
Efficient Synthesis of Fluorescent Coumarins and Phosphorous-Containing Coumarin-Type Heterocycles via Palladium Catalyzed Cross-Coupling Reactions
 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Theoretical Investigation of Substituted 3-Phosphonocoumarin Structures—Evaluation of the Fluorescent Properties
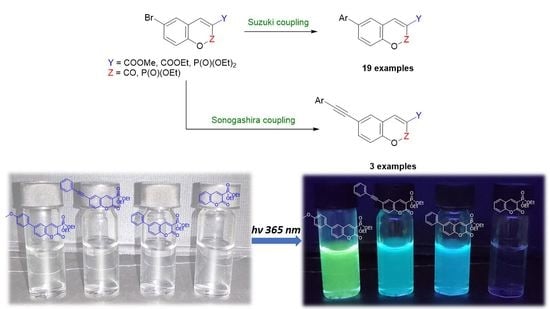
2.2. Synthesis of Fluorescent Coumarins and Phosphorous Containing Coumarin-Type Heterocycles
2.3. Photophysical Properties
3. Materials and Methods
3.1. Starting Materials
- 4-Hydroxy-[1,1’-biphenyl]-3-carbaldehyde, 2
- Diethyl (6-bromo-2-oxo-2H-chromen-3-yl)phosphonate, 5
- Ethyl 6-bromo-2-ethoxybenzo[e][1,2]oxaphosphinine-3-carboxylate 2-oxide, 6
3.2. Procedure for Suzuki Reaction—Preliminary Study
3.2.1. Procedure for Suzuki Reaction with PEPPSI, Pd(PPh3)4, and IMesPd(dmba)Cl
3.2.2. Procedure for Suzuki Reaction with Pd(PPh3)2Cl2
3.3. General Procedure for the Preparative Suzuki Reaction
- Diethyl (2-oxo-6-phenyl-2H-chromen-3-yl)phosphonate, 3a

- Diethyl (6-(2-methoxyphenyl)-2-oxo-2H-chromen-3-yl)phosphonate, 3b

- Diethyl (6-(4-methoxyphenyl)-2-oxo-2H-chromen-3-yl)phosphonate, 3c

- Diethyl (6-(4-fluorophenyl)-2-oxo-2H-chromen-3-yl)phosphonate, 3d

- Diethyl (6-mesityl-2-oxo-2H-chromen-3-yl)phosphonate, 3e

- Ethyl 2-oxo-6-phenyl-2H-chromene-3-carboxylate, 9a

- Ethyl 6-(2-methoxyphenyl)-2-oxo-2H-chromene-3-carboxylate, 9b

- Ethyl 6-(4-methoxyphenyl)-2-oxo-2H-chromene-3-carboxylate, 9c

- Ethyl 6-(4-fluorophenyl)-2-oxo-2H-chromene-3-carboxylate, 9d

- Ethyl 6-mesityl-2-oxo-2H-chromene-3-carboxylate, 9e

- Methyl 2-oxo-6-phenyl-2H-chromene-3-carboxylate, 10a

- Methyl 6-(2-methoxyphenyl)-2-oxo-2H-chromene-3-carboxylate, 10b

- Methyl 6-(4-methoxyphenyl)-2-oxo-2H-chromene-3-carboxylate, 10c

- Methyl 6-(4-fluorophenyl)-2-oxo-2H-chromene-3-carboxylate, 10d

- Methyl 6-mesityl-2-oxo-2H-chromene-3-carboxylate, 10e

- Ethyl 2-ethoxy-6-phenylbenzo[e][1,2]oxaphosphinine-3-carboxylate-2-oxide, 11a

- Ethyl 2-ethoxy-6-(2-methoxyphenyl)benzo[e][1,2]oxaphosphinine-3-carboxylate-2-oxide, 11b

- Ethyl 2-ethoxy-6-(4-methoxyphenyl)benzo[e][1,2]oxaphosphinine-3-carboxylate-2-oxide, 11c

- Ethyl 2-ethoxy-6-(4-fluorophenyl)benzo[e][1,2]oxaphosphinine-3-carboxylate-2-oxide, 11d

3.4. General Procedure for the Sonogashira Reaction
- Diethyl (2-oxo-6-(phenylethynyl)-2H-chromen-3-yl)phosphonate, 12a

- Ethyl 2-oxo-6-(phenylethynyl)-2H-chromene-3-carboxylate, 12b

- Diethyl (6-((4-methoxyphenyl)ethynyl)-2-oxo-2H-chromen-3-yl)phosphonate, 12d

4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koleva, A.I.; Petkova-Yankova, N.I.; Nikolova, R.D. Synthesis and Chemical Properties of 3-Phosphonocoumarins and 1,2-Benzoxaphosphorins as Precursors for Bioactive Compounds. Molecules 2019, 24, 2030. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, R.; Pathania, S.; Singh, V.; Rawalet, R.K. Metal-catalyzed synthetic strategies toward coumarin derivatives. Chem. Heterocycl. Comp. 2018, 54, 280–291. [Google Scholar] [CrossRef]
- Szwaczko, K. Coumarins Synthesis and Transformation via C–H Bond Activation—A Review. Inorganics 2022, 10, 23. [Google Scholar] [CrossRef]
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities. Int. J. Mol. Sci. 2020, 21, 4618. [Google Scholar] [CrossRef] [PubMed]
- Cheke, R.S.; Patel, H.M.; Patil, V.M.; Ansari, I.A.; Ambhore, J.P.; Shinde, S.D.; Kadri, A.; Snoussi, M.; Adnan, M.; Kharkar, P.S.; et al. Molecular Insights into Coumarin Analogues as Antimicrobial Agents: Recent Developments in Drug Discovery. Antibiotics 2022, 11, 566. [Google Scholar] [CrossRef]
- Yañez, O.; Osorio, M.I.; Uriarte, E.; Areche, C.; Tiznado, W.; Pérez-Donoso, J.M.; García-Beltrán, O.; González-Nilo, F. In Silico Study of Coumarins and Quinolines Derivatives as Potent Inhibitors of SARS-CoV-2 Main Protease. Front. Chem. 2021, 8, 595097. [Google Scholar] [CrossRef]
- Abdelmohsen, A.R.; Albohy, A.; Abdulrazik, B.S.; Bayoumi, S.A.L.; Malak, L.G.; Khallaf, I.S.A.; Bringmann, G.; Salwa, F.; Farag, S.F. Natural coumarins as potential anti-SARS-CoV-2 agents supported by docking analysis. RSC Adv. 2021, 11, 16970. [Google Scholar] [CrossRef]
- Sinkeldam, R.W.; Greco, N.J.; Tor, Y. Fluorescent Analogs of Biomolecular Building Blocks: Design, Properties, and Applications. Chem. Rev. 2010, 110, 2579. [Google Scholar] [CrossRef] [Green Version]
- Traven, V.F.; Cheptsov, D.A.; Solovjova, N.P.; Chibisova, T.A.; Voronov, I.I.; Dolotov, S.M.; Ivanov, I.V. Photoinduced Formation of the Laser Dye Coumarin 6 from Its Dihydro Derivatives. Dyes Pigments 2017, 146, 159. [Google Scholar] [CrossRef]
- Signore, G.; Nifosì, R.; Albertazzi, L.; Storti, B.; Bizzarri, R. Polarity-Sensitive Coumarins Tailored to Live Cell Imaging. J. Am. Chem. Soc. 2010, 132, 1276. [Google Scholar] [CrossRef]
- Bayrakçeken, F.; Yaman, A.; Hayvalı, M. Photophysical and Photochemical Study of Laser-Dye Coumarin-481 and Its Photoproduct in Solution. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2005, 61, 983. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, H.; Vekariya, R.H.; Patel, H.D. Recent Advances in the Synthesis of Coumarin Derivatives via Knoevenagel Condensation: A Review. Synth. Commun. 2014, 44, 2756. [Google Scholar] [CrossRef]
- Ansary, I.; Taher, A. One-Pot Synthesis of Coumarin Derivatives. In Phytochemicals in Human Health; Intech Open: London, UK, 2019. [Google Scholar]
- Gulati, S.; Singh, R.; Sangwan, S. A review on convenient synthesis of substituted coumarins using reuseable solid acid catalysts. RSC Adv. 2021, 11, 29130. [Google Scholar] [CrossRef] [PubMed]
- Kanchana, U.S.; Diana, E.J.; Mathew, T.V.; Anilkumar, G. Palladium-catalyzed cross-coupling reactions of coumarin derivatives: An overview. Appl Organomet. Chem. 2020, 34, e5983. [Google Scholar] [CrossRef]
- Wu, J.; Zhanga, L.; Xia, H. Palladium-catalyzed Suzuki–Miyaura couplings of potassium aryl trifluoroborates with 4-tosyloxycoumarins or 4-tosyloxyquinolin-2(1H)-one. Tetrahedron Letters 2006, 47, 1525. [Google Scholar] [CrossRef]
- Li, K.; Zeng, Y.; Neuenswander, B.; Tunge, J.A. Sequential Pd(II)-Pd(0) Catalysis for the Rapid Synthesis of Coumarins. J. Org. Chem. 2005, 70, 6515. [Google Scholar] [CrossRef]
- Yin, Q.; Yan, H.; Zhang, Y.; Wang, Y.; Zhang, G.; He, Y.; Zhang, W. Palladium-catalyzed synthesis of 8-allyl or 8-prenylcoumarins by using organotin reagents as multicoupling nucleophiles. Appl. Organometal. Chem. 2013, 27, 85. [Google Scholar] [CrossRef]
- Mao, W.; Wang, T.; Zeng, H.; Wang, Z.; Chen, J.; Shen, J. Synthesis and evaluation of novel substituted 5-hydroxycoumarin and pyranocoumarin derivatives exhibiting significant antiproliferative activity against breast cancer cell lines. Bioorg. Med. Chem. Lett. 2009, 19, 4570. [Google Scholar] [CrossRef]
- Bhaumick, P.; Jana, A.; Choudhury, L.H. Synthesis of novel coumarin containing conjugated fluorescent polymers by Suzuki cross-coupling reactions and their chemosensing studies for iron and mercury ions. Polymer 2021, 218, 123415. [Google Scholar] [CrossRef]
- Reddy, K.H.V.; Brion, J.; Messaoudi, S.; Alami, M. Synthesis of Biheterocycles Based on Quinolinone, Chromone, and Coumarin Scaffolds by Palladium-Catalyzed Decarboxylative Couplings. J. Org. Chem. 2016, 81, 424. [Google Scholar] [CrossRef]
- Balalas, T.; Abdul-Sada, A.; Hadjipavlou-Litina, D.J.; Litinas, K.E. Pd-Catalyzed Efficient Synthesis of Azacoumestans Via Intramolecular Cross Coupling of 4-(Arylamino)coumarins in the Presence of Copper Acetate under Microwaves. Synthesis 2017, 49, 2575. [Google Scholar] [CrossRef]
- Cocco, A.; Caria, P.; Sanna, G.; Stagi, L.; Cadoni, E.; Corpino, R.; Ricci, R.C.; Carbonaro, C.M.; Secci, F. Synthesis and Photophysical Properties of Fluorescent 6-Aryl-D-π-A Coumarin Derivatives. ACS Omega 2021, 6, 33708. [Google Scholar] [CrossRef] [PubMed]
- Starčević, Š.; Brožič, P.; Turk, S.; Cesar, J.; Rižner, T.L.; Stanislav Gobec, S. Synthesis and Biological Evaluation of (6- and 7-Phenyl) Coumarin Derivatives as Selective Nonsteroidal Inhibitors of 17β-Hydroxysteroid Dehydrogenase Type 1. J. Med. Chem. 2011, 54, 248. [Google Scholar] [CrossRef] [PubMed]
- Thanzeel, F.Y.; Wolf, C. Chemoselective bioconjugation based on modular click chemistry with 4-halocoumarins and arylsulfonates. RSC Adv. 2021, 11, 18960. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989; ISBN 9780195092769. [Google Scholar]
- Salahub, D.R.; Zerner, M.C. The Challenge of d and f Electrons. In Chemical Congress of North America; American Chemical Society: Washington, DC, USA, 1989; ISBN 9780841216280. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845. [Google Scholar] [CrossRef]
- Laurent, A.D.; Adamo, C.; Jacquemin, D. Dye chemistry with time-dependent density functional theory. Phys. Chem. Chem. Phys. 2014, 16, 14334. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Revision, B; Gaussian. Inc.: Pittsburgh, PA, USA, 2016. [Google Scholar]
- Oberhofer, K.E.; Musheghyan, M.; Wegscheider, S.; Wörle, M.; Iglev, E.D.; Nikolova, R.D.; Reinhard Kienberger, R.; Pekov, P.S.; Iglev, H. Individual control of singlet lifetime and triplet yield in halogen-substituted coumarin derivatives. RSC Adv. 2020, 10, 27096. [Google Scholar] [CrossRef]
- Wagner, M.S.; Ilieva, E.D.; Petkov, P.S.; Nikolova, R.D.; Kienbergera, R.; Iglev, H. Ultrafast hydrogen bond dynamics and partial electron transfer after photoexcitation of diethyl ester of 7-(diethylamino)-coumarin-3-phosphonic acid and its benzoxaphosphorin analog. Phys. Chem. Chem. Phys. 2015, 17, 9919. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999. [Google Scholar] [CrossRef] [PubMed]
- Raju, B.C.; Tiwari, A.K.; Kumar, J.A.; Ali, A.Z.; Agawane, S.B.; Saidachary, G.; Madhusudana, K. a-Glucosidase inhibitory antihyperglycemic activity of substituted chromenone derivatives. Bioorganic Med. Chem. 2010, 18, 358. [Google Scholar] [CrossRef] [PubMed]
- Bojilova, A.; Nikolova, R.; Ivanov, C.; Rodios, N.; Terzis, A.; Raptopoulou, C. A comparative study of the interaction of salicylaldehydes with phosphonoacetates under Knoevenagel reaction conditions. Synthesis of 1,2-benzoxaphosphorines and their dimers. Tetrahedron 1996, 52, 12597–12612. [Google Scholar] [CrossRef]
- Reeves, E.K.; Olivia, R.; Bauman, O.R.; Mitchem, G.B.; Neufeldt, S.R. Solvent Effects on the Selectivity of Palladium-Catalyzed Suzuki-Miyaura Couplings. Isr. J. Chem. 2019, 59, 1. [Google Scholar] [CrossRef]
- Carrow, B.P.; Hartwig, J.F. Distinguishing Between Pathways for Transmetalation in Suzuki-Miyaura Reactions. J. Am. Chem. Soc. 2011, 133, 2116. [Google Scholar] [CrossRef] [Green Version]
- Amatore, C.; Jutand, A.; Le Duc, G. Kinetic Data for the Transmetalation/Reductive Elimination in Palladium-Catalyzed Suzuki–Miyaura Reactions: Unexpected Triple Role of Hydroxide Ions Used as Base. Chem. Eur. J. 2011, 17, 2492. [Google Scholar] [CrossRef]
- Bojilova, A.; Kostadinova, T.; Ivanov, C. Interaction of Nitromethane with Esters and Amides of 2-Oxo-2H-1-benzopyrane-3-carboxylic Acid. Synth. Commun. 1989, 19, 2963. [Google Scholar] [CrossRef]
- Grant, J.A.; Bonnick, T.; Gossell-Williams, M.; Clayton, T.; Cook, J.M.; Jackson, Y.A. Synthesis, pharmacological studies and molecular modeling of some tetracyclic 1,3-diazepinium chlorides. Bioorganic Med. Chem. 2010, 18, 909. [Google Scholar] [CrossRef]
- O’Brien, C.J.; Kantchev, E.A.B.; Valente, C.; Hadei, N.; Chass, G.A.; Lough, A.; Hopkinson, A.C.; Michael, G.; Organ, M.G. Easily Prepared Air- and Moisture-Stable Pd–NHC (NHC=N-Heterocyclic Carbene) Complexes: A Reliable, User-Friendly, Highly Active Palladium Precatalyst for the Suzuki–Miyaura Reaction. Chem. A Eur. J. 2006, 12, 4743. [Google Scholar] [CrossRef]
- Kantchev, E.A.B.; Peh, G.; Zhang, C.; Ying, J.Y. Practical Heck−Mizoroki Coupling Protocol for Challenging Substrates Mediated by an N-Heterocyclic Carbene-Ligated Palladacycle. Org. Lett. 2008, 10, 3949. [Google Scholar] [CrossRef] [PubMed]
- Clique, B.; Fabritius, C.; Couturier, C.; Monteiro, N.; Balme, G. Unexpected isolation, and structural characterization, of a β-hydrogen-containing σ-alkylpalladium halide complex in the course of an intramolecular Heck reaction. Synthesis of polycyclic isoquinoline derivatives. Chem. Commun. 2003, 272–273. [Google Scholar] [CrossRef] [PubMed]
- Lázaro-Milla, C.; Quirós, M.T.; Cárdenas, D.J.; Almendros, P. Triflyl-assisted reductive Pd-catalyzed Tsuji–Trost type reaction. Chem. Commun. 2020, 56, 6070. [Google Scholar] [CrossRef] [PubMed]
- Kagalwala, H.N.; Tong, L.; Zong, R.; Kohler, L.; Ahlquist, M.S.G.; Fan, T.; Gagnon, K.J.; Thummel, R.P. Evidence for Oxidative Decay of a Ru-Bound Ligand during Catalyzed Water Oxidation. ACS Catal. 2017, 7, 2607. [Google Scholar] [CrossRef]
- Bojilova, A.; Kostadinova, T.; Ivanov, C. Allylation-Assisted Addition of Nitromethane to 2H-1-Benzopyran-2-ones Substituted in Position 3. Liebigs Ann. Chem. 1989, 1041–1043. [Google Scholar] [CrossRef]
- Ilieva, E.D.; Petkova, N.I.; Nikolova, R.D. Ring Opening Reactions of 3-Phosphonocoumarin Under Michael Reaction Conditions. Phosphorus Sulfur Silicon 2012, 187, 39, published online December 2011. [Google Scholar] [CrossRef]
- Jackson, Y.A.; Williams, M.F. Heterocycles from 2-Aminopyridine and Derivatives of 3-Methylbenzofuran-2-carboxylic Acid. Heterocycles 1997, 45, 787. [Google Scholar] [CrossRef]
- Skarga, V.V.; Matrosov, A.A.; Nichugovskiy, A.I.; Negrebetsky, V.V.; Maslov, M.A.; Boldyrev, I.A.; Malakhov, M.V. pH-Dependent Photoinduced Interconversion of Furocoumaric and Furocoumarinic Acids. Molecules 2021, 26, 2800. [Google Scholar] [CrossRef]
- Khoobi, M.; Molaverdi, F.; Alipour, M.; Jafarpour, F.; Foroumadi, A.; Shafiee, A. Palladium-catalyzed domino protodecarboxylation/oxidative Heck reaction: Regioselective arylation of coumarin-3-carboxylic acids. Tetrahedron 2013, 69, 11164. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, T.; Fan, R.; Wu, J. General and Efficient Route for the Synthesis of 3,4-Disubstituted Coumarins via Pd-Catalyzed Site-Selective Cross-Coupling Reactions. J. Org. Chem. 2007, 72, 7279. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Mondal, S. A new strategy for the synthesis of coumarin- and quinolone-annulated pyrroles via Pd(0) mediated cross-coupling followed by Cu(I) catalyzed heteroannulation. Tetrahedron Lett. 2008, 49, 2418. [Google Scholar] [CrossRef]
- Elenkova, D.; Lyapchev, R.; Romanova, J.; Morgenstern, B.; Dimitrova, Y.; Dimov, D.; Tsvetkov, M.; Zaharieva, J. Luminescent Complexes of Europium (III) with 2-(Phenylethynyl)-1,10-phenanthroline: The Role of the Counterions. Molecules 2021, 26, 7272. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, B.; Haizhu, Y.H.; Zhu, L.; Feng, Y.; Zhu, M.; Guo, Q.; Meng, X. Two-Photon Fluorescent Probes for Biological Mg2+ Detection Based on 7-Substituted Coumarin. J. Org. Chem. 2015, 80, 4306. [Google Scholar] [CrossRef]
- Arsenyan, P.; Vasiljeva, J.; Domracheva, I.; Kanepe-Lapsa, I.; Gulbe, A. Selenopheno[2,3-f]coumarins: Novel scaffolds with antimetastatic activity against melanoma and breast cancer. N. J. Chem. 2019, 43, 11851. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2017, 71, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3. [Google Scholar] [CrossRef] [Green Version]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281. [Google Scholar] [CrossRef] [Green Version]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed reaction of 1-alkenylboronates with vinylic halides: (1Z,3E)-1-phenyl-1,3-octadiene. Org. Synth. 1990, 68, 130. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calculated Models (CM) | Structure | λabs, (nm) | fabs | λem, (nm) | fem | Stokes Shift, (nm) |
|---|---|---|---|---|---|---|
| CM-1 |  | 295 | 0.2800 | 344 | 0.4989 | 49 |
| CM-2 |  | 309 | 0.3305 | 378 | 0.2419 | 69 |
| CM-3 |  | 312 | 0.2574 | 386 | 0.1802 | 74 |
| CM-4 |  | 299 | 0.4665 | 357 | 0.3821 | 58 |
| CM-5 |  | 320 | 0.2703 | 404 | 0.2480 | 84 |
| CM-6 |  | 310 | 0.3446 | 378 | 0.2513 | 68 |
| CM-7 |  | 299 | 0.5267 | 372 | 0.2611 | 73 |
| CM-8 |  | 319 | 0.3404 | 384 | 0.3114 | 65 |
| CM-9 |  | 327 | 0.2703 | 402 | 0.2480 | 75 |
| CM-10 |  | 309 | 0.3569 | 374 | 0.2826 | 66 |
| CM-11 |  | 304 | 0.4667 | 363 | 0.3832 | 59 |
| CM-12 |  | 304 | 0.4195 | 364 | 0.3230 | 61 |
| Entry | Solvent | 5 | 3a | 7 | Catalyst |
|---|---|---|---|---|---|
| 1 | PhCH3 | traces | >99 | traces | PdCl2(PPh3)2 |
| 2 | PhCH3:H2O | traces | >99 | traces | PdCl2(PPh3)2 |
| 3 | Dioxane | ~14 | ~86 | traces | PdCl2(PPh3)2 |
| 4 | Dioxane:H2O | traces | >99 | traces | PdCl2(PPh3)2 |
| 5 | EtOH:H2O:PhCH3 | traces | >99 | traces | PdCl2(PPh3)2 |
| 6 | PhCH3 | ~54 | ~45 | traces | Pd(PPh3)4 |
| 7 | PhCH3:H2O | 18 | 74 | 7 | Pd(PPh3)4 |
| 8 | Dioxane | ~54 | ~45 | traces | Pd(PPh3)4 |
| 9 | Dioxane:H2O | 25 | 68 | 6 | Pd(PPh3)4 |
| 10 | EtOH:H2O:PhCH3 | ~34 | ~65 | traces | Pd(PPh3)4 |
| 11 | PhCH3 | ~57 | ~42 | traces | IMesPd(dmba)Cl |
| 12 | PhCH3:H2O | traces | >99 | - | IMesPd(dmba)Cl |
| 13 | Dioxane | ~56 | ~43 | traces | IMesPd(dmba)Cl |
| 14 | Dioxane:H2O | traces | >99 | traces | IMesPd(dmba)Cl |
| 15 | EtOH:H2O:PhCH3 | traces | >99 | traces | IMesPd(dmba)Cl |
| 16 | PhCH3 | 30 | 70 | - | PEPPSI-type |
| 17 | PhCH3:H2O | - | 100 | - | PEPPSI-type |
| 18 | Dioxane | 24 | 76 | - | PEPPSI-type |
| 19 | Dioxane:H2O | - | 100 | - | PEPPSI-type |
| 20 | EtOH | ? | ? | ? | PEPPSI-type |
| 21 | EtOH:H2O | traces | ~77 | ~23 | PEPPSI-type |
| 22 | EtOH:H2O:PhCH3 | traces | 94 | 5 | PEPPSI-type |
| Entry | Boronic Acid | Product | [Pd] | Time, [h] | Yield *, [%] |
|---|---|---|---|---|---|
| 1 |  |  | PEPPSI-type | 17 h | 90% |
| 2 |  |  | PEPPSI-type | 120 h | 41% |
| 3 | Pd(PPh3)2Cl2 | 48 h | 63% | ||
| 4 |  |  | PEPPSI-type | 48 h | 95% |
| 5 |  |  | PEPPSI-type | 48 h | 61% |
| 6 |  |  | PEPPSI-type | 48 h | 19% |
| 7 | Pd(PPh3)2Cl2 | 48 h | 45% |
| Entry | Boronic Acid | Product | [Pd] | Time | Yield *, [%] |
|---|---|---|---|---|---|
| 1 |  |  | PEPPSI-type | 30 min | 90% |
| 2 | PEPPSI-type | 60 min | 97% | ||
| 3 |  |  | PEPPSI-type | 48 h | 20% |
| 4 | Pd(PPh3)2Cl2 | 24 h | 68% | ||
| 5 | Pd(PPh3)2Cl2 | 21 h | 83% | ||
| 6 |  |  | PEPPSI-type | 45 min | 92% |
| 7 |  | PEPPSI-type | 45 min | 99% | |
| 8 |  |  | PEPPSI-type | 45 min | 80% |
| 9 | PEPPSI-type | 60 min | 99% | ||
| 10 |  |  | Pd(PPh3)2Cl2 | 72 h | 29% |
| 11 | Pd(PPh3)2Cl2 | 90 h | 18% | ||
| 12 |  |  | Pd(PPh3)2Cl2 | 72 h | complex mixture |
| 13 | Pd(PPh3)2Cl2 | 72 h | complex mixture |
| Entry | Boronic Acid | Product | [Pd] | Time, [h] | Yield *, [%] |
|---|---|---|---|---|---|
| 1 |  |  | PEPPSI-type | 16 h | 89% |
| 2 |  |  | Pd(PPh3)2Cl2 | 42 h | 45% |
| 3 |  |  | PEPPSI-type | 3 h | 87% |
| 4 |  |  | PEPPSI-type | 2.5 h | 89% |
| Entry | Alkyne | Product | [Pd] | Time, [h] | Yield *, [%] |
|---|---|---|---|---|---|
| 1 |  |  | Pd(PPh3)2Cl2 | 20 h | 82% |
| 2 | Pd(PPh3)2Cl2 | 22 h | 52% | ||
| 3 | Pd(PPh3)2Cl2 | 48 h | complex mixture | ||
| 4 |  |  | PEPPSI-type | 30 h | 56% |
| 5 | Pd(PPh3)2Cl2 | 48 h | complex mixture |
| Compounds | Solvent | λabs, nm | λem, nm | Stokes Shift, nm |
|---|---|---|---|---|
| 7 | MeCN | 324 | 411 | 87 |
| 3a | DMSO | 345 | 460 | 115 |
| MeOH | 346 | 459 | 113 | |
| MeCN | 343 | 453 | 110 | |
| DCM | 344 | 452 | 108 | |
| EtOAc | 343 | 447 | 104 | |
| 3b | MeCN | 345 | 493 | 148 |
| 3c | MeCN | 356 | 535 | 179 |
| 3d | MeCN | 345 | 456 | 148 |
| 3e | MeCN | 333 | 451 | 118 |
| 12a | MeCN | 351 | 461 | 110 |
| 12d | MeCN | 356 | 543 | 187 |
| 9-H * | MeCN | 326 | 412 | 86 |
| 9a | MeOH | 355 | 470 | 115 |
| MeCN | 355 | 463 | 108 | |
| DCM | 360 | 457 | 97 | |
| 9b | MeCN | 353 | 503 | 150 |
| 9c | MeCN | 366 | 546 | 180 |
| 9d | MeCN | 366 | 458 | 92 |
| 9e | MeCN | 343 | 458 | 115 |
| 12b | MeCN | 357 | 472 | 115 |
| 10-H * | MeCN | 326 | 411 | 85 |
| 10a | MeOH | 357 | 472 | 115 |
| MeCN | 353 | 466 | 113 | |
| DCM | 358 | 457 | 99 | |
| 10b | MeCN | 351 | 504 | 153 |
| 10c | MeCN | 363 | 543 | 180 |
| 10d | MeCN | 354 | 466 | 112 |
| 10e | MeCN | 340 | 460 | 120 |
| 11-H * | MeCN | 324 | 410 | 86 |
| 11a | MeOH | 343 | 468 | 125 |
| MeCN | 342 | 459 | 117 | |
| DCM | 342 | 452 | 110 | |
| 11b | MeCN | 340 | 500 | 160 |
| 11c | MeCN | 350 | 541 | 191 |
| 11d | MeCN | 341 | 460 | 119 |
| Exp | CM | |||||
|---|---|---|---|---|---|---|
| 7 | CM-1 | 324 | 0 | 411 | 0 | −38 |
| 3a | CM-2 | 339 | −4 | 452 | 1 | −41 |
| 3b | CM-3 | 343 | −2 | 461 | 32 | −74 |
| 3c | CM-5 | 351 | −5 | 483 | 52 | −95 |
| 3d | CM-6 | 340 | −5 | 452 | 41 | −43 |
| 3e | CM-7 | 328 | −5 | 444 | 7 | −45 |
| 12a | CM-8 | 350 | −1 | 459 | 2 | −45 |
| 12d | CM-9 | 359 | 3 | 480 | 62 | −112 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyapchev, R.; Koleva, A.I.; Koleva, I.Z.; Subev, K.; Madzharova, I.; Simeonova, K.B.; Petkova-Yankova, N.; Morgenstern, B.; Lozanova, V.; Petrov, P.Y.; et al. Efficient Synthesis of Fluorescent Coumarins and Phosphorous-Containing Coumarin-Type Heterocycles via Palladium Catalyzed Cross-Coupling Reactions. Molecules 2022, 27, 7649. https://doi.org/10.3390/molecules27217649
Lyapchev R, Koleva AI, Koleva IZ, Subev K, Madzharova I, Simeonova KB, Petkova-Yankova N, Morgenstern B, Lozanova V, Petrov PY, et al. Efficient Synthesis of Fluorescent Coumarins and Phosphorous-Containing Coumarin-Type Heterocycles via Palladium Catalyzed Cross-Coupling Reactions. Molecules. 2022; 27(21):7649. https://doi.org/10.3390/molecules27217649
Chicago/Turabian StyleLyapchev, Rumen, Ana I. Koleva, Iskra Z. Koleva, Kristian Subev, Ivelina Madzharova, Kristina B. Simeonova, Nevena Petkova-Yankova, Bernd Morgenstern, Vesela Lozanova, Petar Y. Petrov, and et al. 2022. "Efficient Synthesis of Fluorescent Coumarins and Phosphorous-Containing Coumarin-Type Heterocycles via Palladium Catalyzed Cross-Coupling Reactions" Molecules 27, no. 21: 7649. https://doi.org/10.3390/molecules27217649