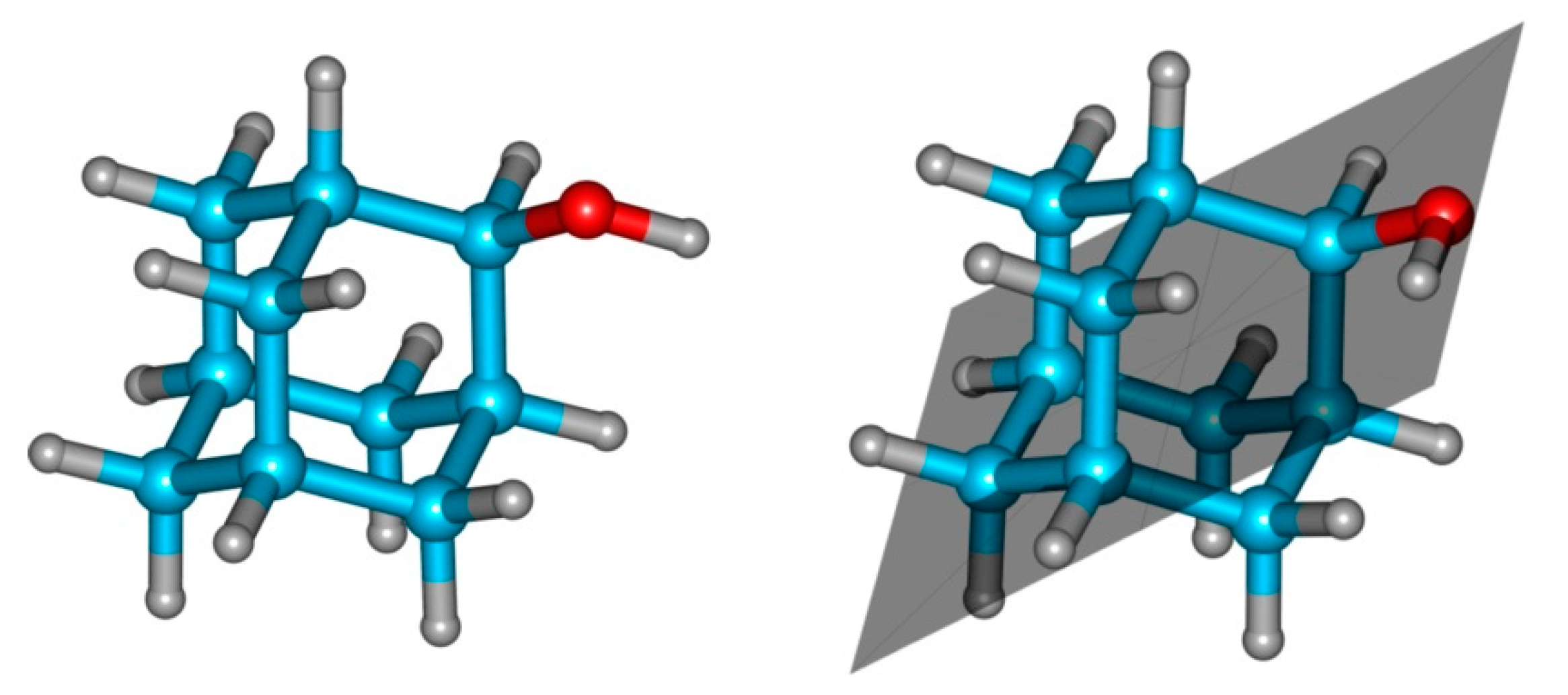
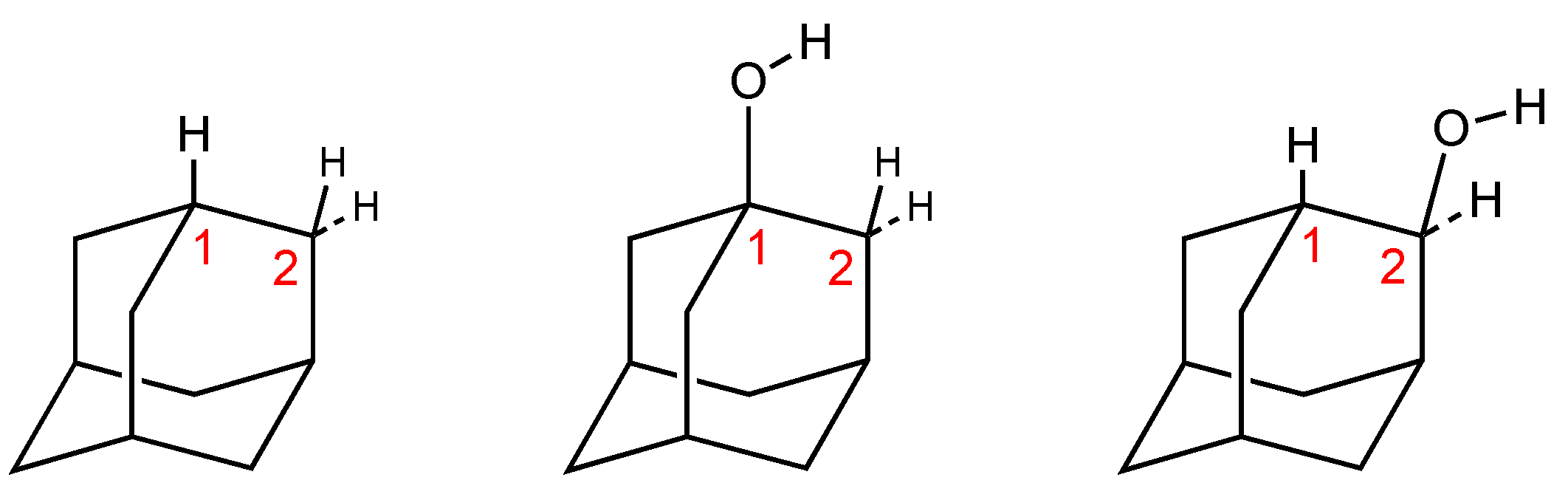
3.1. 2-Adamantanol Monomer
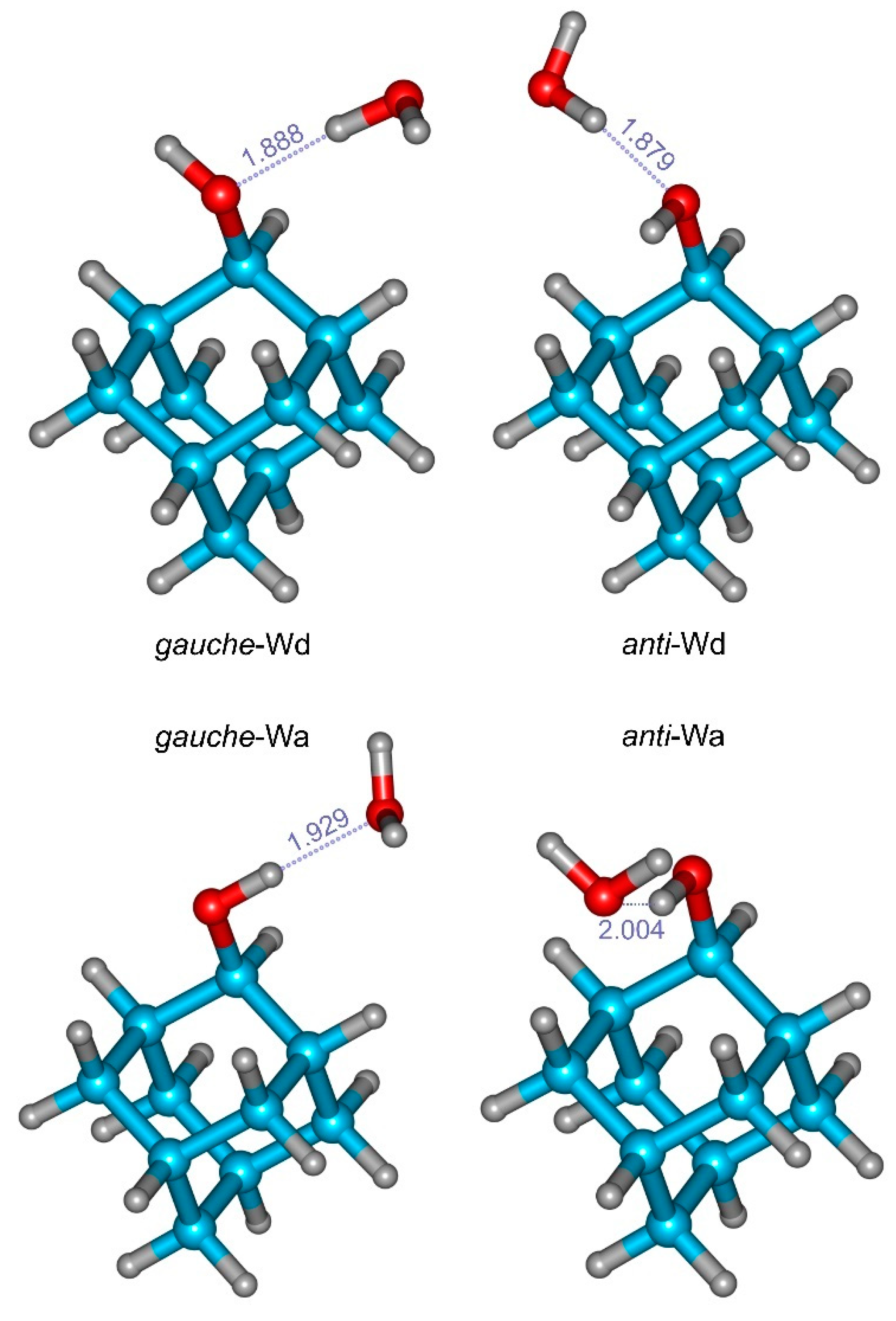
3.2. 2-Adamantanol Monohydrate
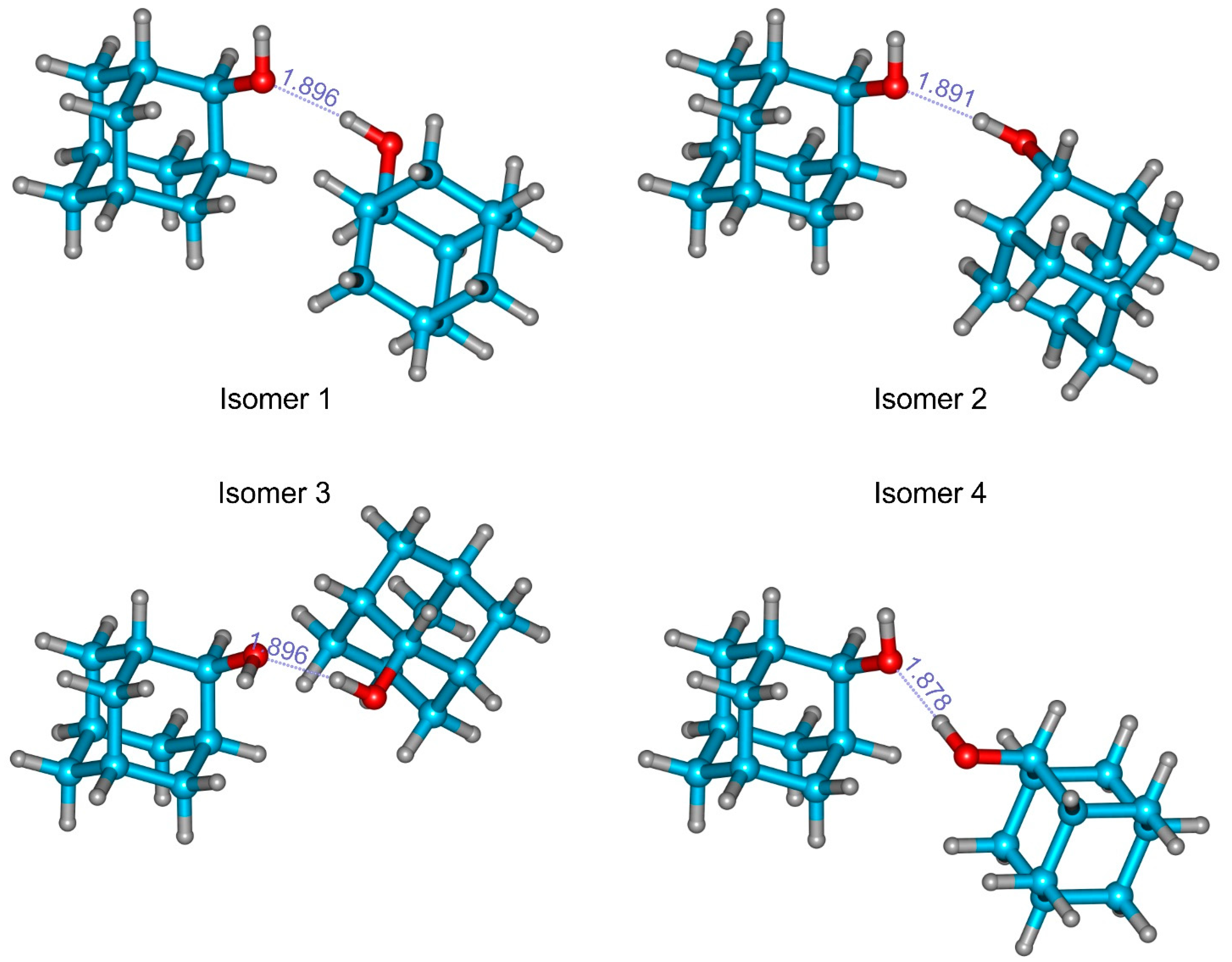
3.3. 2-Adamantanol Dimer
3.4. Non-Covalent Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levy, D.H. Laser Spectroscopy of Cold Gas-Phase Molecules. Annu. Rev. Phys. Chem. 1980, 31, 197–225. [Google Scholar] [CrossRef]
- Schermann, J.-P. Spectroscopy and Modeling of Biomolecular Building Blocks; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 9780444527080. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, UK, 1997; ISBN 0-19-509549-9. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond; Oxford University Press: Oxford, UK, 2009; ISBN 9780199558964. [Google Scholar]
- Hobza, P.; Muller-Dethlefs, K. Non-Covalent Interactions; Hobza, P., Muller-Dethlefs, K., Eds.; Theoretical and Computational Chemistry Series; Royal Society of Chemistry: Cambridge, UK, 2009; ISBN 978-1-84755-853-4. [Google Scholar]
- Challenges and Advances in Computational Chemistry and Physics. In Noncovalent Forces; Scheiner, S. (Ed.) Springer International Publishing: Cham, Switzerland, 2015; Volume 19, ISBN 978-3-319-14162-6. [Google Scholar]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: Oxford, UK, 1997; ISBN 0-19-509011-X. [Google Scholar]
- Panwaria, P.; Das, A. Exploring Non-covalent Interactions by Jet-Cooled Electronic and Vibrational Spectroscopy. In Modern Techniques of Spectroscopy; Singh, D.K., Pradhan, M., Materny, A., Eds.; Springer Nature: Berlin/Heidelberg, Germany, 2021; pp. 57–86. ISBN 978-981-33-6083-9. [Google Scholar]
- Caminati, W.; Grabow, J.-U. Advancements in Microwave Spectroscopy. In Frontiers and Advances in Molecular Spectroscopy; Laane, J., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 569–598. ISBN 9780128112212. [Google Scholar]
- Juanes, M.; Saragi, R.T.; Caminati, W.; Lesarri, A. The Hydrogen Bond and Beyond: Perspectives for Rotational Investigations of Non-Covalent Interactions. Chem.-A Eur. J. 2019, 25, 11402–11411. [Google Scholar] [CrossRef] [PubMed]
- Steber, A.L.; Li, W.; Pate, B.H.; Lesarri, A.; Pérez, C. The First Stages of Nanomicelle Formation Captured in the Sevoflurane Trimer. J. Phys. Chem. Lett. 2022; in press. [Google Scholar] [CrossRef]
- Evangelisti, L.; Caminati, W. Internal dynamics in complexes of water with organic molecules. Details of the internal motions in tert-butylalcohol-water. Phys. Chem. Chem. Phys. 2010, 12, 14433–14441. [Google Scholar] [CrossRef]
- Stockman, P.A.; Blake, G.A.; Lovas, F.J.; Suenram, R.D. Microwave rotation-tunneling spectroscopy of the water-methanol dimer: Direct structural proof for the strongest bound conformation. J. Chem. Phys. 1997, 107, 3782–3790. [Google Scholar] [CrossRef]
- Nedić, M.; Wassermann, T.N.; Xue, Z.; Zielke, P.; Suhm, M.A. Raman spectroscopic evidence for the most stable water/ethanol dimer and for the negative mixing energy in cold water/ethanol trimers. Phys. Chem. Chem. Phys. 2008, 10, 5953–5956. [Google Scholar] [CrossRef]
- Finneran, I.A.; Carroll, P.B.; Allodi, M.A.; Blake, G.A. Hydrogen bonding in the ethanol-water dimer. Phys. Chem. Chem. Phys. 2015, 17, 24210–24214. [Google Scholar] [CrossRef] [Green Version]
- Evangelisti, L.; Gou, Q.; Feng, G.; Caminati, W.; Mead, G.J.; Finneran, I.A.; Carroll, P.B.; Blake, G.A. Conformational equilibrium and internal dynamics in the iso-propanol-water dimer. Phys. Chem. Chem. Phys. 2017, 19, 568–573. [Google Scholar] [CrossRef] [Green Version]
- Melandri, S.; Maris, A.; Favero, P.G.; Caminati, W. Free jet absorption millimetre-wave spectrum and model calculations of phenol-water. Chem. Phys. 2002, 283, 185–192. [Google Scholar] [CrossRef]
- León, I.; Cocinero, E.J.; Millán, J.; Jaeqx, S.; Rijs, A.M.; Lesarri, A.; Castaño, F.; Fernández, J.A.; Leon, I.; Cocinero, E.J.; et al. Exploring microsolvation of the anesthetic propofol. Phys. Chem. Chem. Phys. 2012, 14, 4398–4409. [Google Scholar] [CrossRef]
- Heger, M.; Scharge, T.; Suhm, M.A. From hydrogen bond donor to acceptor: The effect of ethanol fluorination on the first solvating water molecule. Phys. Chem. Chem. Phys. 2013, 15, 16065–16073. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.; Li, W.; Spada, L.; Evangelisti, L.; Lesarri, A.; Caminati, W. Internal dynamics of cyclohexanol and the cyclohexanol-water adduct. Phys. Chem. Chem. Phys. 2019, 21, 3676–3682. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.; Usabiaga, I.; León, I.; Evangelisti, L.; Fernández, J.A.; Lesarri, A. The Six Isomers of the Cyclohexanol Dimer: A Delicate Test for Dispersion Models. Angew. Chemie Int. Ed. 2020, 59, 14081–14085. [Google Scholar] [CrossRef] [PubMed]
- Seifert, N.A.; Steber, A.L.; Neill, J.L.; Pérez, C.; Zaleski, D.P.; Pate, B.H.; Lesarri, A. The interplay of hydrogen bonding and dispersion in phenol dimer and trimer: Structures from broadband rotational spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 11468–11477. [Google Scholar] [CrossRef] [PubMed]
- Medel, R.; Camiruaga, A.; Saragi, R.T.; Pinacho, P.; Pérez, C.; Schnell, M.; Lesarri, A.; Suhm, M.A.; Fernández, J.A. Rovibronic signatures of molecular aggregation in the gas phase: Subtle homochirality trends in the dimer, trimer and tetramer of benzyl alcohol. Phys. Chem. Chem. Phys. 2021, 23, 23610–23624. [Google Scholar] [CrossRef] [PubMed]
- Altnöder, J.; Oswald, S.; Suhm, M.A. Phenyl- vs. cyclohexyl-substitution in methanol: Implications for the OH conformation and for dispersion-affected aggregation from vibrational spectra in supersonic jets. J. Phys. Chem. A 2014, 118, 3266–3279. [Google Scholar] [CrossRef]
- Juanes, M.; Lesarri, A.; Pinacho, R.; Charro, E.; Rubio, J.E.; Enríquez, L.; Jaraíz, M. Sulfur Hydrogen Bonding in Isolated Monohydrates: Furfuryl Mercaptan versus Furfuryl Alcohol. Chem.-A Eur. J. 2018, 24, 6564–6571. [Google Scholar] [CrossRef]
- Juanes, M.; Saragi, R.T.; Pérez, C.; Enríquez, L.; Jaraiz, M.; Lesarri, A. Torsional Chirality and Molecular Recognition: The Homo and Heterochiral Dimers of Thenyl and Furfuryl Alcohol. Phys. Chem. Chem. Phys. 2022, in press. [Google Scholar] [CrossRef]
- Juanes, M.; Saragi, R.T.; Pinacho, R.; Rubio, J.E.; Lesarri, A. Sulfur hydrogen bonding and internal dynamics in the monohydrates of thenyl mercaptan and thenyl alcohol. Phys. Chem. Chem. Phys. 2020, 22, 12412–12421. [Google Scholar] [CrossRef]
- Kanters, J.A.; Hooft, R.W.W.; Duisenberg, A.J.M. Low-temperature, high-resolution crystal structure study of tricyclo[3.3.1.13,7]decan-2-ol (adamantanol-2). J. Crystallogr. Spectrosc. Res. 1990, 20, 123–131. [Google Scholar] [CrossRef]
- Corbelli, G.; Degli Esposti, A.; Favero, L.; Lister, D.G.; Cervellati, R. Large-amplitude vibrations and microwave band spectra. Part 2.-1-Adamantarnine. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1987, 83, 2235–2246. [Google Scholar] [CrossRef]
- Cézard, C.; Rice, C.A.; Suhm, M.A. OH-stretching red shifts in bulky hydrogen-bonded alcohols: Jet spectroscopy and modeling. J. Phys. Chem. A 2006, 110, 9839–9848. [Google Scholar] [CrossRef] [PubMed]
- Baran, J.; Wierzejewska-Hnat, M.; Lutz, E.T.G.; Van Der Maas, J.H. Polarized FT—IR spectra of adamantanol derivatives. J. Mol. Struct. 1991, 244, 87–101. [Google Scholar] [CrossRef]
- Charapennikau, M.B.; Blokhin, A.V.; Kabo, G.J.; Sevruk, V.M.; Krasulin, A.P. Thermodynamic properties of three adamantanols in the ideal gas state. Thermochim. Acta 2003, 405, 85–91. [Google Scholar] [CrossRef]
- Neill, J.L.; Shipman, S.T.; Alvarez-Valtierra, L.; Lesarri, A.; Kisiel, Z.; Pate, B.H. Rotational spectroscopy of iodobenzene and iodobenzene-neon with a direct digital 2-8 GHz chirped-pulse Fourier transform microwave spectrometer. J. Mol. Spectrosc. 2011, 269, 21–29. [Google Scholar] [CrossRef]
- Pérez, C.; Lobsiger, S.; Seifert, N.A.; Zaleski, D.P.; Temelso, B.; Shields, G.C.; Kisiel, Z.; Pate, B.H. Broadband Fourier transform rotational spectroscopy for structure determination: The water heptamer. Chem. Phys. Lett. 2013, 571, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Shipman, S.T.; Pate, B.H. New Techniques in Microwave Spectroscopy. In Handbook of High-Resolution Spectroscopy; Merkt, F., Quack, M., Eds.; Major Reference Works; ohn Wiley & Sons, Ltd.: New York, NY, USA, 2011; pp. 801–828. ISBN 9780470749593. [Google Scholar]
- Grabow, J.-U. Fourier Transform Microwave Spectroscopy Measurement and Instrumentation. In Handbook of High-Resolution Spectroscopy; Merkt, F., Quack, M., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 2011; pp. 723–799. ISBN 978-0-470-74959-3. [Google Scholar]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, rev. C.01, Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Ohno, K.; Maeda, S. A scaled hypersphere search method for the topography of reaction pathways on the potential energy surface. Chem. Phys. Lett. 2004, 384, 277–282. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.K.G. Aspects of Quartic and Sextic Centrifugal Effects on Rotational Energy Levels. In Vibrational Spectra and Structure; Durig, J.R., Ed.; Elsevier, B.V.: Amsterdam, The Netherlands, 1977; Volume 6, pp. 1–89. [Google Scholar]
- Godfrey, P.D.; Brown, R.D. Proportions of species observed in jet spectroscopy-vibrational-energy effects: Histamine tautomers and conformers. J. Am. Chem. Soc. 1998, 120, 10724–10732. [Google Scholar] [CrossRef]
- Florio, G.M.; Christie, R.A.; Jordan, K.D.; Zwier, T.S. Conformational preferences of jet-cooled melatonin: Probing trans- and cis-amide regions of the potential energy surface. J. Am. Chem. Soc. 2002, 124, 10236–10247. [Google Scholar] [CrossRef]
- Lesarri, A.; Pinacho, R.; Enríquez, L.; Rubio, J.E.; Jaraíz, M.; Abad, J.L.; Gigosos, M.A. Rotational spectra of tetracyclic quinolizidine alkaloids: Does a water molecule flip sparteine? Phys. Chem. Chem. Phys. 2017, 19, 17553–17559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, N.A.; Finneran, I.A.; Pérez, C.; Zaleski, D.P.; Neill, J.L.; Steber, A.L.; Suenram, R.D.; Lesarri, A.; Shipman, S.T.; Pate, B.H. AUTOFIT, an automated fitting tool for broadband rotational spectra, and applications to 1-hexanal. J. Mol. Spectrosc. 2015, 312, 13–21. [Google Scholar] [CrossRef]
- Xie, F.; Seifert, N.A.; Heger, M.; Thomas, J.; Jäger, W.; Xu, Y. The rich conformational landscape of perillyl alcohol revealed by broadband rotational spectroscopy and theoretical modelling. Phys. Chem. Chem. Phys. 2019, 21, 15408–15416. [Google Scholar] [CrossRef]
- Ruoff, R.S.; Klots, T.D.; Emilsson, T.; Gutowsky, H.S. Relaxation of conformers and isomers in seeded supersonic jets of inert gases. J. Chem. Phys. 1990, 93, 3142–3150. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Cole, W.T.S.; Saykally, R.J. The water dimer I: Experimental characterization. Chem. Phys. Lett. 2015, 633, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Pérez, C.; Muckle, M.T.; Zaleski, D.P.; Seifert, N.A.; Temelso, B.; Shields, G.C.; Kisiel, Z.; Pate, B.H. Structures of Cage, Prism, and Book Isomers of Water Hexamer from Broadband Rotational Spectroscopy. Science 2012, 336, 897–901. [Google Scholar] [CrossRef] [Green Version]
- Pérez, C.; Zaleski, D.P.; Seifert, N.A.; Temelso, B.; Shields, G.C.; Kisiel, Z.; Pate, B.H. Hydrogen Bond Cooperativity and the Three-Dimensional Structures of Water Nonamers and Decamers. Angew. Chemie Int. Ed. 2014, 53, 14368–14372. [Google Scholar] [CrossRef] [Green Version]
- Saragi, R.T.; Juanes, M.; Pinacho, R.; Rubio, J.E.; Fernández, J.A.; Lesarri, A. Molecular Recognition, Transient Chirality and Sulfur Hydrogen Bonding in the Benzyl Mercaptan Dimer. Symmetry 2021, 13, 2022. [Google Scholar] [CrossRef]
- Saragi, R.T.; Juanes, M.; Pérez, C.; Pinacho, P.; Tikhonov, D.S.; Caminati, W.; Lesarri, A. Switching hydrogen bonding to π-stacking: The thiophenol dimer and trimer. J. Phys. Chem. Lett. 2021, 12, 1367–1373. [Google Scholar] [CrossRef]
- Das, A.; Mandal, P.K.; Lovas, F.J.; Medcraft, C.; Walker, N.R.; Arunan, E. The H2S Dimer is Hydrogen-Bonded: Direct Confirmation from Microwave Spectroscopy. Angew. Chem. Int. Ed. 2018, 57, 15199–15203. [Google Scholar] [CrossRef] [Green Version]
- Gou, Q.; Spada, L.; Vallejo-López, M.; Lesarri, A.; Cocinero, E.J.; Caminati, W. Interactions between alkanes and aromatic molecules: A rotational study of pyridine–methane. Phys. Chem. Chem. Phys. 2014, 16, 13041–13046. [Google Scholar] [CrossRef] [PubMed]
- Jurečka, P.; Šponer, J.; Černý, J.; Hobza, P. Benchmark database of accurate (MP2 and CCSD (T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
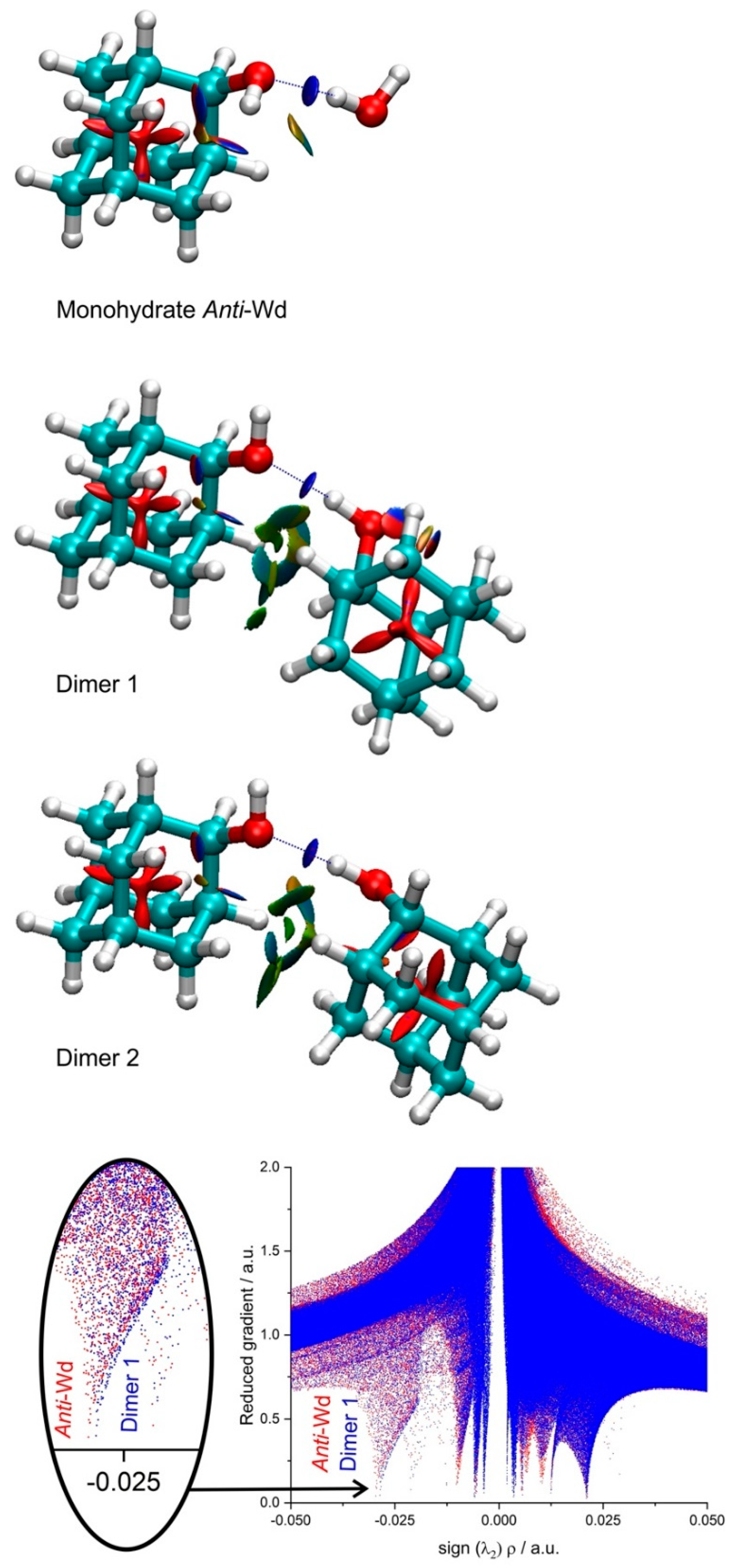{kind=link}
{kind=link}
| Experiment | Theory B2PLYP-D3(BJ) | ||
|---|---|---|---|
| Gauche | Gauche | Anti | |
| A/MHz a | 1680.6888(30) d | 1691.9 | 1689.5 |
| B/MHz | 1197.8532(20) | 1204.5 | 1197.5 |
| C/MHz | 1195.3429(18) | 1202.3 | 1192.6 |
| κ | −0.990(4) | −0.991 | −0.980 |
| DJ/kHz | [ 0.] e | 0.0322 | 0.0310 |
| DJK/kHz | 0.280(89) | 0.0303 | 0.0299 |
| DK/kHz | −0.204(61) | −0.0094 | −0.0080 |
| d1/kHz | [ 0.] | 0.0005 | 0.0010 |
| d2/kHz | [ 0.] | 0.0000 | 0.0006 |
| |μa|/D | 0.48 | 1.75 | |
| |μb|/D | 0.93 | 0.72 | |
| |μc|/D | 0.96 | 0.00 | |
| ΔEZPE/kJ mol−1 b | 0.00 | 2.75 | |
| ΔG/kJ mol−1 | 0.00 | 2.47 | |
| Nc | 25 | ||
| σ/kHz | 7.5 | ||
| Experiment | Theory B2PLYP-D3(BJ) | ||||
|---|---|---|---|---|---|
| Gauche-Wd | Anti-Wd | Gauche-Wa | Anti-Wa | ||
| A/MHz a | 1511.8092(12) d | 1534.02 | 1524.14 | 1561.90 | 1481.44 |
| B/MHz | 690.17508(75) | 684.93 | 694.66 | 654.28 | 738.62 |
| C/MHz | 662.22912(72) | 659.66 | 666.48 | 635.52 | 696.20 |
| κ | −0.934(1) | −0.942 | −0.934 | −0.959 | −0.892 |
| DJ/kHz | 0.3846(79) | 0.2399 | −0.6722 | 0.1968 | 0.1643 |
| DJK/kHz | 1.732(24) | 7.2570 | 0.1143 | 0.3920 | 2.9012 |
| DK/kHz | −1.721(36) | −7.2804 | −0.7008 | −0.3013 | −2.8952 |
| d1/kHz | −0.0366(65) | −0.0291 | −3.4672 | −0.0221 | −0.0178 |
| d2/kHz | 0.0067(13) | 0.0316 | −0.6228 | 0.0018 | 0.0177 |
| |μa|/D | 2.68 | 2.13 | 2.97 | 1.59 | |
| |μb|/D | 1.21 | 1.20 | 1.24 | 0.84 | |
| |μc|/D | 0.45 | 0.37 | 0.32 | 0.00 | |
| ΔEZPE/kJ mol−1 b | 0.00 | 1.98 | 3.25 | 6.34 | |
| ΔG/kJ mol−1 | 0.00 | 4.24 | 2.50 | 11.12 | |
| ΔEc/kJ mol−1 | −25.31 | −27.11 | −22.34 | −21.51 | |
| Nc | 60 | ||||
| σ/kHz | 10.6 | ||||
| Experiment | Theory | |||||||
|---|---|---|---|---|---|---|---|---|
| Isomer A | Isomer B | Isomer 1-CS1 | Isomer 2-CS4 | Isomer 3 | Isomer 4 | Isomer 5 | Isomer 6 | |
| A/MHz a | 701.117(17) e | 702.79(27) | 703.95 | 702.74 | 701.46 | 709.22 | 701.84 | 712.05 |
| B/MHz | 138.34738(18) | 136.84142(21) | 140.03 | 139.42 | 141.81 | 139.83 | 145.30 | 138.32 |
| C/MHz | 135.18537(18) | 133.80126(23) | 136.84 | 136.13 | 138.49 | 137.08 | 142.18 | 135.81 |
| κ | −0.9888(4) | −0.9893(8) | −0.989 | −0.988 | −0.988 | −0.990 | −0.989 | −0.991 |
| DJ/kHz | 0.00643(18) | 0.00471(23) | 0.0063 | 0.0045 | 0.0061 | 0.0046 | 0.0057 | 0.0065 |
| DJK/kHz | −0.0169(54) | −0.0150 | 0.0142 | −0.0067 | 0.0037 | −0.0121 | −0.0222 | |
| DK/kHz | 0.0532 | 0.0104 | 0.0361 | 0.0203 | 0.0426 | 0.0808 | ||
| d1/kHz | −0.0003 | −0.0002 | −0.0003 | −0.0002 | −0.0003 | −0.0004 | ||
| d2/kHz | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | ||
| |μa|/D | 3.12 | 2.66 | 2.61 | 2.60 | 2.33 | 2.86 | ||
| |μb|/D | 0.23 | 0.31 | 0.73 | 0.24 | 1.11 | 0.46 | ||
| |μc|/D | 1.10 | 1.21 | 0.79 | 1.84 | 2.07 | 1.83 | ||
| Nb | 124 | 88 | ||||||
| σ/kHz | 13.2 | 10.5 | ||||||
| B3LYP-D3 c | ||||||||
| ΔEZPE/kJ mol−1 | 0.00 | 0.00 | 1.00 | 0.69 | 1.09 | 0.83 | ||
| ΔG298 K/kJ mol−1 | 0.97 | 0.00 | 2.03 | 1.68 | 3.18 | 0.69 | ||
| ΔEc/kJ mol−1 | −35.77 | −35.61 | −36.86 | −35.19 | −34.64 | −34.48 | ||
| B2PLYP-D3 d | ||||||||
| ΔEZPE/kJ mol−1 | 0.00 | 0.04 | 3.05 | 0.43 | 2.32 | 0.32 | ||
| ΔEc/kJ mol−1 | −32.72 | −32.47 | −33.76 | −32.17 | −31.09 | −31.46 | ||
| Cluster | ΔEelec | ΔEdisp | ΔEind | ΔEexch | ΔEtotal | ΔES22 |
|---|---|---|---|---|---|---|
| (Benzyl alcohol)2 a | −58.7 [44.5%] k | −54.6 [41.4%] | −18.6 [14.1%] | 89.8 | −42.1 | |
| (Furfuryl alcohol)2 b | −61.7 [49.7%] | −21.5 [17.3%] | −41.0 [33.0%] | 85.7 | −38.5 | |
| (Benzyl mercaptan)2 c | −39.3 [34.4%] | −61.1 [53.6%] | −13.6 [12.0%] | 78.7 | −35.3 | |
| (2-Adamantanol)2 Isomer 1 d | −44.8 [46.6% ] | −16.2 [19.2%] | −35.2 [51.7%] | 63.3 | −32.9 | |
| (2-Adamantanol)2 Isomer 2 d | −45.6 [46.9%] | −16.7 [19.8%] | −35.2 [51.9%] | 64.9 | −32.9 | |
| (Cyclohexanol)2 e | −46.5 [51.8%] | −16.6 [18.5%] | −26.6 [29.7%] | 60.5 | −29.2 | |
| (Phenol)2 f | −41.8 [48.3%] | −28.8 [18.4%] | −15.9 [33.3%] | 58.9 | −27.6 | −29.5 l |
| (Thiophenol)2 PD1-trans g | −24.9 [31.0%] | −47.9 [59.5%] | −7.7 [9.5%] | 54.6 | −25.9 | |
| 2-Adamantanol ··· H2O d | −42.3 [57.6%] | −14.6 [26.7%] | −16.5 [41.3%] | 49.9 | −23.5 | |
| (H2O)2 h | −35.7 [63.5%] | −9.5 [16.8%] | −11.1 [19.8%] | 37.7 | −18.6 | −21.0 l |
| (H2S)2 i | −12.1 [49.0%] | −7.8 [31.7%] | −4.7 [19.3%] | 19.2 | −5.4 | |
| Pyridine–methane j | −3.0 [20.6%] | −10.9 [74.6%] | −0.7 [4.8%] | 9.4 | −5.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juanes, M.; Saragi, R.T.; Pérez, C.; Evangelisti, L.; Enríquez, L.; Jaraíz, M.; Lesarri, A. Hydrogen Bonding in the Dimer and Monohydrate of 2-Adamantanol: A Test Case for Dispersion-Corrected Density Functional Methods. Molecules 2022, 27, 2584. https://doi.org/10.3390/molecules27082584
Juanes M, Saragi RT, Pérez C, Evangelisti L, Enríquez L, Jaraíz M, Lesarri A. Hydrogen Bonding in the Dimer and Monohydrate of 2-Adamantanol: A Test Case for Dispersion-Corrected Density Functional Methods. Molecules. 2022; 27(8):2584. https://doi.org/10.3390/molecules27082584
Chicago/Turabian StyleJuanes, Marcos, Rizalina Tama Saragi, Cristóbal Pérez, Luca Evangelisti, Lourdes Enríquez, Martín Jaraíz, and Alberto Lesarri. 2022. "Hydrogen Bonding in the Dimer and Monohydrate of 2-Adamantanol: A Test Case for Dispersion-Corrected Density Functional Methods" Molecules 27, no. 8: 2584. https://doi.org/10.3390/molecules27082584