
Coumarin-Resveratrol-Inspired Hybrids as Monoamine Oxidase B Inhibitors: 3-Phenylcoumarin versus trans-6-Styrylcoumarin

 , , , , , ,
, , , , , , 
Abstract
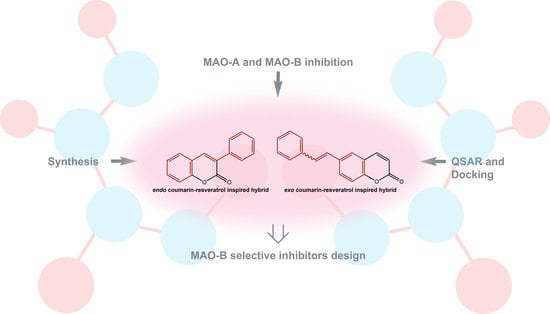
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Pharmacology
2.3. Computer-Aided Studies Using an Internal Database of Coumarins and Chromone Analogs
2.3.1. D-QSAR Studies
Steric Map of MAO-B Inhibitors
Electrostatic Map of MAO-B Inhibitors
Hydrophobic Map of MAO-B Inhibitors
Hydrogen-Bond Donor Map of MAO-B Inhibitors
2.3.2. Molecular Docking
2.4. Computational Studies for the New Coumarin-Resveratrol-Inspired Hybrids
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Schneider, A.; Sari, A.T.; Alhaddad, H.; Sari, Y. Overview of Therapeutic Drugs and Methods for the Treatment of Parkinson′s Disease. CNS Neurol. Disord. Drug Targets 2020, 19, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Santin, Y.; Resta, J.; Parini, A.; Mialet-Perez, J. Monoamine oxidases in age-associated diseases: New perspectives for old enzymes. Ageing Res. Rev. 2021, 66, 101256. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Möhr, J.D. Pharmacokinetics of monoamine oxidase B inhibitors in Parkinson′s disease: Current status. Expert Opin. Drug Metab. Toxicol. 2019, 15, 429–435. [Google Scholar] [CrossRef]
- Higuchi, Y.; Soga, T.; Parhar, I.S. Regulatory Pathways of Monoamine Oxidase A during Social Stress. Front. Neurosci. 2017, 11, 604. [Google Scholar] [CrossRef] [Green Version]
- Ishiki, H.M.; Filho, J.M.B.; da Silva, M.S.; Scotti, M.T.; Scotti, L. Computer-aided Drug Design Applied to Parkinson Targets. Curr. Neuropharmacol. 2018, 16, 865–880. [Google Scholar] [CrossRef]
- Guardado, Y.E.; Santana, L.; Uriarte, E.; Borges, F.; Matos, M.J. Computer-aided design of coumarins for neurodegenerative diseases: Trends of the last decade. Curr. Top. Med. Chem. 2021, 21, 2245–2257. [Google Scholar]
- Mellado, M.; González, C.; Mella, J.; Aguilar, L.F.; Viña, D.; Uriarte, E.; Cuellar, M.; Matos, M.J. Combined 3D-QSAR and docking analysis for the design and synthesis of chalcones as potent and selective monoamine oxidase B inhibitors. Bioorg. Chem. 2021, 108, 104689. [Google Scholar] [CrossRef]
- Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriarte, E. Simple coumarins and analogues in medicinal chemistry: Occurrence, synthesis and biological activity. Curr. Med. Chem. 2005, 12, 887–916. [Google Scholar] [CrossRef]
- Carneiro, A.; Matos, M.J.; Uriarte, E.; Santana, L. Trending Topics on Coumarin and Its Derivatives in 2020. Molecules 2021, 26, 501. [Google Scholar] [CrossRef]
- Matos, M.J.; Santana, L.; Uriarte, E.; Abreu, O.A.; Molina, E.; Yordi, E.M.A.E.G. Coumarins—An Important Class of Phytochemicals. In Phytochemicals—Isolation, Characterisation and Role in Human Health; Rao, A.V., Rao, L.G., Eds.; IntechOpen: London, UK, 2015; pp. 113–140. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Liu, J. Resveratrol: A review of plant sources, synthesis, stability, modification and food application. J. Sci. Food Agric. 2020, 100, 1392–1404. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.-F.; Wang, X.-B.; Xie, S.-S.; Lia, S.-Y.; Kong, L.-Y. Multitarget-directed resveratrol derivatives: Anti-cholinesterases, anti-β-amyloid aggregation and monoamine oxidase inhibition properties against Alzheimer′s disease. Med. Chem. Commun. 2014, 5, 609–616. [Google Scholar] [CrossRef]
- Matos, M.J.; Uriarte, E.; Santana, L. 3-Phenylcoumarins as a privileged scaffold in Medicinal Chemistry: The landmarks of the past decade. Molecules 2021, 26, 6755. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Terán, C.; Pérez-Castillo, Y.; Uriarte, E.; Santana, L.; Viña, D. Synthesis and Study of a Series of 3-Arylcoumarins as Potent and Selective Monoamine Oxidase B Inhibitors. J. Med. Chem. 2011, 54, 7127–7137. [Google Scholar] [CrossRef]
- Matos, M.J.; Santana, L.; Uriarte, E. 3-Phenylcoumarin. Acta Cryst. 2012, E68, o2645. [Google Scholar] [CrossRef] [Green Version]
- Cushman, M.; Nagarathnam, D.; Gopal, D.; Chakraborti, A.K.; Lin, C.M.; Hamel, E. Synthesis and evaluation of stilbene and dihydrostilbene derivatives as potential anticancer agents that inhibit tubulin polymerization. J. Med. Chem. 1991, 34, 2579–2588. [Google Scholar] [CrossRef]
- Mellado, M.; Mella, J.; González, C.; Viña, D.; Uriarte, E.; Matos, M.J. 3-Arylcoumarins as highly potent and selective monoamine oxidase B inhibitors: Which chemical features matter? Bioorg. Chem. 2020, 101, 103964. [Google Scholar] [CrossRef]
- Matos, M.J.; Viña, D.; Quezada, E.; Picciau, C.; Delogu, G.; Orallo, F.; Santana, L.; Uriarte, E. A new series of 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3268–3270. [Google Scholar] [CrossRef]
- Matos, M.J.; Viña, D.; Janeiro, P.; Borges, F.; Santana, L.; Uriarte, E. New halogenated 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 5157–5160. [Google Scholar] [CrossRef]
- Serra, S.; Ferino, G.; Matos, M.J.; Vázquez-Rodríguez, S.; Delogu, G.; Viña, D.; Cadoni, E.; Santana, L.; Uriarte, E. Hydroxycoumarins as selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 258–261. [Google Scholar] [CrossRef]
- Viña, D.; Matos, M.J.; Ferino, G.; Cadoni, E.; Laguna, R.; Borges, F.; Uriarte, E.; Santana, L. 8-Substituted 3-Arylcoumarins as Potent and Selective MAO-B Inhibitors: Synthesis, Pharmacological Evaluation, and Docking Studies. ChemMedChem 2012, 7, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Viña, D.; Matos, M.J.; Yáñez, M.; Santana, L.; Uriarte, E. 3-Substituted coumarins as dual inhibitors of AChE and MAO for the treatment of Alzheimer’s disease. MedChemComm 2012, 3, 213–218. [Google Scholar] [CrossRef]
- Ferino, G.; Cadoni, E.; Matos, M.J.; Quezada, E.; Uriarte, E.; Santana, L.; Vilar, S.; Tatonetti, N.P.; Yáñez, M.; Viña, D.; et al. MAO inhibitory activity of 2-arylbenzofurans versus 3-arylcoumarins: Synthesis, in vitro study, and docking calculations. ChemMedChem 2013, 8, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Vilar, S.; Gonzalez-Franco, R.M.; Uriarte, E.; Santana, L.; Friedman, C.; Tatonetti, N.P.; Viña, D.; Fontenla, J.A. Novel (coumarin-3-yl)carbamates as selective MAO-B inhibitors: Synthesis, in vitro and in vivo assays, theoretical evaluation of ADME properties and docking study. Eur. J. Med. Chem. 2013, 63, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Vilar, S.; García-Morales, V.; Tatonetti, N.P.; Uriarte, E.; Santana, L.; Viña, D. Insight into the Functional and Structural Properties of 3-Arylcoumarin as an Interesting Scaffold in Monoamine Oxidase B Inhibition. CemMedChem 2014, 9, 1488–1500. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Janeiro, P.; González-Franco, R.M.; Vilar, S.; Tatonetti, N.P.; Santana, L.; Uriarte, E.; Borges, F.; Fontenla, J.A.; Viña, D. Synthesis, pharmacological study and docking calculations of new benzo[f]coumarin derivatives as dual inhibitors of enzymatic systems involved in neurodegenerative diseases. Fut. Med. Chem. 2014, 6, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Rodríguez-Enríquez, F.; Vilar, S.; Santana, L.; Uriarte, E.; Hripcsak, G.; Estrada, M.; Rodríguez-Franco, M.I.; Viña, D. Potent and selective MAO-B inhibitory activity: Amino- versus nitro-3-arylcoumarin derivatives. Bioorg. Med. Chem. Lett. 2015, 25, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.J.; Rodríguez-Enríquez, F.; Borges, F.; Santana, L.; Uriarte, E.; Estrada, M.; Rodríguez-Franco, M.I.; Laguna, R.; Viña, D. 3-Amidocoumarins as Potential Multifunctional Agents against Neurodegenerative Diseases. ChemMedChem 2015, 10, 2071–2079. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, A.; Matos, M.J.; Reis, J.; Duarte, Y.; Gutiérrez, M.; Santana, L.; Uriarte, E.; Borges, F. Exploring coumarin potentialities: Development of new enzymatic inhibitors based on the 6-methyl-3-carboxamidocoumarin scaffold. RSC Adv. 2016, 6, 49764–49768. [Google Scholar] [CrossRef]
- Fonseca, A.; Reis, J.; Silva, T.; Matos, M.J.; Bagetta, D.; Ortuso, F.; Alcaro, S.; Uriarte, E.; Borges, F. Coumarin versus Chromone Monoamine Oxidase B Inhibitors: Quo Vadis? J. Med. Chem. 2017, 60, 7206–7212. [Google Scholar] [CrossRef]
- Rodríguez-Enríquez, F.; Viña, D.; Uriarte, E.; Fontenla, J.A.; Matos, M.J. Discovery and optimization of 3-thiophenylcoumarins as novel agents against Parkinson’s disease: Synthesis, in vitro and in vivo studies. Bioorg. Chem. 2020, 101, 103986. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Herrera Ibatá, D.M.; Uriarte, E.; Viña, D. Coumarin-Rasagiline Hybrids as Potent and Selective hMAO-B Inhibitors, Antioxidants, and Neuroprotective Agents. ChemMedChem 2020, 15, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enríquez, F.; Viña, D.; Uriarte, E.; Laguna, R.; Matos, M.J. 7-Amidocoumarins as Multitarget Agents against Neurodegenerative Diseases: Substitution Pattern Modulation. ChemMedChem 2021, 16, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Chakraborty, P.; Mitra, I.; Kumar Ojha, P.; Kar, S.; Narayan Das, R. Some case studies on application of “rm2” metrics for judging quality of quantitative structure–activity relationship predictions: Emphasis on scaling of response data. J. Comp. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, C.; Newton-Vinson, P.; Hubálek, F.; Edmonson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L.C.C. The PyMOL Molecular Graphics System; 40; Schrödinger, L.C.C.: New York, NY, USA, 2017. [Google Scholar]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Mellado, M.; Salas, C.O.; Uriarte, E.; Viña, D.; Jara-Gutiérrez, C.; Matos, M.J.; Cuellar, M. Design, Synthesis and Docking Calculations of Prenylated Chalcones as Selective Monoamine Oxidase B Inhibitors with Antioxidant Activity. ChemistrySelect 2019, 4, 7698–7703. [Google Scholar] [CrossRef]
- Abad, E.; Zenn, R.K.; Kästner, J. Reaction Mechanism of Monoamine Oxidase from QM/MM Calculations. J. Phys. Chem. B 2013, 117, 14238–14246. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Caflisch, A. Molecular dynamics in drug design. Eur. J. Med. Chem. 2015, 91, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Celik, I.; Erol, M.; Duzgun, Z. In silico evaluation of potential inhibitory activity of remdesivir, favipiravir, ribavirin and galidesivir active forms on SARS-CoV-2 RNA polymerase. Mol. Divers. 2021, 1–14. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 hMAO-A (µM) | IC50 hMAO-B (µM) | S.I. * |
|---|---|---|---|
| 3-Phenylcoumarin | >100 | 11.81 | >8.47 |
| trans-6-Styrylcoumarin | >100 | 0.110 | >909.09 |
| trans-Resveratrol | 17.41 | 29.37 | 0.59 |
| Selegiline | 67.25 | 0.020 | 3362.5 |
| Iproniazid | 6.56 | 7.54 | 0.87 |
| Compounds | pIC50 MAO-A | ||||
|---|---|---|---|---|---|
| Exp. | CoMFA | Res | CoMSIA | Res | |
| 3-Phenylcoumarin | <4.000 | - | - | - | - |
| trans-6-Styrylcoumarin | <4.000 | - | - | - | - |
| trans-Resveratrol | 4.759 | 4.767 | 0.008 | 4.759 | −0.008 |
| Compounds | pIC50 MAO-B | ||||
|---|---|---|---|---|---|
| Exp. | CoMFA | Res | CoMSIA | Res | |
| 3-Phenylcoumarin | 4.928 | 5.309 | −0.381 | 5.700 | −0.772 |
| trans-6-Styrylcoumarin | 6.959 | 7.212 | −0.254 | 7.000 | −0.041 |
| trans-Resveratrol | 4.532 | 5.173 | −0.641 | 5.362 | −0.830 |
| Parameters (Energy) | Enzyme-Ligand Complexes | |||
|---|---|---|---|---|
| MAO-B and 3-phenylcoumarin (kJ/mol) | MAO-B and 3-(3′-bromophenyl)-6-Methylcoumarin (kJ/mol) | MAO-B and trans-6-styrylcoumarin (kJ/mol) | MAO-B and trans-resveratrol (kJ/mol) | |
| Van der Waals | −145.335 ± 6.596 | −137.996 ± 8.218 | −152.618 ± 9.322 | −178.535 ± 8.654 |
| Electrostatic | −2.253 ± 4.536 | −4.754 ± 7.797 | −31.762 ± 4.914 | 0.826 ± 4.532 |
| Polar solvation | 99.294 ± 7.536 | 133.773 ± 19.676 | 137.238 ± 8.533 | 94.308 ± 7.084 |
| SASA | −13.845 ± 0.623 | −17.202 ± 0.826 | −14.723 ± 0.709 | −15.502 ± 0.767 |
| Binding free | −62.139 ± 9.680 | −26.179 ± 16.851 | −61.865 ± 10.412 | −98.903 ± 9.527 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mellado, M.; González, C.; Mella, J.; Aguilar, L.F.; Celik, I.; Borges, F.; Uriarte, E.; Delogu, G.; Viña, D.; Matos, M.J. Coumarin-Resveratrol-Inspired Hybrids as Monoamine Oxidase B Inhibitors: 3-Phenylcoumarin versus trans-6-Styrylcoumarin. Molecules 2022, 27, 928. https://doi.org/10.3390/molecules27030928
Mellado M, González C, Mella J, Aguilar LF, Celik I, Borges F, Uriarte E, Delogu G, Viña D, Matos MJ. Coumarin-Resveratrol-Inspired Hybrids as Monoamine Oxidase B Inhibitors: 3-Phenylcoumarin versus trans-6-Styrylcoumarin. Molecules. 2022; 27(3):928. https://doi.org/10.3390/molecules27030928
Chicago/Turabian StyleMellado, Marco, César González, Jaime Mella, Luis F. Aguilar, Ismail Celik, Fernanda Borges, Eugenio Uriarte, Giovanna Delogu, Dolores Viña, and Maria J. Matos. 2022. "Coumarin-Resveratrol-Inspired Hybrids as Monoamine Oxidase B Inhibitors: 3-Phenylcoumarin versus trans-6-Styrylcoumarin" Molecules 27, no. 3: 928. https://doi.org/10.3390/molecules27030928