
Amino Alcohols as Potential Antibiotic and Antifungal Leads

 ,
,  , , and
, , and
Abstract
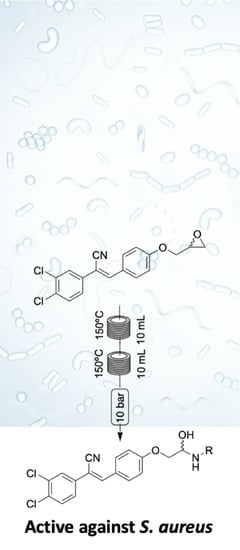
:
1. Introduction
2. Results
3. Discussion
4. Conclusions
5. Experimental
5.1. Biology
5.2. Antibacterial Assays
5.3. Antifungal Assay
5.4. Chemistry
General Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Shore, C.K.; Coukell, A. Roadmap for antibiotic discovery. Nat. Microbiol. 2016, 1, 16083. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2019; US Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019. [CrossRef] [Green Version]
- Report E Joint Technical. The Bacterial Challenge: Time to React 2009. Available online: http://ecdc.europa.eu/en/publications/Publications/0909_TER_The_Bacterial_Challenge_Time_to_React.pdf (accessed on 12 December 2021).
- Silver, L.L. Challenges of Antibacterial Discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [Green Version]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J. Design and Discovery of New Antibacterial Agents: Advances, Perspectives, Challenges. Curr. Med. Chem. 2019, 25, 4972–5006. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Cooper, M.A. Antibiotics in the clinical pipeline in 2011. J. Antibiot. 2011, 64, 413–425. [Google Scholar] [CrossRef]
- Silver, L.L. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 2007, 6, 41–55. [Google Scholar] [CrossRef]
- Plackett, B. No money for new drugs. Nature 2020, 586, S50–S52. [Google Scholar] [CrossRef]
- McKenna, M. The antibiotic gamble. Nature 2020, 584, 338–341. [Google Scholar] [CrossRef]
- CarB-X. Available online: http://www.carb-x.org (accessed on 12 December 2021).
- León-Buitimea, A.; Garza-Cárdenas, C.R.; Garza-Cervantes, J.A.; Lerma-Escalera, J.A.; Morones-Ramírez, J.R. The Demand for New Antibiotics: Antimicrobial Peptides, Nanoparticles, and Combinatorial Therapies as Future Strategies in Antibacterial Agent Design. Front. Microbiol. 2020, 11, 1669. [Google Scholar] [CrossRef]
- Fuller, A.A.; Dounay, A.B.; Schirch, D.; Rivera, D.G.; Hansford, K.A.; Elliott, A.G.; Zuegg, J.; Cooper, M.A.; Blaskovich, M.A.T.; Hitchens, J.R.; et al. Multi-Institution Research and Education Collaboration Identifies New Antimicrobial Compounds. ACS Chem. Biol. 2020, 15, 3187–3196. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.R.; Russell, C.C.; Gilbert, J.; McCluskey, A.; Sakoff, J.A. Amino alcohol acrylonitriles as broad spectrum and tumour selective cytotoxic agents. RSC Med. Chem. 2021, 12, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.C.; Stevens, A.; Young, K.A.; Baker, J.R.; McCluskey, S.N.; Khazandi, M.; Pi, H.; Ogunniyi, A.; Page, S.W.; Trott, D.J.; et al. Discovery of 4,6-bis(2-((E)-benzylidene)hydrazinyl)pyrimidin-2-Amine with Antibiotic Activity. Chemistryopen 2019, 8, 896–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, C.C.; Stevens, A.; Pi, H.; Khazandi, M.; Ogunniyi, A.D.; Young, K.A.; Baker, J.R.; McCluskey, S.N.; Page, S.W.; Trott, D.J.; et al. Gram-Positive and Gram-Negative Antibiotic Activity of Asymmetric and Monomeric Robenidine Analogues. ChemMedChem 2018, 13, 2573–2580. [Google Scholar] [CrossRef]
- Pi, H.; Nguyen, H.; Venter, H.; Boileau, A.R.; Woolford, L.; Garg, S.; Page, S.W.; Russell, C.C.; Baker, J.R.; McCluskey, A.; et al. In vitro Activity of Robenidine Analog NCL195 in Combination With Outer Membrane Permeabilizers Against Gram-Negative Bacterial Pathogens and Impact on Systemic Gram-Positive Bacterial Infection in Mice. Front. Microbiol. 2020, 11, 1556. [Google Scholar] [CrossRef]
- Baker, J.R.; Pollard, B.L.; Lin, A.J.S.; Gilbert, J.; Paula, S.; Zhu, X.; Sakoff, J.A.; McCluskey, A. Modelling and Phenotypic Screening of NAP-6 and 10-Cl-BBQ, AhR Ligands Displaying Selective Breast Cancer Cytotoxicity in Vitro. ChemMedChem 2021, 16, 1499–1512. [Google Scholar] [CrossRef]
- Sun, J.; Baker, J.R.; Russell, C.C.; Cossar, P.J.; Pham, H.N.T.; Sakoff, J.A.; Scarlett, C.J.; McCluskey, A. Novel Cytotoxic 1,2,3-Triazoles as Potential new Leads Targeting the S100A2-p53 Complex. ChemMedChem 2021, 16. [Google Scholar] [CrossRef]
- Baker, J.R.; Russell, C.C.; Gilbert, J.; Sakoff, J.A.; McCluskey, A. Amino Alcohol Acrylonitriles as Activators of the Aryl Hydrocarbon Receptor Pathway: An Unexpected MTT Phenotypic Screening Outcome. ChemMedChem 2020, 15, 490–505. [Google Scholar] [CrossRef]
- Stanton, D.T.; Baker, J.R.; McCluskey, A.; Paula, S. Development and interpretation of a QSAR model for in vitro breast cancer (MCF-7) cytotoxicity of 2-phenylacrylonitriles. J. Comput. Aid. Mol. Des. 2021, 35, 613–628. [Google Scholar] [CrossRef]
- Baker, J.R.; Sakoff, J.A.; McCluskey, A. The aryl hydrocarbon receptor (AhR) as a breast cancer drug target. Med. Res. Rev. 2020, 40, 972–1001. [Google Scholar] [CrossRef]
- Mishra, S.; Patel, S. Design, Synthesis, and Anti-bacterial Activity of Novel Deoxycholic Acid-Amino Alcohol Conjugates. Med Chem. 2020, 16, 385–391. [Google Scholar] [CrossRef]
- Myers, A.G.; Clark, R.B. Discovery of Macrolide Antibiotics Effective against Multi-Drug Resistant Gram-Negative Pathogens. ACC Chem Res. 2021, 54, 1635–1645. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Ke, M.; Liu, M.; Zhan, P.; Xiao, Y.C.; Chen, F. Unified Strategy to Amphenicol Antibiotics: Asymmetric Synthesis of (-)-Chloramphenicol, (-)-Azidamphenicol, and (+)-Thiamphenicol and Its (+)-3-Floride. J. Org. Chem. 2020, 85, 15360–15367. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.T.; Hansford, K.A.; Gong, Y.; Butler, M.S.; Muldoon, C.; Huang, J.X.; Ramu, S.; Silva, A.B.; Cheng, M.; Kavanagh, A.M.; et al. Protein-inspired antibiotics active against vancomycin- and daptomycin-resistant bacteria. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossar, P.J.; Baker, J.; Cain, N.; McCluskey, A. In situ epoxide generation by dimethyldioxirane oxidation and the use of epichlorohydrin in the flow synthesis of a library of β-amino alcohols. R. Soc. Open Sci. 2018, 5, 171190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, Structure–Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef]
- Liebler, D.C. Protein Damage by Reactive Electrophiles: Targets and Consequences. Chem. Res. Toxicol. 2008, 21, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.C.; Watt, A.P.; Nicoll-Griffith, D.A.; Baillie, T.A. Drug−Protein Adducts: An Industry Perspective on Minimizing the Potential for Drug Bioactivation in Drug Discovery and Development. Chem. Res. Toxicol. 2004, 17, 3–16, Erratum in Chem. Res. Toxicol. 2005, 18, 1777. [Google Scholar] [CrossRef]
- Baillie, T.A. Metabolism and Toxicity of Drugs. Two Decades of Progress in Industrial Drug Metabolism. Chem. Res. Toxicol. 2008, 21, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Hansford, K.A.; Blaskovich, M.A.; Cooper, M.A. Chemical philanthropy: A path forward for antibiotic discovery? Future Med. Chem. 2016, 8, 925–929. [Google Scholar] [CrossRef] [Green Version]
- Blaskovich, M.A.T.; Zuegg, J.; Elliott, A.; Cooper, M. Helping Chemists Discover New Antibiotics. ACS Infect. Dis. 2015, 1, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Desselle, M.; Neale, R.; Hansford, K.A.; Zuegg, J.; Elliott, A.; Cooper, M.A.; Blaskovich, M.A. Institutional profile: Community for Open Antimicrobial Drug Discovery—Crowdsourcing new antibiotics and antifungals. Future Sci. OA 2017, 3, FSO171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossar, P.J.M.C.; Gordon, C.P.; Ambrus, J.I.; Lewis, P.J.; McCluskey, A. Identification and validation of small molecule modulators of the NusB-NusE interaction. Bioorg. Med. Chem. Lett. 2017, 27, 162–167. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.; Daniel, J.A.; Hadzic, G.; Chau, N.; Clayton, E.L.; Mariana, A.; Whiting, A.; Gorgani, N.N.; Lloyd, J.R.; Quan, A.; et al. Building a Better Dynasore: The Dyngo Compounds Potently Inhibit Dynamin and Endocytosis. Traffic 2013, 14, 1272–1289. [Google Scholar] [CrossRef]
- Baker, J.R.; Gilbert, J.; Paula, S.; Zhu, X.; Sakoff, J.A.; McCluskey, A. Dichlorophenylacrylonitriles as AhR Ligands That Display Selective Breast Cancer Cytotoxicity in vitro. ChemMedChem 2018, 13, 1447–1458. [Google Scholar] [CrossRef]
- Tarleton, M.; Gilbert, J.; Robertson, M.J.; McCluskey, A.; Sakoff, J.A. Library synthesis and cytotoxicity of a family of 2-phenylacrylonitriles and discovery of an estrogen dependent breast cancer lead compound. MedChemComm 2011, 2, 31–37. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structure | S. aureusa | E. colib | K. pneumoniaec | P. aeruginosad | A. baumanniie | C. albicansf | C. neoformansg |
 1a | 14.2 | <10 | <10 | 22.3 | <10 | 14.7 | <10 |
 1b | 16.2 | <10 | <10 | 11.4 | 13.9 | 11.8 | 10.7 |
 1c | 13.2 | <10 | 10.5 | 16.2 | <10 | <10 | <10 |
 1d | 13.5 | <10 | 13.6 | 19.0 | 15.0 | <10 | <10 |
 1e | 15.9 | <10 | 12.6 | 27.8 | <10 | 11.8 | <10 |
 1f | <10 | <10 | 17.1 | 14.6 | 13.2 | 10 | 22.3 |
 1g | <10 | <10 | 11.9 | 10.4 | 37.0 | 17.2 | <10 |
 1h | 13.6 | <10 | <10 | 23.4 | 10 | <10 | <10 |
 1i | <10 | <10 | <10 | 16.6 | <10 | 12.2 | <10 |
 1j | <10 | <10 | 10.2 | 18.5 | 27.5 | <10 | <10 |
 1k | 12.7 | <10 | 17.5 | 17.7 | 10.3 | <10 | 10.3 |
 1l | 10 | <10 | <10 | 21.6 | <10 | <10 | <10 |
 1m | 14.9 | <10 | <10 | 13.0 | <10 | 63.6 | 10 |
Structure | S. aureus a | E. coli b | K. pneumoniae c | P. aeruginosa d | A. baumannii e | C. albicans f | C. neoformansg |
 1n | <10 | <10 | <10 | 17.5 | <10 | 10 | <10 |
 1o | <10 | <10 | 10.1 | 17.6 | <10 | 37.1 | <10 |
 1p | 32.0 | <10 | 19.0 | 22.8 | <10 | 10.3 | <10 |
 1q | 24.4 | <10 | 14.0 | 20.9 | <10 | 10 | <10 |
 1r | 13.8 | <10 | <10 | 26.8 | 14.5 | 16.6 | <10 |
 | |||||||
 1s | <10 | <10 | <10 | 11.1 | 15.9 | <10 | 23.6 |
 1t | <10 | <10 | <10 | 11.3 | 19.7 | <10 | 28.6 |
 | |||||||
 1u | 12.9 | <10 | <10 | 13.2 | 14.3 | <10 | 25.9 |
 1v | 16.1 | <10 | 16.3 | 20.1 | 12.0 | 10.1 | <10 |
 1w | 22.7 | <10 | <10 | <10 | <10 | <10 | 21.8 |
 | |||||||
 1x | <10 | <10 | 11.01 | 10.4 | <10 | <10 | 18.5 |
 1y | <10 | <10 | 21.3 | 17.3 | 48.2 | <10 | 23.8 |
Structure | S. aureus a | E. coli b | K. pneumoniae c | P. aeruginosa d | A. baumannii e | C. albicans f | C. neoformansg |
 6 | <10 | <10 | <10 | 24.5 | <10 | <10 | <10 |
 8a | 14.5 | <10 | 17.1 | 22.7 | 11.6 | <10 | <10 |
 8b | <10 | <10 | <10 | <10 | <10 | 14.1 | <10 |
 8c | <10 | <10 | <10 | 11.7 | 19.3 | 10 | <10 |
 8d | <10 | <10 | 10 | 23.2 | <10 | 17.3 | <10 |
Structure | S. aureus a | E. coli b | K. pneumoniae c | P. aeruginosa d | A. baumannii e | C. albicans f | C. neoformansg |
 8e | 88.3 | <10 | 13.1 | 20.8 | 13.8 | 11.3 | <10 |
 8f | 54.9 | <10 | <10 | <10 | 15.1 | <10 | <10 |
 8g | 67.2 | <10 | <10 | 22.3 | <10 | 50.1 | <10 |
 8h | 92 | <10 | <10 | 13.2 | <10 | <10 | <10 |
 8i | 90.3 | <10 | <10 | 26.5 | 11.8 | 13.3 | <10 |
 8j | 90.1 | <10 | 16.9 | 27.5 | <10 | 19.1 | <10 |
 8k | 16.2 | <10 | <10 | 24.4 | <10 | 53.0 | <10 |
 8l | 15.9 | <10 | <10 | 21.5 | <10 | 35.0 | <10 |
Structure | S. aureusa | E. colib | K. pneumoniaec | P. aeruginosad | A. baumanniie | C. albicansf | C. neoformansg |
 8m | <10 | <10 | <10 | 11.2 | <10 | <10 | <10 |
 8n | <10 | <10 | <10 | 19.0 | <10 | <10 | <10 |
 8o | 11.6 | <10 | 10.8 | 23.6 | <10 | 12.5 | <10 |
 8p | 12.8 | <10 | <10 | 16.1 | <10 | 12.5 | 96.3 |
 8q | <10 | <10 | <10 | 15.2 | <10 | <10 | -<10 |
 8r | 15.3 | <10 | 28.4 | 22.8 | 10 | <10 | <10 |
 8s | 13.9 | <10 | 12.3 | 20.6 | <10 | 64.6 | 97.1 |
 8t | 12.8 | <10 | <10 | 10 | <10 | <10 | <10 |
 8u | 10 | <10 | 15.1 | 27.7 | 11.2 | 10.6 | <10 |
 8v | 12.6 | <10 | 13.1 | 23.1 | <10 | <10 | <10 |
 8w | <10 | <10 | <10 | 18.8 | <10 | 11.7 | <10 |
 8x | <10 | <10 | 10.3 | 16.0 | <10 | <10 | 98.4 |
Structure | S. aureus a | E. coli b | K. pneumoniae c | P. aeruginosa d | A. baumannii e | C. albicans f | C. neoformansg |
| Amoxicillin | 16 | 8 | >32 | >32 | >32 | - h | - |
| Colistin | 32 | 8 | >32 | >32 | >32 | - | - |
| Amphotericin B | - | - | - | - | - | 1.56 | 1.56 |
| 8e | 32 | - | - | - | - | - | - |
| 8h | 32 | - | - | - | - | - | - |
| 8i | 32 | - | - | - | - | - | - |
| 8j | 32 | - | - | - | - | - | - |
| 8q | - | - | - | - | - | - | - |
| 8s | - | - | - | - | - | - | - |
| 8x | - | - | - | - | - | - | 32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baker, J.R.; Cossar, P.J.; Blaskovich, M.A.T.; Elliott, A.G.; Zuegg, J.; Cooper, M.A.; Lewis, P.J.; McCluskey, A. Amino Alcohols as Potential Antibiotic and Antifungal Leads. Molecules 2022, 27, 2050. https://doi.org/10.3390/molecules27072050
Baker JR, Cossar PJ, Blaskovich MAT, Elliott AG, Zuegg J, Cooper MA, Lewis PJ, McCluskey A. Amino Alcohols as Potential Antibiotic and Antifungal Leads. Molecules. 2022; 27(7):2050. https://doi.org/10.3390/molecules27072050
Chicago/Turabian StyleBaker, Jennifer R., Peter J. Cossar, Mark A. T. Blaskovich, Alysha G. Elliott, Johannes Zuegg, Matthew A. Cooper, Peter J. Lewis, and Adam McCluskey. 2022. "Amino Alcohols as Potential Antibiotic and Antifungal Leads" Molecules 27, no. 7: 2050. https://doi.org/10.3390/molecules27072050