
Design, Synthesis, and Mechanistic Study of 2-Pyridone-Bearing Phenylalanine Derivatives as Novel HIV Capsid Modulators
 , , , , and
, , , , and
Abstract
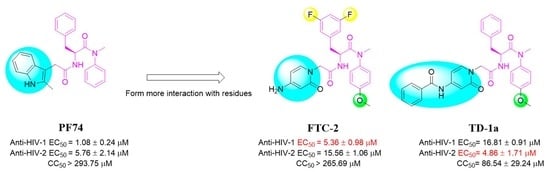
:
1. Introduction
2. Chemistry
3. Results and Discussion
3.1. In Vitro Anti-HIV Assays and SAR Analysis for HIV-1 Potency
3.2. Solubility of Representative Compounds
3.3. Binding Mode of FTC-2 within the Interprotomer Pocket
3.4. Computational Assessment of Drug-like Properties and Metabolic Stability
4. Conclusions
5. Experimental Section
5.1. Chemistry
5.1.1. General Procedure for the Synthesis of 2a and 2b
tert-butyl (S)-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (2a)
tert-butyl (S)-(3-(3,5-difluorophenyl)-1-((4-methoxyphenyl) (methyl)amino)-1-oxopropan-2-yl)carbamate (2b)
5.1.2. General Procedure for the Synthesis of 3a and 3b
(S)-2-amino-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (3a)
(S)-2-amino-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methylpropanamide (3b)
5.1.3. General Procedure for the Synthesis of 4a and 4b
(S)-2-(2-bromoacetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (4a)
(S)-2-(2-bromoacetamido)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methylpropanamide (4b)
5.1.4. General Procedure for the Synthesis of FTC-1 and TC-1
tert-butyl (S)-(1-(2-((3-(3,5-difluorophenyl)-1-((4-methoxyphenyl)(methyl)amino)-1-oxopropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)carbamate (FTC-1)
tert-butyl (S)-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)carbamate (TC-1)
5.1.5. General Procedure for the Synthesis of FTC-2 and TC-2
(S)-2-(2-(4-amino-2-oxopyridin-1(2H)-yl)acetamido)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methylpropanamide (FTC-2)
(S)-2-(2-(4-amino-2-oxopyridin-1(2H)-yl)acetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (TC-2)
5.1.6. General Procedure for the Synthesis of TD-1a–1l
(S)-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1a)
(S)-4-fluoro-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1b)
(S)-3-fluoro-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1c)
(S)-2-fluoro-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1d)
(S)-4-chloro-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1e)
(S)-4-bromo-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1f)
(S)-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)-4-methylbenzamide (TD-1g)
(S)-4-methoxy-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1h)
methyl (S)-4-((1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)carbamoyl)benzoate (TD-1i)
(S)-4-cyano-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1j)
(S)-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)-2-naphthamide (TD-1k)
(S)-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)-4-nitrobenzamide (TD-1l)
5.1.7. Procedure for the Synthesis of TD-1m
(S)-4-amino-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1m)
5.2. In Vitro Anti-HIV Assay in MT-4 Cells
5.3. Solubility Studies
5.4. Molecular Docking Studies
5.5. Computational Assessment of Drug-like Properties, Metabolic Stability, and Toxicity
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef]
- Campbell-Yesufu, O.T.; Gandhi, R.T. Update on human immunodeficiency virus (HIV)-2 infection. Clin. Infect. Dis. 2011, 52, 780–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyamweya, S.; Hegedus, A.; Jaye, A.; Rowland-Jones, S.; Flanagan, K.L.; Macallan, D.C. Comparing HIV-1 and HIV-2 infection: Lessons for viral immunopathogenesis. Rev. Med. Virol. 2013, 23, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.; Logan, R.; Sterne, J.A.; Hernández-Díaz, S.; Robins, J.M.; Sabin, C.; Bansi, L.; van Sighem, A.; de Wolf, F.; Costagliola, D.; et al. The effect of combined antiretroviral therapy on the overall mortality of HIV-infected individuals. Aids 2010, 24, 123–137. [Google Scholar]
- Wang, Z.; Cherukupalli, S.; Xie, M.; Wang, W.; Jiang, X.; Jia, R.; Pannecouque, C.; De Clercq, E.; Kang, D.; Zhan, P.; et al. Contemporary medicinal chemistry strategies for the discovery and development of novel HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2022, 65, 3729–3757. [Google Scholar] [CrossRef]
- Du, J.; Guo, J.; Kang, D.; Li, Z.; Wang, G.; Wu, J.; Zhang, Z.; Fang, H.; Hou, X.; Huang, Z.; et al. New techniques and strategies in drug discovery. Chin. Chem. Lett. 2020, 31, 1695–1708. [Google Scholar] [CrossRef]
- Rossi, E.; Meuser, M.E.; Cunanan, C.J.; Cocklin, S. Structure, Function, and Interactions of the HIV-1 Capsid Protein. Life 2021, 11, 100. [Google Scholar] [CrossRef]
- Xu, S.; Sun, L.; Zalloum, W.A.; Zhang, X.; Huang, T.; Ding, D.; Tao, Y.; Zhao, F.; Gao, S.; Kang, D.; et al. From design to biological mechanism evaluation of phenylalanine-bearing HIV-1 capsid inhibitors targeting a vital assembly interface. Chin. Chem. Lett. 2022. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Xu, S.; Huang, T.; Song, S.; Cherukupalli, S.; Zhan, P.; Liu, X. An insight on medicinal aspects of novel HIV-1 capsid protein inhibitors. Eur. J. Med. Chem. 2021, 217, 113380. [Google Scholar] [CrossRef]
- Dick, R.A.; Mallery, D.L.; Vogt, V.M.; James, L.C. IP6 Regulation of HIV Capsid Assembly, Stability, and Uncoating. Viruses 2018, 10, 640. [Google Scholar] [CrossRef] [Green Version]
- Gruenke, P.R.; Aneja, R.; Welbourn, S.; Ukah, O.B.; Sarafianos, S.G.; Burke, D.H.; Lange, M.J. Selection and identification of an RNA aptamer that specifically binds the HIV-1 capsid lattice and inhibits viral replication. Nucleic Acids Res. 2022, 50, 1701–1717. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, S.; Sun, L.; Ding, D.; Tao, Y.; Kang, D.; Liu, X.; Zhan, P. HIV-1 capsid inhibitors: A sword to destroy the virus. Future Med. Chem. 2022, 14, 605–607. [Google Scholar] [CrossRef]
- Rihn, S.J.; Wilson, S.J.; Loman, N.J.; Alim, M.; Bakker, S.E.; Bhella, D.; Gifford, R.J.; Rixon, F.J.; Bieniasz, P.D. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog. 2013, 9, e1003461. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Dick, A.; Meuser, M.E.; Huang, T.; Zalloum, W.A.; Chen, C.H.; Cherukupalli, S.; Xu, S.; Ding, X.; Gao, P.; et al. Design, synthesis, and mechanism study of benzenesulfonamide-containing Phenylalanine derivatives as novel HIV-1 capsid inhibitors with improved antiviral activities. J. Med. Chem. 2020, 63, 4790–4810. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, L.; Meuser, M.E.; Zalloum, W.A.; Xu, S.; Huang, T.; Cherukupalli, S.; Jiang, X.; Ding, X.; Tao, Y.; et al. Design, synthesis, and mechanism study of dimerized phenylalanine derivatives as novel HIV-1 capsid inhibitors. Eur. J. Med. Chem. 2021, 226, 113848. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Sun, L.; Dick, A.; Zalloum, W.A.; Huang, T.; Meuser, M.E.; Zhang, X.; Tao, Y.; Cherukupalli, S.; Ding, D.; et al. Design, synthesis, and mechanistic investigations of phenylalanine derivatives containing a benzothiazole moiety as HIV-1 capsid inhibitors with improved metabolic stability. Eur. J. Med. Chem. 2022, 227, 113903. [Google Scholar] [CrossRef] [PubMed]
- Vernekar, S.K.V.; Sahani, R.L.; Casey, M.C.; Kankanala, J.; Wang, L.; Kirby, K.A.; Du, H.; Zhang, H.; Tedbury, P.R.; Xie, J.; et al. Toward Structurally Novel and Metabolically Stable HIV-1 Capsid-Targeting Small Molecules. Viruses 2020, 12, 452. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Casey, M.C.; Vernekar, S.K.V.; Sahani, R.L.; Kirby, K.A.; Du, H.; Zhang, H.; Tedbury, P.R.; Xie, J.; Sarafianos, S.G.; et al. Novel PF74-like small molecules targeting the HIV-1 capsid protein: Balance of potency and metabolic stability. Acta Pharm. Sin. B 2021, 11, 810–822. [Google Scholar] [CrossRef]
- Meuser, M.E.; Reddy, P.A.N.; Dick, A.; Maurancy, J.M.; Salvino, J.M.; Cocklin, S. Rapid optimization of the metabolic stability of a human immunodeficiency virus type-1 capsid inhibitor using a multistep computational workflow. J. Med. Chem. 2021, 64, 3747–3766. [Google Scholar] [CrossRef]
- Xu, J.P.; Francis, A.C.; Meuser, M.E.; Mankowski, M.; Ptak, R.G.; Rashad, A.A.; Melikyan, G.B.; Cocklin, S. Exploring modifications of an HIV-1 capsid inhibitor: Design, synthesis, and mechanism of action. J. Drug Des. Res. 2018, 5, 1070. [Google Scholar]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F.; et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef]
- Rasaiyaah, J.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.A.; Selwood, D.L.; James, L.C.; Noursadeghi, M.; et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503, 402–405. [Google Scholar] [CrossRef] [Green Version]
- Price, A.J.; Fletcher, A.J.; Schaller, T.; Elliott, T.; Lee, K.; KewalRamani, V.N.; Chin, J.W.; Towers, G.J.; James, L.C. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012, 8, e1002896. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Yücel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013, 9, e1003693. [Google Scholar] [CrossRef] [Green Version]
- Di Nunzio, F.; Fricke, T.; Miccio, A.; Valle-Casuso, J.C.; Perez, P.; Souque, P.; Rizzi, E.; Severgnini, M.; Mavilio, F.; Charneau, P.; et al. Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 2013, 440, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Lelek, M.; Di Nunzio, F.; Henriques, R.; Charneau, P.; Arhel, N.; Zimmer, C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc. Natl. Acad. Sci. USA 2012, 109, 8564–8569. [Google Scholar] [CrossRef] [Green Version]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hué, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef] [PubMed]
- Matreyek, K.A.; Engelman, A. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol. 2011, 85, 7818–7827. [Google Scholar] [CrossRef] [PubMed]
- Bichel, K.; Price, A.J.; Schaller, T.; Towers, G.J.; Freund, S.M.; James, L.C. HIV-1 capsid undergoes coupled binding and isomerization by the nuclear pore protein NUP358. Retrovirology 2013, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, A.; Meuser, M.E.; Cocklin, S. Clade-Specific Alterations within the HIV-1 Capsid Protein with Implications for Nuclear Translocation. Biomolecules 2022, 12, 695. [Google Scholar] [CrossRef] [PubMed]
- Dick, A.; Cocklin, S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules 2020, 25, 1687. [Google Scholar] [CrossRef] [Green Version]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010, 6, e1001220. [Google Scholar] [CrossRef] [Green Version]
- Rankovic, S.; Ramalho, R.; Aiken, C.; Rousso, I. PF74 Reinforces the HIV-1 Capsid To Impair Reverse Transcription-Induced Uncoating. J. Virol. 2018, 92, e00845-18. [Google Scholar] [CrossRef] [Green Version]
- Jacques, D.A.; McEwan, W.A.; Hilditch, L.; Price, A.J.; Towers, G.J.; James, L.C. HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature 2016, 536, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef]
- Bester, S.M.; Wei, G.; Zhao, H.; Adu-Ampratwum, D.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Annamalai, A.S.; Singh, P.K.; Shkriabai, N.; et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science 2020, 370, 360–364. [Google Scholar] [CrossRef]
- Margot, N.A.; Naik, V.; VanderVeen, L.; Anoshchenko, O.; Singh, R.; Dvory-Sobol, H.; Rhee, M.S.; Callebaut, C. Resistance analyses in Highly Treatment-Experienced People with HIV Treated with the Novel Capsid HIV Inhibitor Lenacapavir. J. Infect. Dis. 2022, jiac364. [Google Scholar] [CrossRef] [PubMed]
- Segall, M.; Champness, E.; Obrezanova, O.; Leeding, C. Beyond profiling: Using ADMET models to guide decisions. Chem. Biodivers. 2009, 6, 2144–2151. [Google Scholar] [CrossRef]
- Tuyishime, M.; Danish, M.; Princiotto, A.; Mankowski, M.K.; Lawrence, R.; Lombart, H.G.; Esikov, K.; Berniac, J.; Liang, K.; Ji, J.; et al. Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorg. Med. Chem. Lett. 2014, 24, 5439–5445. [Google Scholar] [CrossRef] [Green Version]
- Karadsheh, R.; Meuser, M.E.; Cocklin, S. Composition and orientation of the core region of novel HIV-1 entry inhibitors influences metabolic stability. Molecules 2020, 25, 1430. [Google Scholar] [CrossRef] [Green Version]
- Tyzack, J.D.; Hunt, P.A.; Segall, M.D. Predicting regioselectivity and lability of cytochrome P450 metabolism using quantum mechanical simulations. J. Chem. Inf. Model. 2016, 56, 2180–2193. [Google Scholar] [CrossRef]
- Hunt, P.A.; Segall, M.D.; Tyzack, J.D. WhichP450: A multi-class categorical model to predict the major metabolising CYP450 isoform for a compound. J. Comput. Aided Mol. Des. 2018, 32, 537–546. [Google Scholar] [CrossRef]
- Sega, G.A. A review of the genetic effects of ethyl methanesulfonate. Mutat. Res. 1984, 134, 113–142. [Google Scholar] [CrossRef]
- Pillans, P.I.; Ghiculescu, R.A.; Lampe, G.; Wilson, R.; Wong, R.; Macdonald, G.A. Severe acute liver injury associated with lumiracoxib. J. Gastroenterol. Hepatol. 2012, 27, 1102–1105. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; Chapter 8, Unit 8.14. [Google Scholar]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 15. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound ID | R1 | R2 | EC50 a (μM) | CC50 b (μM) | SI c | ||
| HIV-1 IIIB | HIV-2 ROD | HIV-1 | HIV-2 | ||||
| FTC-2 | F | H | 5.36 ± 0.98 | 15.56 ± 1.06 | >265.69 | >49.57 | >17.08 |
| TC-2 | H | H | 29.07 ± 4.23 | 19.72 ± 6.42 | 261.13 ± 33.14 | 8.98 | 13.24 |
| TD-1a | H |  | 16.81 ± 0.91 | 4.86 ± 1.71 | 86.54 ± 29.24 | 5.15 | 17.81 |
| TD-1b | H |  | >34.80 | 16.22 ± 17.16 | 34.80 ± 5.75 | N.D.d | 2.15 |
| TD-1c | H |  | 18.90 ± 2.66 | 6.59 ± 1.63 | 61.65 ± 8.44 | 3.26 | 9.36 |
| TD-1d | H |  | 17.88 ± 2.68 | 7.92 ± 0.83 | 81.42 ± 22.66 | 4.55 | 10.28 |
| TD-1e | H |  | >25.48 | >25.48 | 25.48 ± 2.90 | N.D. | N.D. |
| TD-1f | H |  | >23.51 | >23.51 | 23.51 ± 2.79 | N.D. | N.D. |
| TD-1g | H |  | 25.60 ± 7.51 | 20.48 ± 8.49 | 58.27 ± 4.85 | 2.28 | 2.85 |
| TD-1h | H |  | 29.62 ± 4.34 | 10.67 ± 3.66 | 53.13 ± 5.12 | 1.79 | 4.98 |
| TD-1i | H |  | 19.09 ± 5.08 | 15.25 ± 0.55 | 96.17 ± 19.83 | 5.04 | 6.31 |
| TD-1j | H |  | 16.77 ± 0.78 | 17.83 ± 2.79 | 137.19 ± 22.50 | 8.18 | 7.69 |
| TD-1k | H |  | >27.59 | >27.59 | 27.59 ± 3.18 | N.D. | N.D. |
| TD-1l | H |  | >31.87 | >31.87 | 31.87 ± 1.73 | N.D. | N.D. |
| TD-1m | H |  | >189.23 | 17.14 ± 2.49 | 189.23 ± 2.19 | N.D. | 11.04 |
| PF74 | - | - | 1.08 ± 0.24 | 5.76 ± 2.14 | >293.75 | >271.99 | >51.00 |
| Compounds | Solubility (μg/mL) a | ||
|---|---|---|---|
| pH 2 | pH 7 | pH 7.4 | |
| TD-1a | 2.69 ± 0.52 | 5.07 ± 2.60 | 8.66 ± 0.41 |
| FTC-2 | 366.57 ± 58.39 | 261.19 ± 130.46 | 436.58 ± 51.87 |
| PF74 | <1.12 b | 1.44 ± 0.10 c | 1.91 ± 0.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Sun, L.; Xu, S.; Shao, X.; Li, Z.; Ding, D.; Jiang, X.; Zhao, S.; Cocklin, S.; Clercq, E.D.; et al. Design, Synthesis, and Mechanistic Study of 2-Pyridone-Bearing Phenylalanine Derivatives as Novel HIV Capsid Modulators. Molecules 2022, 27, 7640. https://doi.org/10.3390/molecules27217640
Zhang X, Sun L, Xu S, Shao X, Li Z, Ding D, Jiang X, Zhao S, Cocklin S, Clercq ED, et al. Design, Synthesis, and Mechanistic Study of 2-Pyridone-Bearing Phenylalanine Derivatives as Novel HIV Capsid Modulators. Molecules. 2022; 27(21):7640. https://doi.org/10.3390/molecules27217640
Chicago/Turabian StyleZhang, Xujie, Lin Sun, Shujing Xu, Xiaoyu Shao, Ziyi Li, Dang Ding, Xiangyi Jiang, Shujie Zhao, Simon Cocklin, Erik De Clercq, and et al. 2022. "Design, Synthesis, and Mechanistic Study of 2-Pyridone-Bearing Phenylalanine Derivatives as Novel HIV Capsid Modulators" Molecules 27, no. 21: 7640. https://doi.org/10.3390/molecules27217640