
Synthesis and Evaluation of 11C-Labeled Triazolones as Probes for Imaging Fatty Acid Synthase Expression by Positron Emission Tomography


 , ,
, , {kind=link}
{kind=link}
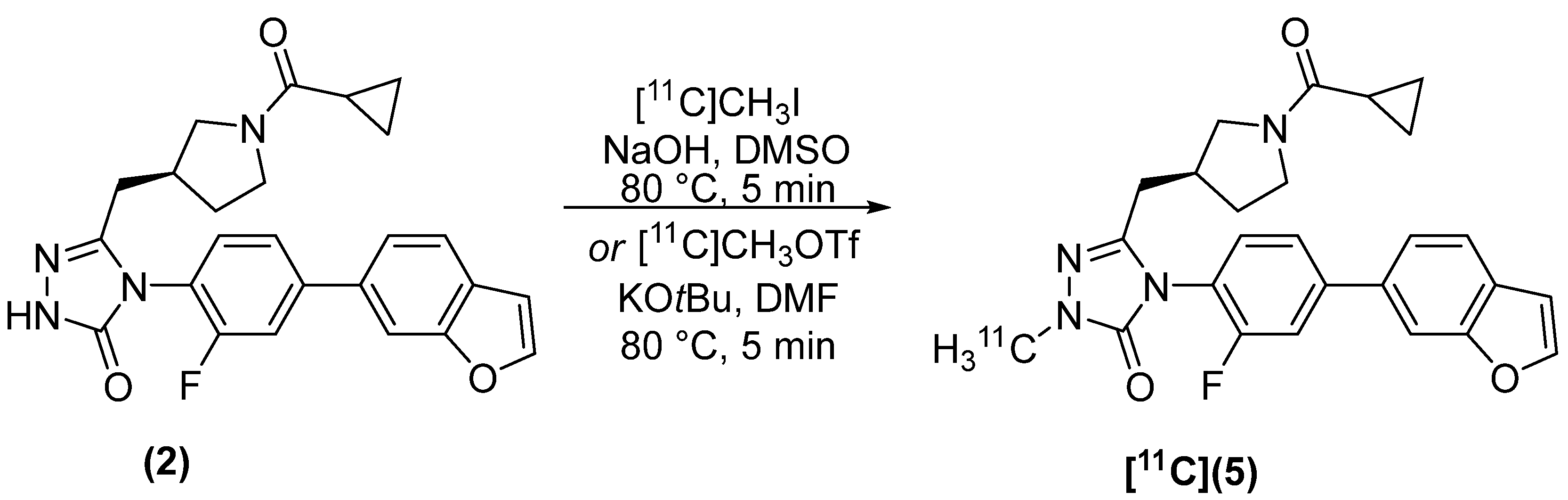{kind=link}
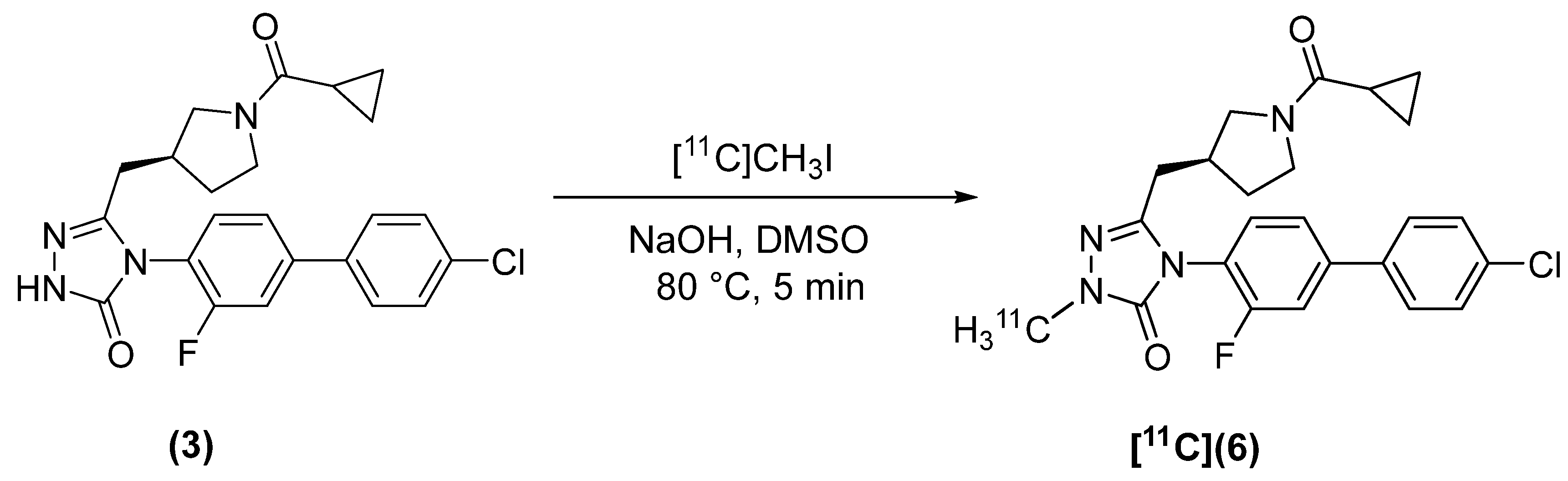{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
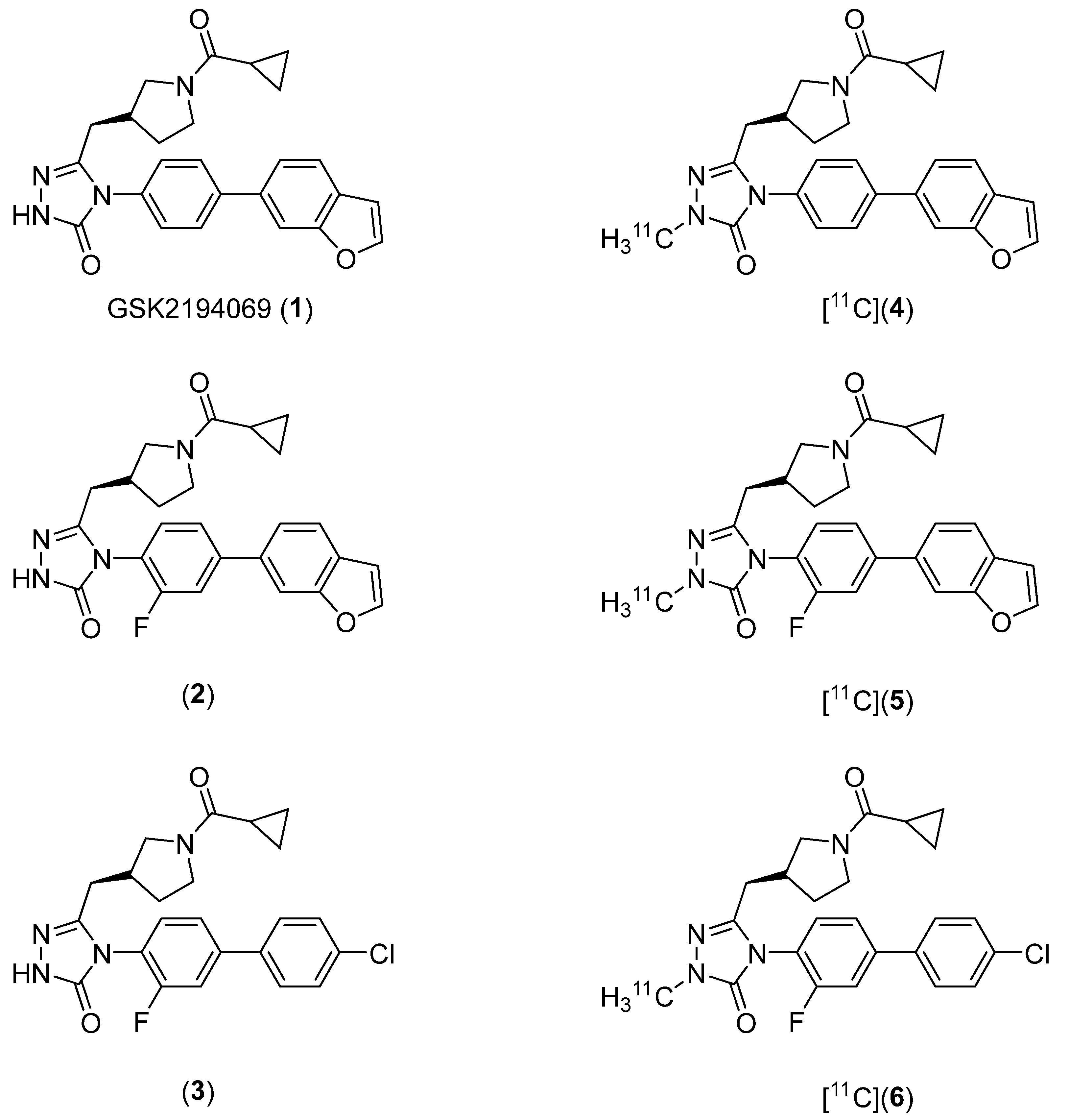
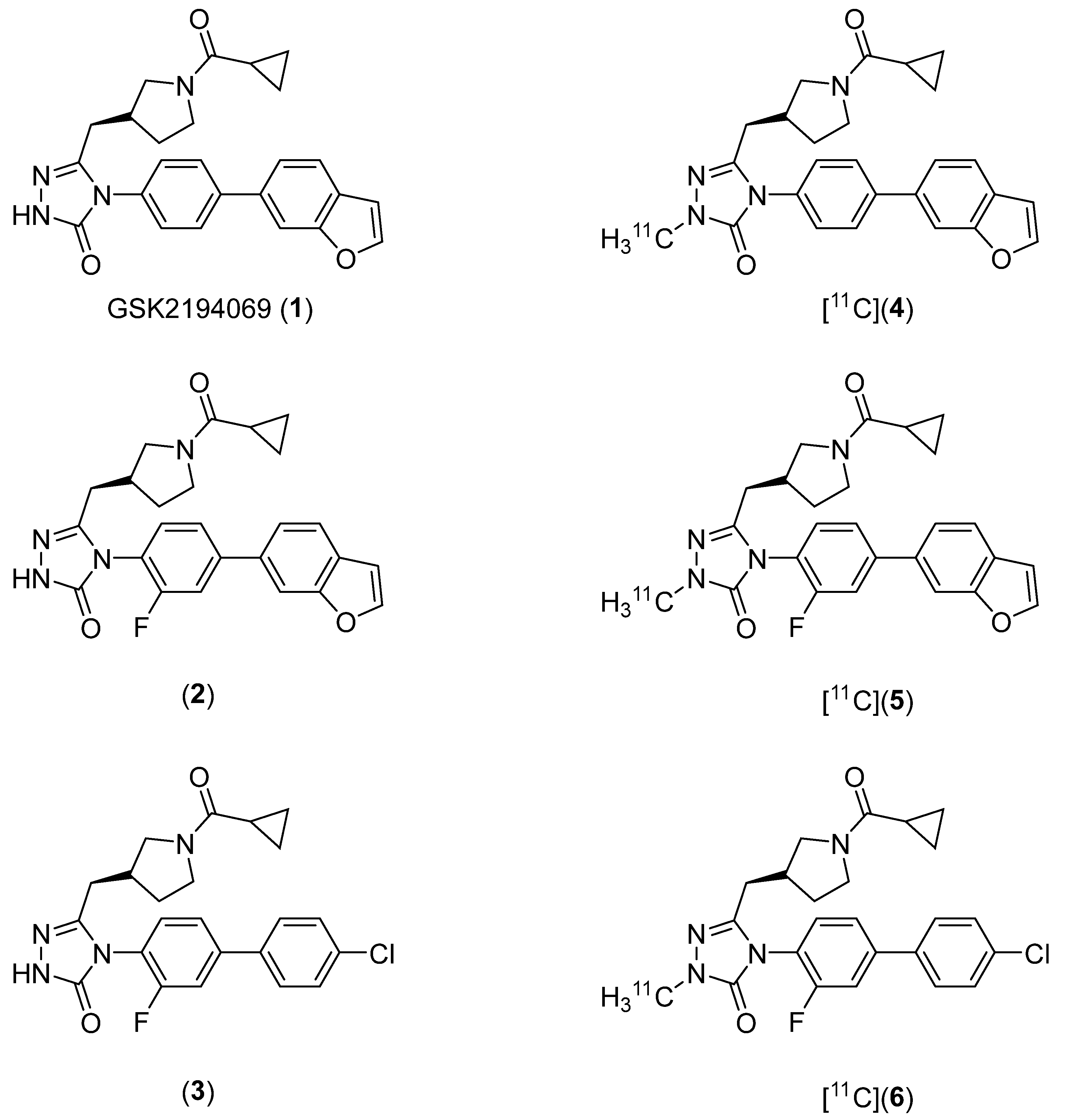
2.1. Lead Compound Identification
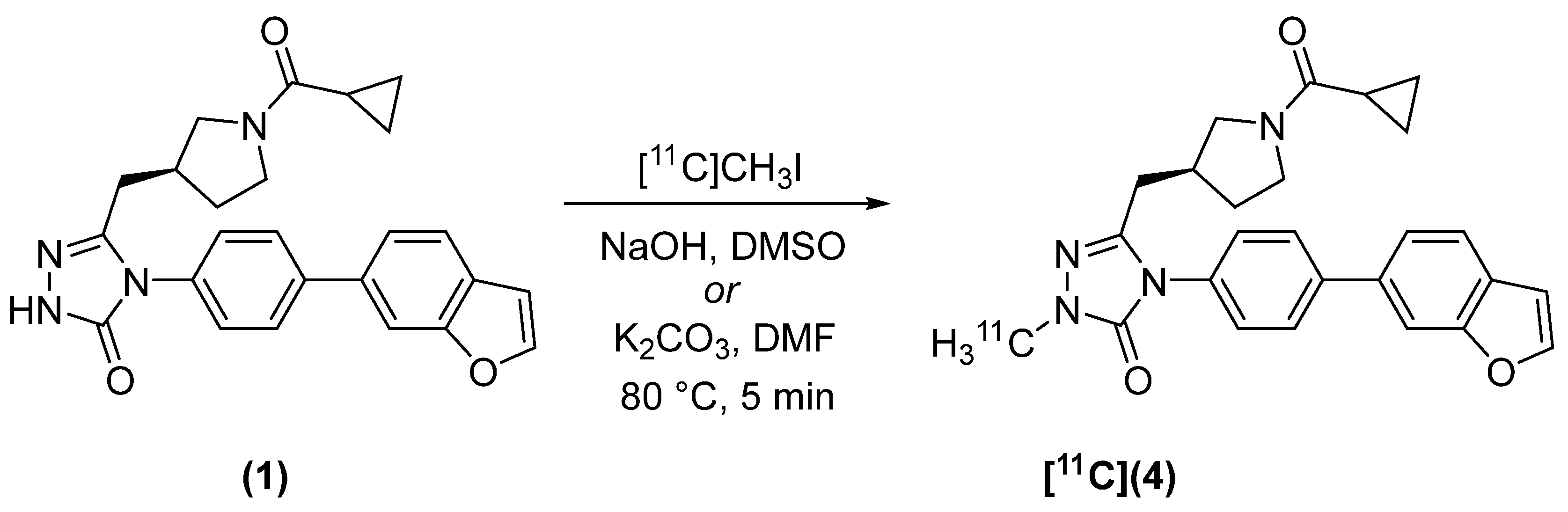
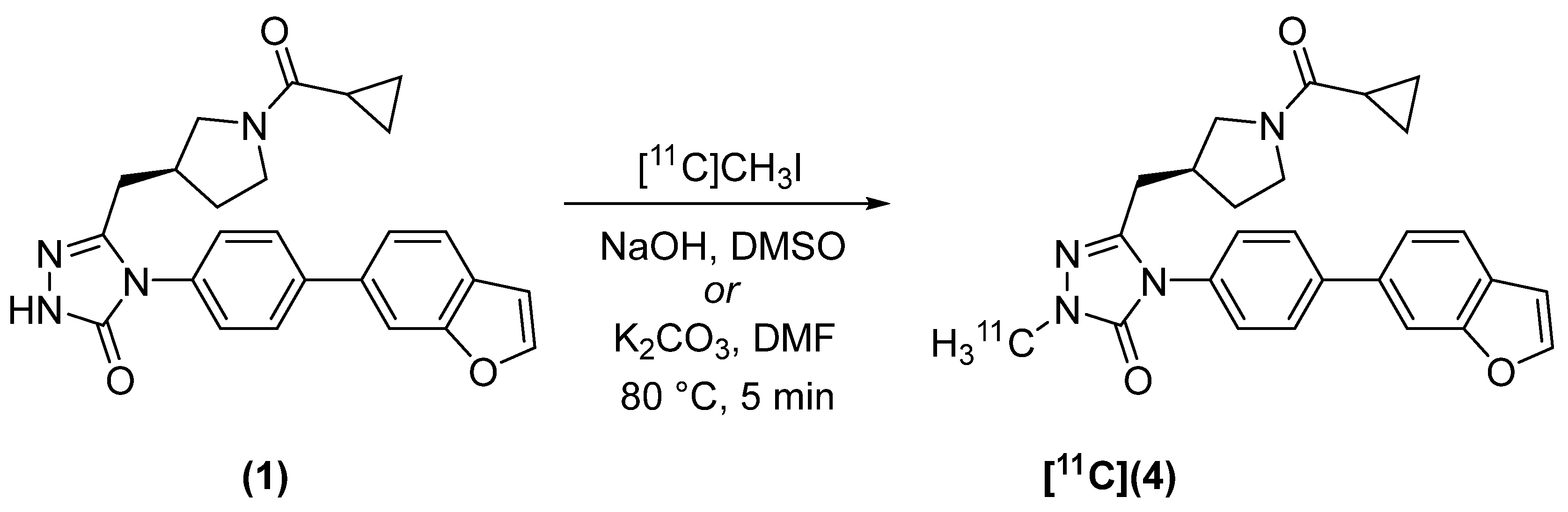
2.2. Radiosynthesis
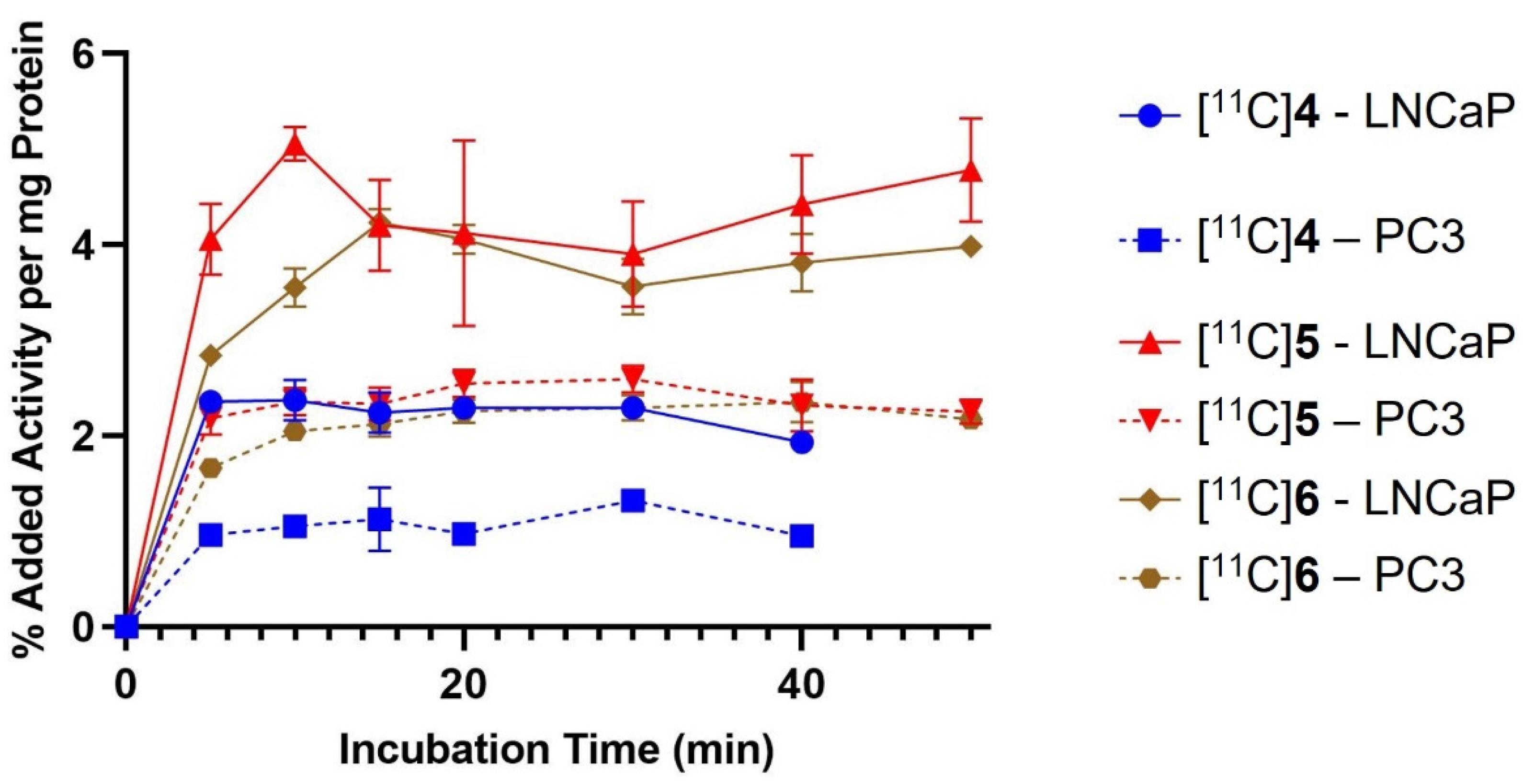
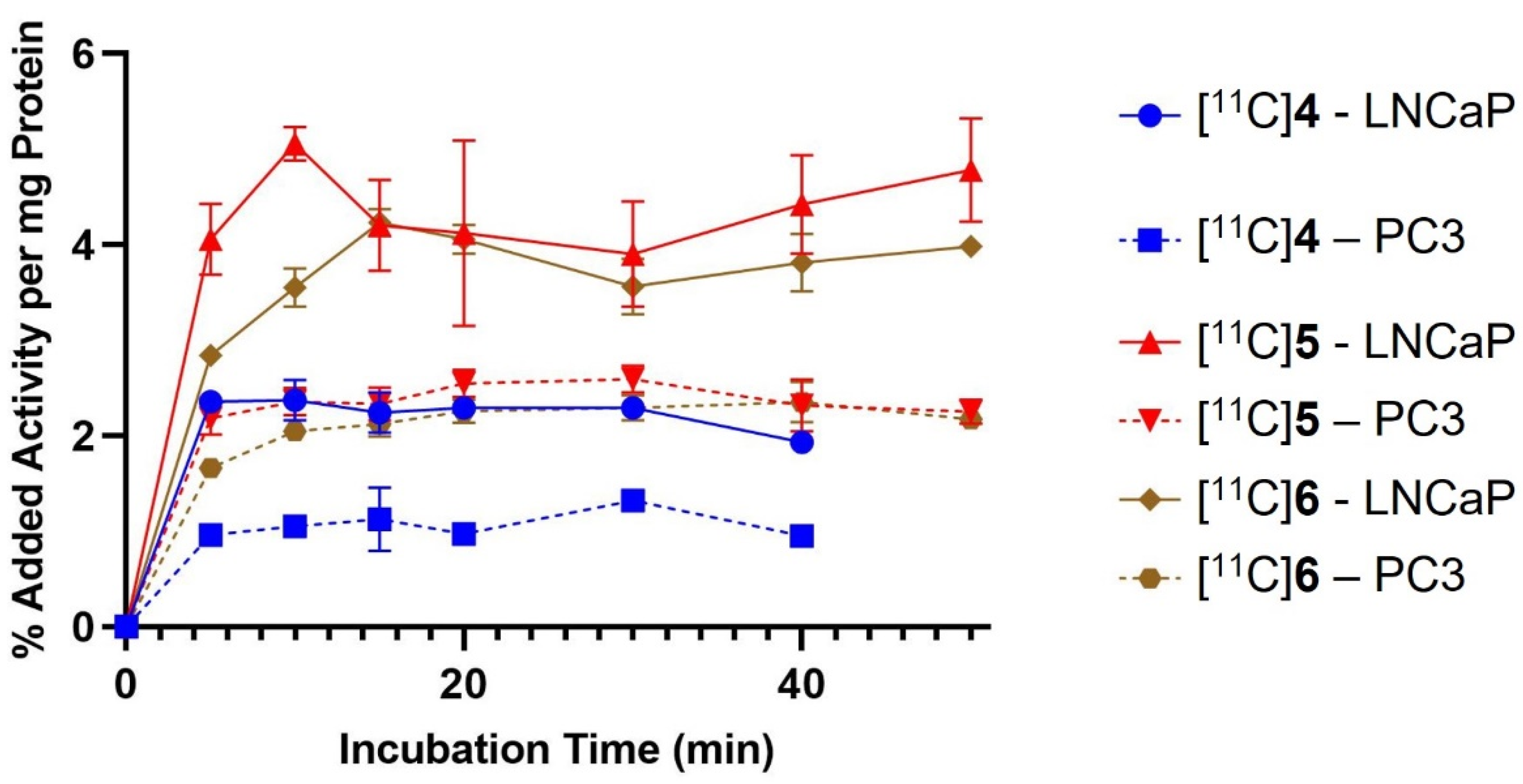
2.3. In Vitro Cell Binding
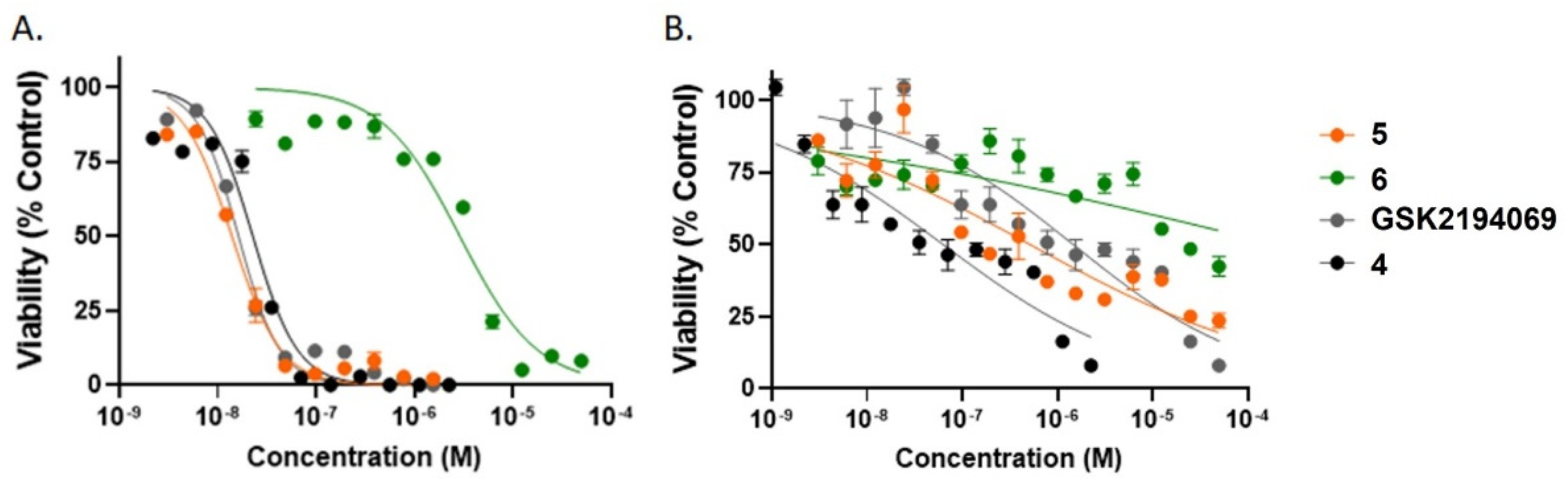
2.4. In Vitro Growth Inhibition Assays
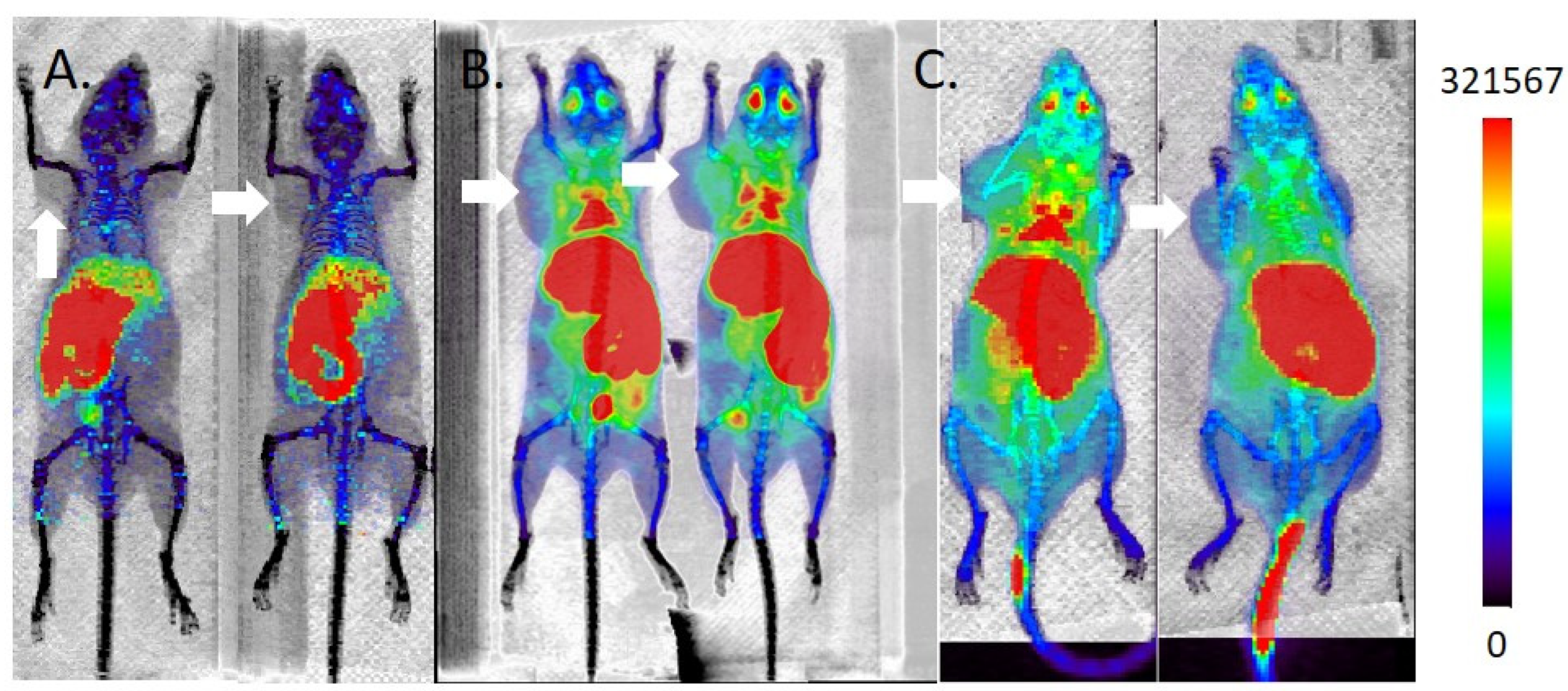
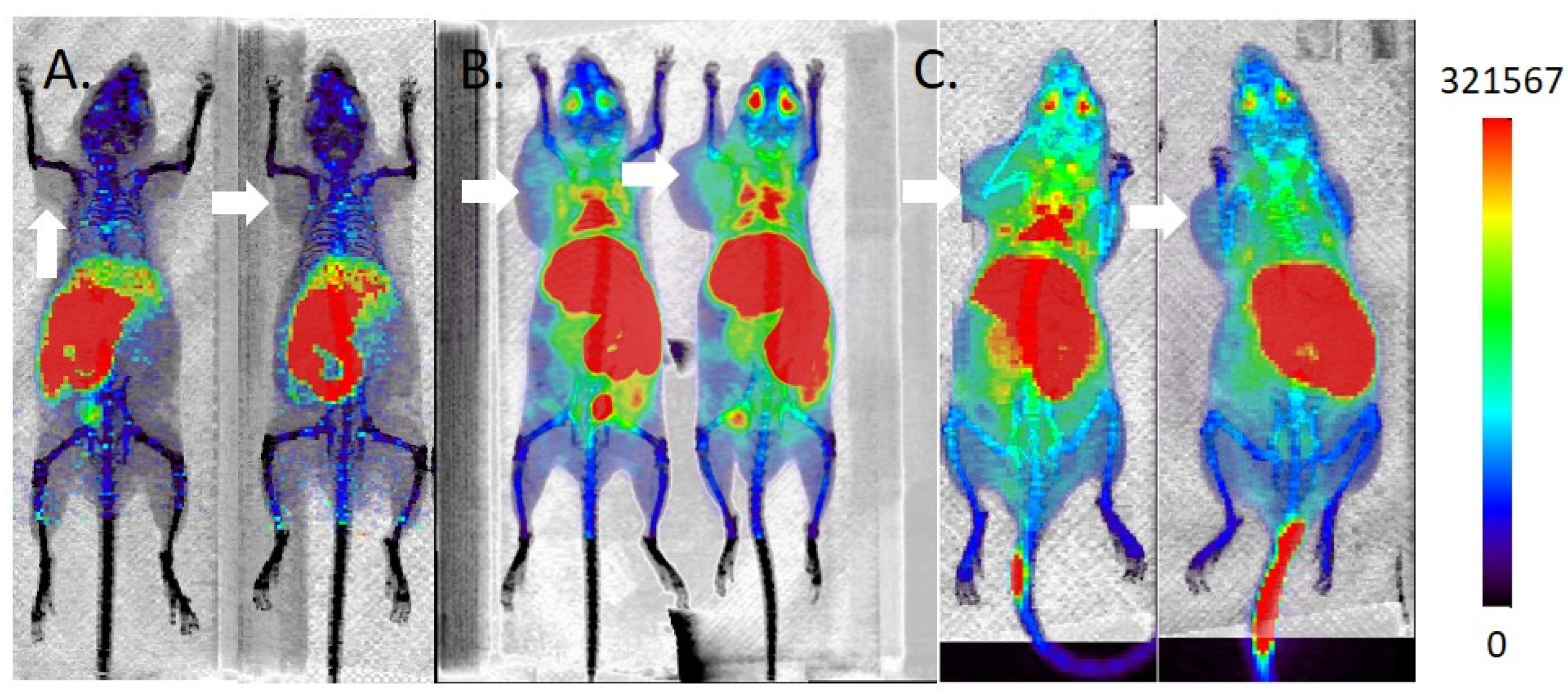
2.5. MicroPET/CT Imaging
3. Discussion
4. Materials and Methods
4.1. Synthesis of Precursors
4.2. Radiosynthesis
4.2.1. (S)-4-(4-(benzofuran-6-yl)phenyl)-5-((1-(cyclopropanecarbonyl)pyrrolidin-3-yl)methyl)-2-[methyl-11C]-2,4-dihydro-3H-1,2,4-triazol-3-one ([11C]4)
4.2.2. (S)-4-(4-(benzofuran-6-yl)-2-fluorophenyl)-5-((1-(cyclopropanecarbonyl)pyrrolidin-3-yl)methyl)-2-[methyl-11C]-2,4-dihydro-3H-1,2,4-triazol-3-one [11C]5
4.2.3. (S)-4-(4′-chloro-3-fluoro-[1,1′-biphenyl]-4-yl)-5-((1-(cyclopropanecarbonyl)pyrrolidin-3-yl)methyl)-2-[methyl-11C]-2,4-dihydro-3H-1,2,4-triazol-3-one ([11C]6)
4.3. Cell Lines
4.4. Western Blotting
4.5. Binding to Prostate Cancer Cell Lines
4.6. Growth Inhibition Assay
4.7. Animal Models
4.8. MicroPET/CT Imaging
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Yellen, P.; Foster, D.A. Inhibition of fatty acid synthase induces pro-survival Akt and ERK signaling in K-Ras-driven cancer cells. Cancer Lett. 2014, 353, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Veldhoven, P.P.V.; Timmermans, L.; Schrijver, E.D.; Brusselmans, K.; Vanderhoydonc, F.; Sande, T.V.d.; Heemers, H.; Heyns, W.; Verhoeven, G. Fatty Acid Synthase Drives the Synthesis of Phospholipids Partitioning Into Detergent-Resistant Membrane Microdomains. Biochem. Biophys. Res. Commun. 2003, 302, 898–903. [Google Scholar] [CrossRef]
- Benedettini, E.; Nguyen, P.; Loda, M. The pathogenesis of prostate cancer: From molecular to metabolic alterations. Diagn. Histopathol. 2008, 14, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Van de Sande, T.; De Schrijver, E.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. Role of the Phosphatidylinositol 3′-Kinase/PTEN/Akt Kinase Pathway in the Overexpression of Fatty Acid Synthase in LNCaP Prostate Cancer Cells. Cancer Res. 2002, 62, 642–646. [Google Scholar]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Rossi, S.; Graner, E.; Febbo, P.; Weinstein, L.; Bhattacharya, N.; Onody, T.; Bubley, G.; Balk, S.; Loda, M. Fatty Acid Synthase Expression Defines Distinct Molecular Signatures in Prostate Cancer. Mol. Cancer Res. 2003, 1, 707–715. [Google Scholar] [PubMed]
- Wang, Y.; Kuhajda, F.P.; Li, J.N.; Pizer, E.S.; Han, W.F.; Sokoll, L.J.; Chan, D.W. Fatty acid synthase (FAS) expression in human breast cancer culture supernatants and in breast cancer patients. Cancer Lett. 2001, 167, 99–104. [Google Scholar] [CrossRef]
- Alo, P.L.; Amini, M.; Piro, F.; Pizzuti, L.; Sebastiani, V.; Botti, C.; Murari, R.; Zotti, G.; Tondo, U.D. Significance of Fatty Acid Synthase in Pancreatic Carcinoma. Anticancer. Res. 2007, 27, 2523–2527. [Google Scholar] [PubMed]
- Rashid, A.; Pizer, E.S.; Moga, M.; Milgraum, L.Z.; Zahurak, M.; Pasternack, G.R.; Kuhajda, F.P.; Hamilton, S.R. Elevated Expression of Fatty Acid Synthase and Fatty Acid Synthetic Activity in Colorectal Neoplasia. Am. J. Pathol. 1997, 150, 201. [Google Scholar] [PubMed]
- Ito, T.; Sato, K.; Maekawa, H.; Sakurada, M.; Orita, H.; Shimada, K.; Daida, H.; Wada, R.; Abe, M.; Hino, O.; et al. Elevated levels of serum fatty acid synthase in patients with gastric carcinoma. Oncol. Lett. 2014, 7, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Myers, R.B.; Oelschlager, D.K.; Weiss, H.L.; Frost, A.R.; Grizzle, W.E. Fatty Acid Synthase: An Early Molecular Marker of Progression of Prostatic Adenocarcinoma to Androgen Independence. J. Urol. 2001, 165, 1027–1032. [Google Scholar] [CrossRef]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef] [Green Version]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef] [Green Version]
- Pizer, E.S.; Wood, F.D.; Heine, H.S.; Romantsev, F.E.; Pasternack, G.R.; Kuhajda, F.P. Inhibition of Fatty Acid Synthesis Delays Disease Progression in a Xenograft Model of Ovarian Cancer. Cancer Res. 1996, 56, 1189–1193. [Google Scholar]
- Pizer, E.S.; Jackisch, C.; Wood, F.D.; Pasternack, G.R.; Davidson, N.E.; Kuhajda, F.P. Inhibition of Fatty Acid Synthase Induces Programmed Cell Death in Human Breast Cancer Cells. Cancer Res. 1996, 56, 2745–2747. [Google Scholar]
- Carvalho, M.A.; Zecchin, K.G.; Seguin, F.; Bastos, D.C.; Agostini, M.; Rangel, A.L.C.A.; Veiga, S.S.; Raposo, H.F.; Oliveira, H.C.F.; Loda, M.; et al. Fatty acid synthase inhibition with Orlistat promots apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int. J. Cancer 2008, 123, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat Is a Novel Inhibitor of Fatty Acid Synthase with Antitumor Activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizer, E.S.; Thupari, J.; Han, W.F.; Pinn, M.L.; Chrest, F.J.; Frehywot, G.L.; Townsend, C.A.; Kuhajda, F.P. Malonyl-Coenzyme-A Is a Potential Mediator of Cytotoxicity Induced by Fatty-Acid Synthase Inhibition in Human Breast Cancer Cells and Xenografts. Cancer Res. 2000, 60, 213–218. [Google Scholar] [PubMed]
- Sadowski, M.C.; Pouwer, R.H.; Gunter, J.H.; Lubik, A.A.; Quinn, R.J.; Nelson, C.C. The fatty acid synthase inhibitor triclsoan: Repurposing an anti-microbial agent for targeting prostate cancer. Oncotarget 2014, 5, 9362–9381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusselmans, K.; Schrijver, E.D.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. Epigallocatechin-3-gallate Is a Potent Natural Inhibitor of Fatty Acid Synthase in Intact Cells and Selectively Induces Apoptosis in Prostate Cancer Cells. Int. J. Cancer 2003, 106, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Relat, J.; Blancafort, A.; Oliveras, G.; Cufi, S.; Haro, D.; Marrero, P.F.; Puig, T. Different fatty acid metabolism effects of (-)-Epigallocatechin-3-Gallate and C75 in Adenocarcinoma lung cancer. BMC Cancer 2012, 12, 280. [Google Scholar] [CrossRef] [Green Version]
- Loftus, T.M.; Jaworsky, D.E.; Frehywot, G.L.; Townsend, C.A.; Ronnet, G.V.; Lane, M.D.; Kuhajda, F.P. Reduced Food Intake and Body Weight in Mice Treated with Fatty Acid Synthase Inhibitors. Science 2000, 288, 2379–2381. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Rychahou, P.G.; Le, A.-T.; Scott, T.L.; Flight, R.M.; Kim, J.T.; Harris, J.; Liu, J.; Wang, C.; Morris, A.J.; et al. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget 2018, 9, 24787–24800. [Google Scholar] [CrossRef] [Green Version]
- Puig, T.; Aguilar, H.; Cuf, S.; Oliveras, G.; Turrado, C.; Ortega-Gutierrez, S.; Benhamú, B.; López-Rodríguez, M.L.; Urruticoechea, A.; Colomer, R. A novel inhibitor of fatty acid synthase show activity against HER2+ breast cancer xenografts and is active in anti-HER2 drug-resistant cell lines. Breast Cancer Res. 2011, 13, R131. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.; Lewis, D.; Boren, J.; Montoya, A.-R.; Bielik, R.; Soloviev, D.; Brindle, K.; Neal, D. Therapeutic Fatty Acid Synthase Inhibition in Prostate Cancer and the Use of 11C-Acetate to Monitor Therapeutic Effects. J. Urol. 2013, 189, e208–e209. [Google Scholar] [CrossRef]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de nov Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, A.; Infante, J.; Patel, M.; Arkenau, H.-T.; Voskoboynik, M.; Borazanci, E.; Falchook, G.; Molife, L.R.; Pant, S.; Dean, E.; et al. First-in-human study of the first-in-class fatty acid synthase (FASN) inhibitor, TVB-2640 as monotherapy or in combination—Final results of dose escalation. Mol. Cancer Ther. 2015, 14, A54. [Google Scholar]
- Falchook, G.; Patel, M.; Infante, J.; Arkenau, H.-T.; Dean, E.; Brenner, A.; Borazanci, E.; Lopez, J.; Moore, K.; Schmid, P.; et al. First in human study of the first-in-class fatty acid synthase (FASN) inhibitor TVB-2640. Cancer Res. 2017, 77, CT153. [Google Scholar]
- Adams, N.D.; Aquino, C.J.; Chaudhari, A.M.; Ghergurovich, J.M.; Kiesow, T.J.; Parrish, C.A.; Reif, A.J.; Wiggall, K. Triazolones as Fatty Acid Synthase Inhibitors. U.S. Patent No 8,802,864 B2, 12 August 2012. [Google Scholar]
- Hardwicke, M.A.; Rendina, A.R.; Williams, S.P.; Moore, M.L.; Wang, L.; Krueger, J.A.; Plant, R.N.; Totoritis, R.D.; Zhang, G.; Briand, J.; et al. A human fatty acid synthase inhibitor binds β-ketoacyl reductase in the keto-substrate site. Nat. Chem. Biol. 2014, 10, 774–782. [Google Scholar] [CrossRef]
- Lockwood, S.F.; O’Malley, S.; Mosher, G.L. Improved aqueous solubility of crystalline astaxanthin (3,3′-dihydroxy-β,β-carotene-4,4′-dione) by Captisol (sulfobutyl ether β-cyclodextrin). J. Pharm. Sci. 2003, 92, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Oyama, N.; Akino, H.; Suzuki, Y.; Kanamaru, H.; Sadato, N.; Yonekura, Y.; Okada, K. The Increased Accumulation of [18F]Fluorodeoxyglucose in Untreated Prostate Cancer. Jpn. J. Clin. Oncol. 1999, 29, 623–629. [Google Scholar] [CrossRef]
- Jadvar, H. FDG PET in Prostate Cancer. PET Clin. 2009, 4, 155–161. [Google Scholar] [CrossRef]
- Spratt, D.E.; Gavane, S.; Tarlinton, L.; Fareedy, S.B.; Doran, M.G.; Zelefsky, M.J.; Osborne, J.R. Utility of FDG-PET in Clinical Neuroendocrine Prostate Cancer. Prostate 2014, 74, 1153–1159. [Google Scholar] [CrossRef] [Green Version]
- Fricke, E.; Machtens, S.; Hofmann, M.; Hoff, J.v.d.; Bergh, S.; Brunkhorst, T.; Meyer, G.J.; Karstens, J.H.; Knapp, W.H.; Boerner, A.R. Positron Emission Tomography With 11C-acetate and 18F-FDG in Prostate Caner Patients. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 607–611. [Google Scholar] [CrossRef]
- Meirelles, G.S.P.; Schöder, H.; Ravizzini, G.C.; Gönen, M.; Fox, J.J.; Humm, J.; Morris, M.J.; Scher, H.I.; Larson, S.M. Prognostic Value of Baseline [18F] Fluorodeoxyglucose Positron Emission Tomography and 99mTc-MDP Bone Scan in Progressing Metastatic Prostate Cancer. Clin. Cancer Res. 2010, 16, 6093–6099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadvar, H. Imaging Evaluation of Prostate Cancer with 18F-fluorodeoxyglucose PET/CT: Utility and Limitations. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Ackerstaff, E.; Glunde, K.; Bhujwalla, Z.M. Choline Phospholipid Metabolism: A Target in Cancer Cells? J. Cell Biochem. 2003, 90, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzenböck, S.; Souvatzoglou, M.; Krause, B.J. Choline PET and PET/CT in Primary Diagnosis and Staging of Prostate Cancer. Theranostics 2012, 2, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Tousignant, K.D.; Rockstroh, A.; Fard, A.T.; Lehman, M.L.; Wang, C.; McPherson, S.J.; Philp, L.K.; Bartonicek, N.; Dinger, M.E.; Nelson, C.C.; et al. Lipid Uptake Is an Androgen-Enhanced Lipid Supply Pathway Associated with Prostate Cancer Disease Progression and Bone Metastasis. Mol. Cancer Res. 2019, 17, 1166–1179. [Google Scholar] [CrossRef] [Green Version]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef]
- Takahiro, T.; Shinichi, K.; Toshimitsu, S. Expression of Fatty Acid Synthase as a Prognostic Indicator in Soft Tissue Sarcomas. Clin. Cancer Res. 2003, 9, 2204–2212. [Google Scholar]
- Yoshii, Y.; Furukawa, T.; Oyama, N.; Hasegawa, Y.; Kiyono, Y.; Nishii, R.; Waki, A.; Tsuji, A.B.; Sogawa, C.; Wakizaka, H.; et al. Fatty Acid Synthase is a Key Target in Multiple Essential Tumor Functions of Prostate Cancer: Uptake of Radiolabeled Acetate as a Predictor of the Targeted Therapy Outcomes. PLoS ONE 2013, 8, e64570. [Google Scholar]
- Vāvere, A.L.; Kridel, S.J.; Wheeler, F.B.; Lewis, J.S. 1-11C-acetate as a PET Radiopharmaceutical for Imaging Fatty Acid Synthase Expression in Prostate Cancer. J. Nucl. Med. 2008, 49, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Che, L.; Wang, C.; Blecha, J.E.; Li, X.; VanBrocklin, H.F.; Calvisi, D.F.; Puchowicz, M.; Chen, X.; Seo, Y. [11C]acetate PET Imaging is not Always Associated with Increased Lipogenesis in Hepatocellular Carcinoma in Mice. Mol. Imaging Biol. 2016, 18, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.Y.; Lee, M.; Whang, S.H.; Kim, J.-W.; Cho, A.; Yun, M. Regulation of Acetate Utilization by Monocarboxylate Transporter 1 (MCT1) in Hepatocellular Carcinoma (HCC). Oncol. Res. 2018, 26, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Czernin, J.; Benz, M.R.; Allen-Auerbach, M.S. PET Imaging of Prostate Cancer Using 11C-Acetate. PET Clin. 2009, 4, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bang, S.-H.; Kim, J.W.; Park, J.Y.; Kim, K.S.; Lee, J.D. The Importance of Acetyl Coenzyme A Synthetase for 11C-acetate Uptake and Cell Survival in Hepatocellular Carcinoma. J. Nucl. Med. 2009, 50, 1222–1228. [Google Scholar]
- Schug, Z.T.; Voorde, J.V.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lin, S.-H.; Ren, F.; Li, J.-T.; Chen, J.-J.; Yao, C.-B.; Yang, H.-B.; Jiang, S.-X.; Yan, G.-Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef] [Green Version]
- Ponde, D.E.; Dence, C.S.; Oyama, N.; Kim, J.; Tai, Y.-C.; Laforest, R.; Siegel, B.A.; Welch, M.J. 18F-Fluoroacetate: A Potential Acetate Analog for Prostate Tumor Imaging—In Vivo Evaluation of 18F-Fluoroacetate Versus 11C-Acetate. J. Nucl. Med. 2007, 48, 420–428. [Google Scholar] [PubMed]
- Pillarsetty, N.; Punzalan, B.; Larson, S.M. 2-18F-Fluoropropionic Acid as a PET Imaging Agent for Prostate Cancer. J. Nucl. Med. 2009, 50, 1709–1714. [Google Scholar] [CrossRef] [Green Version]
- Lindhe, Ö.; Sun, A.; Ulin, J.; Rahman, O.; Långström, B.; Sörensen, J. [18F]Fluoroacetate is not a functional analogue of [11C]acetate in normal physiology. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1453–1459. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Ma, H.; Nie, D.; Wen, F.; Zhao, J.; Sun, A.; Yuan, G.; Su, S.; Xiang, X.; et al. Validation of R-2-[18F]Fluoropropionic Acid as a Potential Tracer for PET Imaging of Liver Cancer. Mol. Imaging Biol. 2019, 21, 1127–1137. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, Z.; Nie, D.; Ma, H.; Yuan, G.; Su, S.; Liu, S.; Liu, S.; Tang, G. PET Imaging of Hepatocellular Carcinomas: 18F-Fluoropropionic Acid as a Complementary Radiotracer for 18F-Fluorodeoxyglucose. Mol. Imaging 2019, 18, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonović, U.P.; Knez, D.; Hrast, M.; Zidar, N.; Proj, M.; Gobec, S.; Kos, J. Structure-activity relationships of triazole-benzodioxine inhibitors of cathepsin X. Eur. J. Med. Chem. 2020, 193, 112218. [Google Scholar] [CrossRef] [PubMed]
- Singha, P.K.; Mäklin, K.; Vihavainen, T.; Laitinen, T.; Nevalainen, T.J.; Patil, M.R.; Tonduru, A.K.; Poso, A.; Laitinen, J.T.; Savinainen, J.R. Evaluation of FASN inhibitors by a versatile tookit reveals differences in pharmacology between human and rodent FASN preparations and in antiproliferative efficacy in vitro vs. in situ in human cancer cells. Eur. J. Pharm. Sci. 2020, 149, 105321. [Google Scholar] [CrossRef] [PubMed]
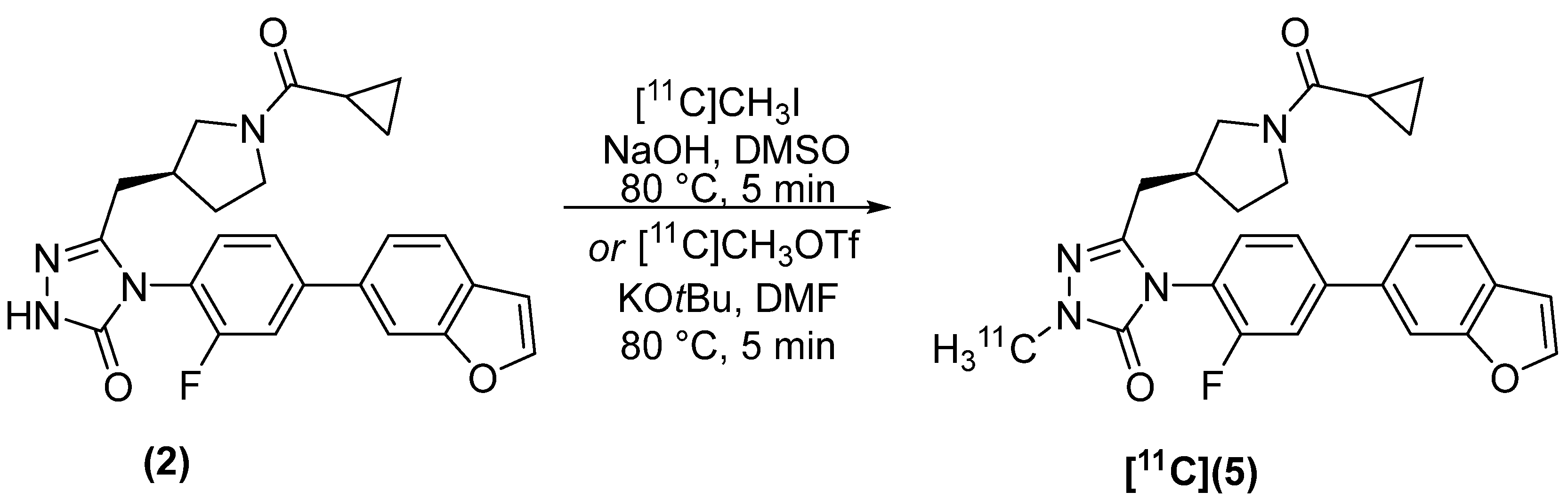


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelly, J.M.; Jeitner, T.M.; Waterhouse, N.N.; Qu, W.; Linstad, E.J.; Samani, B.; Williams, C., Jr.; Nikolopoulou, A.; Amor-Coarasa, A.; DiMagno, S.G.; et al. Synthesis and Evaluation of 11C-Labeled Triazolones as Probes for Imaging Fatty Acid Synthase Expression by Positron Emission Tomography. Molecules 2022, 27, 1552. https://doi.org/10.3390/molecules27051552
Kelly JM, Jeitner TM, Waterhouse NN, Qu W, Linstad EJ, Samani B, Williams C Jr., Nikolopoulou A, Amor-Coarasa A, DiMagno SG, et al. Synthesis and Evaluation of 11C-Labeled Triazolones as Probes for Imaging Fatty Acid Synthase Expression by Positron Emission Tomography. Molecules. 2022; 27(5):1552. https://doi.org/10.3390/molecules27051552
Chicago/Turabian StyleKelly, James M., Thomas M. Jeitner, Nicole N. Waterhouse, Wenchao Qu, Ethan J. Linstad, Banafshe Samani, Clarence Williams, Jr., Anastasia Nikolopoulou, Alejandro Amor-Coarasa, Stephen G. DiMagno, and et al. 2022. "Synthesis and Evaluation of 11C-Labeled Triazolones as Probes for Imaging Fatty Acid Synthase Expression by Positron Emission Tomography" Molecules 27, no. 5: 1552. https://doi.org/10.3390/molecules27051552