
Biodereplication of Antiplasmodial Extracts: Application of the Amazonian Medicinal Plant Piper coruscans Kunth

 , , , ,
, , , , 
Abstract
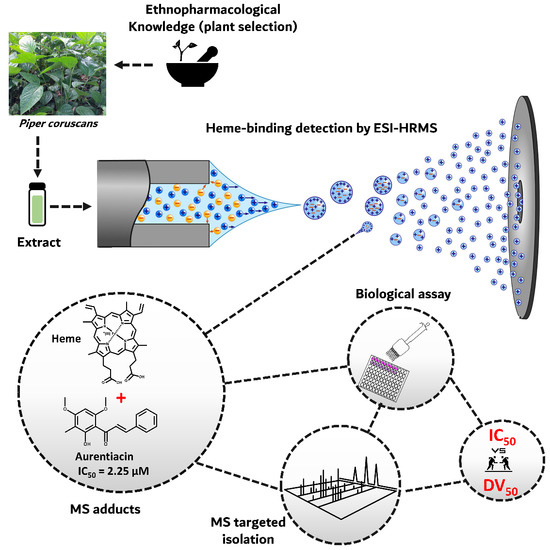
:
1. Introduction
2. Results and Discussion
2.1. Biodereplication as a New Strategy of Antiplasmodial Drug Discovery
2.2. Scientific Validation of the Traditional Use of P. coruscans as an Antimalarial Remedy Using Biodereplication
3. Materials and Methods
3.1. Biological Material
3.2. Heme-Binding Assay by Mass Spectrometry (MS)
3.3. Collision-Induced Dissociation
3.4. Visualization of Adduct Fragmentation via Molecular Networking (MN)
3.5. LC–MS of Extract and Compounds Annotation
3.6. Isolation of Targeted Compounds
3.7. Antiplasmodial Activity
3.7.1. Plasmodium falciparum Culture
3.7.2. In Vitro Antiplasmodial Activity on Plasmodium falciparum
3.8. Cytotoxicity Studies
3.9. ADMET Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Technical Strategy for Malaria 2016–2030. 2015. Available online: https://books.google.com.sg/books?hl=en&lr=&id=LV40DgAAQBAJ&oi=fnd&pg=PA1&dq=Global+Technical+Strategy+for+Malaria+2016%E2%80%932030&ots=kfupEXBADf&sig=8b0jfrcDTzjU4lEA-2rJoCrYiQ0#v=onepage&q=Global%20Technical%20Strategy%20for%20Malaria%202016%E2%80%932030&f=false (accessed on 13 November 2018).
- Cui, L.; Mharakurwa, S.; Ndiaye, D.; Rathod, P.K.; Rosenthal, P.J. Antimalarial Drug Resistance: Literature Review and Activities and Findings of the ICEMR Network. Am. J. Trop. Med. Hyg. 2015, 93, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olliaro, P. Drug Resistance Hampers Our Capacity to Roll Back Malaria. Clin. Infect. Dis. 2005, 41, S247–S257. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.; Nuzillard, J.-M.; Renault, J.-H. Dereplication Strategies in Natural Product Research: How Many Tools and Methodologies behind the Same Concept? Phytochem. Rev. 2017, 16, 55–95. [Google Scholar] [CrossRef]
- Beutler, J.A.; Alvarado, A.B.; Schaufelberger, D.E.; Andrews, P.; McCloud, T.G. Dereplication of Phorbol Bioactives: Lyngbya majuscula and Croton cuneatus. J. Nat. Prod. 1990, 53, 867–874. [Google Scholar] [CrossRef]
- Garg, N.; Kapono, C.A.; Lim, Y.W.; Koyama, N.; Vermeij, M.J.A.; Conrad, D.; Rohwer, F.; Dorrestein, P.C. Mass Spectral Similarity for Untargeted Metabolomics Data Analysis of Complex Mixtures. Int. J. Mass Spectrom. 2015, 377, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Wan, K.X.; Vidavsky, I.; Gross, M.L. Comparing Similar Spectra: From Similarity Index to Spectral Contrast Angle. J. Am. Soc. Mass Spectrom. 2002, 13, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular Networking as a Dereplication Strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef] [Green Version]
- Hook, D.J.; Pack, E.J.; Yacobucci, J.J.; Guss, J. Approaches to Automating the Dereplication of Bioactive Natural Products—The Key Step in High Throughput Screening of Bioactive Materials from Natural Sources. J. Biomol. Screen. 1997, 2, 145–152. [Google Scholar] [CrossRef]
- Potterat, O.; Hamburger, M. Concepts and Technologies for Tracking Bioactive Compounds in Natural Product Extracts: Generation of Libraries, and Hyphenation of Analytical Processes with Bioassays. Nat. Prod. Rep. 2013, 30, 546–564. [Google Scholar] [CrossRef]
- Pashynska, V.A.; Van den Heuvel, H.; Claeys, M.; Kosevich, M.V. Characterization of Noncovalent Complexes of Antimalarial Agents of the Artemisinin-Type and FE(III)-Heme by Electrospray Mass Spectrometry and Collisional Activation Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1181–1190. [Google Scholar] [CrossRef]
- Gligorijevic, B.; Bennett, T.; McAllister, R.; Urbach, J.S.; Roepe, P.D. Spinning Disk Confocal Microscopy of Live, Intraerythrocytic Malarial Parasites. 2. Altered Vacuolar Volume Regulation in Drug Resistant Malaria. Biochemistry 2006, 45, 12411–12423. [Google Scholar] [CrossRef] [PubMed]
- Hayward, R.; Saliba, K.J.; Kirk, K. The PH of the Digestive Vacuole of Plasmodium falciparum Is Not Associated with Chloroquine Resistance. J. Cell Sci. 2006, 119, 1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, J.; Rohrbach, P.; Dalton, J.P. The Malaria Digestive Vacuole. Front. Biosci. Sch. Ed. 2012, 4, 1424–1448. [Google Scholar]
- Olliaro, P.L.; Goldberg, D.E. The Plasmodium Digestive Vacuole: Metabolic Headquarters and Choice Drug Target. Parasitol. Today 1995, 11, 294–297. [Google Scholar] [CrossRef]
- Oliveira, M.F.; d’Avila, J.C.P.; Tempone, A.J.; Correâ Soares, J.B.R.; Rumjanek, F.D.; Ferreira-Pereira, A.; Ferreira, S.T.; Oliveira, P.L. Inhibition of Heme Aggregation by Chloroquine Reduces Schistosoma mansoni Infection. J. Infect. Dis. 2004, 190, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Tekwani, B.L.; Walker, L.A. Targeting the Hemozoin Synthesis Pathway for New Antimalarial Drug Discovery: Technologies for in Vitro Beta-Hematin Formation Assay. Comb. Chem. High Throughput Screen. 2005, 8, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Novelli, J.; Foster, J.; Vaisvila, R.; Conway, L.; Ingram, J.; Ganatra, M.; Rao, A.U.; Hamza, I.; Slatko, B. The Heme Biosynthetic Pathway of the Obligate Wolbachia Endosymbiont of Brugia Malayi as a Potential Anti-Filarial Drug Target. PLoS Negl. Trop. Dis. 2009, 3, e475. [Google Scholar] [CrossRef] [Green Version]
- Buller, R.; Peterson, M.L.; Almarsson, Ö.; Leiserowitz, L. Quinoline Binding Site on Malaria Pigment Crystal: A Rational Pathway for Antimalaria Drug Design. Cryst. Growth Des. 2002, 2, 553–562. [Google Scholar] [CrossRef]
- Kappe, S.H.I.; Vaughan, A.M.; Boddey, J.A.; Cowman, A.F. That Was Then but This Is Now: Malaria Research in the Time of an Eradication Agenda. Science 2010, 328, 862. [Google Scholar] [CrossRef]
- Adams, P.A.; Berman, P.A.M.; Egan, T.J.; Marsh, P.J.; Silver, J. The Iron Environment in Heme and Heme-Antimalarial Complexes of Pharmacological Interest. J. Inorg. Biochem. 1996, 63, 69–77. [Google Scholar] [CrossRef]
- Parapini, S.; Basilico, N.; Pasini, E.; Egan, T.J.; Olliaro, P.; Taramelli, D.; Monti, D. Standardization of the Physicochemical Parameters to Assess in Vitro the β-Hematin Inhibitory Activity of Antimalarial Drugs. Exp. Parasitol. 2000, 96, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Basilico, N.; Pagani, E.; Monti, D.; Olliaro, P.; Taramelli, D. A Microtitre-Based Method for Measuring the Haem Polymerization Inhibitory Activity (HPIA) of Antimalarial Drugs. J. Antimicrob. Chemother. 1998, 42, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, A.; Coppel, Y.; Meunier, B. NMR Characterization of Covalent Adducts Obtained by Alkylation of Heme with the Antimalarial Drug Artemisinin. Protag. Chem. Helmut Sigel 2002, 339, 488–496. [Google Scholar] [CrossRef]
- Muñoz-Durango, K.; Maciuk, A.; Harfouche, A.; Torijano-Gutiérrez, S.; Jullian, J.-C.; Quintin, J.; Spelman, K. Detection, Characterization, and Screening of Heme-Binding Molecules by Mass Spectrometry for Malaria Drug Discovery. Anal. Chem. 2012, 84, 3324–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, S.; Dali-Yahia, K.; Vásquez-Ocmín, P.; Grougnet, R.; Grellier, P.; Michel, S.; Maciuk, A.; Boutefnouchet, S. Heme-Binding Activity of Methoxyflavones from Pentzia Monodiana Maire (Asteraceae). Fitoterapia 2017, 118, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Hamdan, S.M.; Dixon, N.E.; Sheil, M.M.; Beck, J.L. Application of Electrospray Ionization Mass Spectrometry to Study the Hydrophobic Interaction between the ɛ and θ Subunits of DNA Polymerase III. Protein Sci. Publ. Protein Soc. 2004, 13, 2878–2887. [Google Scholar] [CrossRef] [Green Version]
- Veenstra, T.D. Electrospray Ionization Mass Spectrometry in the Study of Biomolecular Non-Covalent Interactions. Biophys. Chem. 1999, 79, 63–79. [Google Scholar] [CrossRef]
- Wright, A.D.; Wang, H.; Gurrath, M.; König, G.M.; Kocak, G.; Neumann, G.; Loria, P.; Foley, M.; Tilley, L. Inhibition of Heme Detoxification Processes Underlies the Antimalarial Activity of Terpene Isonitrile Compounds from Marine Sponges. J. Med. Chem. 2001, 44, 873–885. [Google Scholar] [CrossRef]
- Krieg, R.; Jortzik, E.; Goetz, A.-A.; Blandin, S.; Wittlin, S.; Elhabiri, M.; Rahbari, M.; Nuryyeva, S.; Voigt, K.; Dahse, H.-M.; et al. Arylmethylamino Steroids as Antiparasitic Agents. Nat. Commun. 2017, 8, 14478. [Google Scholar] [CrossRef] [Green Version]
- Vásquez-Ocmín, P.; Cojean, S.; Rengifo, E.; Suyyagh-Albouz, S.; Amasifuen Guerra, C.A.; Pomel, S.; Cabanillas, B.; Mejía, K.; Loiseau, P.M.; Figadère, B.; et al. Antiprotozoal Activity of Medicinal Plants Used by Iquitos-Nauta Road Communities in Loreto (Peru). J. Ethnopharmacol. 2018, 210, 372–385. [Google Scholar] [CrossRef]
- Vásquez-Ocmín, P.G.; Gadea, A.; Cojean, S.; Marti, G.; Pomel, S.; Van Baelen, A.-C.; Ruiz-Vásquez, L.; Ruiz Mesia, W.; Figadère, B.; Ruiz Mesia, L.; et al. Metabolomic Approach of the Antiprotozoal Activity of Medicinal Piper Species Used in Peruvian Amazon. J. Ethnopharmacol. 2021, 264, 113262. [Google Scholar] [CrossRef] [PubMed]
- Olafson, K.N.; Rimer, J.D.; Vekilov, P.G. Growth of Large Hematin Crystals in Biomimetic Solutions. Cryst. Growth Des. 2014, 14, 2123–2127. [Google Scholar] [CrossRef] [PubMed]
- Ketchum, M.A.; Olafson, K.N.; Petrova, E.V.; Rimer, J.D.; Vekilov, P.G. Hematin Crystallization from Aqueous and Organic Solvents. J. Chem. Phys. 2013, 139, 121911. [Google Scholar] [CrossRef] [PubMed]
- Vekilov, P.G.; Rimer, J.D.; Olafson, K.N.; Ketchum, M.A. Lipid or Aqueous Medium for Hematin Crystallization? CrystEngComm 2015, 17, 7790–7800. [Google Scholar] [CrossRef]
- Stiebler, R.; Hoang, A.N.; Egan, T.J.; Wright, D.W.; Oliveira, M.F. Increase on the Initial Soluble Heme Levels in Acidic Conditions Is an Important Mechanism for Spontaneous Heme Crystallization in Vitro. PLoS ONE 2010, 5, e12694. [Google Scholar] [CrossRef] [Green Version]
- Kapishnikov, S.; Berthing, T.; Hviid, L.; Dierolf, M.; Menzel, A.; Pfeiffer, F.; Als-Nielsen, J.; Leiserowitz, L. Aligned Hemozoin Crystals in Curved Clusters in Malarial Red Blood Cells Revealed by Nanoprobe X-Ray Fe Fluorescence and Diffraction. Proc. Natl. Acad. Sci. USA 2012, 109, 11184–11187. [Google Scholar] [CrossRef] [Green Version]
- Kapishnikov, S.; Weiner, A.; Shimoni, E.; Guttmann, P.; Schneider, G.; Dahan-Pasternak, N.; Dzikowski, R.; Leiserowitz, L.; Elbaum, M. Oriented Nucleation of Hemozoin at the Digestive Vacuole Membrane in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2012, 109, 11188–11193. [Google Scholar] [CrossRef] [Green Version]
- Egan, T.J. Haemozoin Formation. Mol. Biochem. Parasitol. 2008, 157, 127–136. [Google Scholar] [CrossRef]
- Egan, T.J. Recent Advances in Understanding the Mechanism of Hemozoin (Malaria Pigment) Formation. J. Inorg. Biochem. 2008, 102, 1288–1299. [Google Scholar] [CrossRef]
- Sangster, J. Octanol-Water Partition Coefficients: Fundamentals and Physical Chemistry; Wiley: Chichester, UK, 1997; Volume 2. [Google Scholar]
- Olafson, K.N.; Ketchum, M.A.; Rimer, J.D.; Vekilov, P.G. Molecular Mechanisms of Hematin Crystallization from Organic Solvent. Cryst. Growth Des. 2015, 15, 5535–5542. [Google Scholar] [CrossRef]
- Huy, N.T.; Uyen, D.T.; Maeda, A.; Trang, D.T.X.; Oida, T.; Harada, S.; Kamei, K. Simple Colorimetric Inhibition Assay of Heme Crystallization for High-Throughput Screening of Antimalarial Compounds. Antimicrob. Agents Chemother. 2007, 51, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Charkin, O.P.; Klimenko, N.M.; Nguyen, P.T.; Charkin, D.O.; Mebel, A.M.; Lin, S.H.; Wang, Y.-S.; Wei, S.-C.; Chang, H.-C. Fragmentation of Heme and Hemin+ with Sequential Loss of Carboxymethyl Groups: A DFT and Mass-Spectrometry Study. Chem. Phys. Lett. 2005, 415, 362–369. [Google Scholar] [CrossRef]
- Ateacha, D.N.; Koch, U.; Engelhard, C. Direct Analysis of Alkaloids in Natural Cinchona Bark and Commercial Extracts Using Time-of-Flight Secondary Ion Mass Spectrometry. Anal. Methods 2018, 10, 950–958. [Google Scholar] [CrossRef]
- Shibuya, H.; Kitamura, C.; Maehara, S.; Nagahata, M.; Winarno, H.; Simanjuntak, P.; Kim, H.-S.; Wataya, Y.; Ohashi, K. Transformation of Cinchona Alkaloids into 1-N-Oxide Derivatives by Endophytic Xylaria sp. Isolated from Cinchona pubescens. Chem. Pharm. Bull. 2003, 51, 71–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissbuch, I.; Leiserowitz, L. Interplay between Malaria, Crystalline Hemozoin Formation, and Antimalarial Drug Action and Design. Chem. Rev. 2008, 108, 4899–4914. [Google Scholar] [CrossRef] [PubMed]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Wang, M.; Leber, C.A.; Nothias, L.-F.; Reher, R.; Kang, K.B.; van der Hooft, J.J.J.; Dorrestein, P.C.; Gerwick, W.H.; Cottrell, G.W. NPClassifier: A Deep Neural Network-Based Structural Classification Tool for Natural Products. J. Nat. Prod. 2021, 84, 2795–2807. [Google Scholar] [CrossRef]
- Harborne, J.B.; Baxter, H. The Handbook of Natural Flavonoids. Volume 1 and Volume 2; John Wiley and Sons: Hoboken, NJ, USA, 1999; ISBN 978-0-471-95893-2. [Google Scholar]
- Díaz, P.; Arias, T.; Joseph-Nathan, P. A Chromene an Isoprenylated Methyl Hydroxybenzoate and a C-Methyl Flavanone from the Bark of Piper hostmannianum. Phytochemistry 1987, 26, 809–811. [Google Scholar] [CrossRef]
- Vasquez-Ocmín, P.; Haddad, M.; Gadea, A.; Jullian, V.; Castillo, D.; Paloque, L.; Cerapio, J.P.; Bourdy, G.; Sauvain, M. A New Phthalide Derivative from Peperomia nivalis. Nat. Prod. Res. 2017, 31, 138–142. [Google Scholar] [CrossRef]
- Hua, S.; Liu, J.; Zhang, Y.; Li, J.; Zhang, X.; Dong, L.; Zhao, Y.; Fu, X. Piperine as a Neuroprotective Functional Component in Rats with Cerebral Ischemic Injury. Food Sci. Nutr. 2019, 7, 3443–3451. [Google Scholar] [CrossRef] [Green Version]
- Mazlan, R.N.A.R.; Rukayadi, Y.; Maulidiani, M.; Ismail, I.S. Solvent Extraction and Identification of Active Anticariogenic Metabolites in Piper Cubeba L. through 1H-NMR-Based Metabolomics Approach. Molecules 2018, 23, 1730. [Google Scholar] [CrossRef] [PubMed]
- Campelo, Y.; Ombredane, A.; Vasconcelos, A.G.; Albuquerque, L.; Moreira, D.C.; Plácido, A.; Rocha, J.; Hilarion Fokoue, H.; Yamaguchi, L.; Mafud, A.; et al. Structure Activity Relationship of Piplartine and Synthetic Analogues against Schistosoma mansoni and Cytotoxicity to Mammalian Cells. Int. J. Mol. Sci. 2018, 19, 1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orjala, J.; Wright, A.D.; Rali, T.; Sticher, O. Aduncamide, a Cytotoxic and Antibacterial b-Phenylethylamine-Derived Amide from Piper aduncum. Nat. Prod. Lett. 1993, 2, 231–236. [Google Scholar] [CrossRef]
- Braga, F.G.; Bouzada, M.L.M.; Fabri, R.L.; de O. Matos, M.; Moreira, F.O.; Scio, E.; Coimbra, E.S. Antileishmanial and Antifungal Activity of Plants Used in Traditional Medicine in Brazil. J. Ethnopharmacol. 2007, 111, 396–402. [Google Scholar] [CrossRef]
- Parmar, V.S.; Jain, S.C.; Bisht, K.S.; Jain, R.; Taneja, P.; Jha, A.; Tyagi, O.D.; Prasad, A.K.; Wengel, J.; Olsen, C.E.; et al. Phytochemistry of the Genus Piper. Int. J. Plant Biochem. Mol. Biol. 1997, 46, 597–673. [Google Scholar] [CrossRef]
- Berthod, A. Comprehensive Analytical Chemistry Volume XXXVIII: Countercurrent Chromatography—The Support-Free Liquid Stationary Phase; Elsevier: Amsterdam, The Netherlands, 2002; Volume 38. [Google Scholar]
- Mi-Ichi, F.; Miyadera, H.; Kobayashi, T.; Takamiya, S.; Waki, S.; Iwata, S.; Shibata, S.; Kita, K. Parasite Mitochondria as a Target of Chemotherapy: Inhibitory Effect of Licochalcone A on the Plasmodium falciparum Respiratory Chain. Ann. N. Y. Acad. Sci. 2005, 1056, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Rao Ravu, R.; Tekwani, B.L.; Li, W.; Liu, W.-B.; Jacob, M.R.; Khan, S.I.; Cai, X.; Peng, C.-Y.; Khan, I.A.; et al. Biological Evaluation of Phytoconstituents from Polygonum hydropiper. Nat. Prod. Res. 2017, 31, 2053–2057. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Li, Y.; Liu, J.; Hu, Z.; Chen, X. Specific Interaction of Chalcone-Protein: Cardamonin Binding Site II on the Human Serum Albumin Molecule. Biopolymers 2005, 79, 48–57. [Google Scholar] [CrossRef]
- Gomes, M.N.; Muratov, E.N.; Pereira, M.; Peixoto, J.C.; Rosseto, L.P.; Cravo, P.V.L.; Andrade, C.H.; Neves, B.J. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Ram, V.J.; Saxena, A.S.; Srivastava, S.; Chandra, S. Oxygenated Chalcones and Bischalcones as Potential Antimalarial Agents. Bioorg. Med. Chem. Lett. 2000, 10, 2159–2161. [Google Scholar] [CrossRef]
- Gan, F.-F.; Kaminska, K.K.; Yang, H.; Liew, C.-Y.; Leow, P.-C.; So, C.-L.; Tu, L.N.; Roy, A.; Yap, C.-W.; Kang, T.-S.; et al. Identification of Michael Acceptor-Centric Pharmacophores with Substituents That Yield Strong Thioredoxin Reductase Inhibitory Character Correlated to Antiproliferative Activity. Antioxid. Redox Signal. 2013, 19, 1149–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Mohanakrishnan, D.; Sharma, U.K.; Kumar, R.; Richa; Sinha, A.K.; Sahal, D. Design, Economical Synthesis and Antiplasmodial Evaluation of Vanillin Derived Allylated Chalcones and Their Marked Synergism with Artemisinin against Chloroquine Resistant Strains of Plasmodium falciparum. Eur. J. Med. Chem. 2014, 79, 350–368. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent Developments in Biological Activities of Chalcones: A Mini Review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef]
- Lobo, L.; Cabral, L.I.L.; Sena, M.I.; Guerreiro, B.; Rodrigues, A.S.; de Andrade-Neto, V.F.; Cristiano, M.L.S.; Nogueira, F. New Endoperoxides Highly Active in Vivo and in Vitro against Artemisinin-Resistant Plasmodium falciparum. Malar. J. 2018, 17, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olafson, K.N.; Ketchum, M.A.; Rimer, J.D.; Vekilov, P.G. Mechanisms of Hematin Crystallization and Inhibition by the Antimalarial Drug Chloroquine. Proc. Natl. Acad. Sci. USA 2015, 112, 4946–4951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-Independent MS/MS Deconvolution for Comprehensive Metabolome Analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Fraisier-Vannier, O.; Chervin, J.; Cabanac, G.; Puech, V.; Fournier, S.; Durand, V.; Amiel, A.; André, O.; Benamar, O.A.; Dumas, B.; et al. MS-CleanR: A Feature-Filtering Workflow for Untargeted LC–MS Based Metabolomics. Anal. Chem. 2020, 92, 9971–9981. [Google Scholar] [CrossRef]
- Vásquez-Ocmín, P.G.; Marti, G.; Bonhomme, M.; Mathis, F.; Fournier, S.; Bertani, S.; Maciuk, A. Cannabinoids vs. Whole Metabolome: Relevance of Cannabinomics in Analyzing Cannabis Varieties. Anal. Chim. Acta 2021, 1184, 339020. [Google Scholar] [CrossRef]
- Vásquez-Ocmín, P.G.; Marti, G.; Gadea, A.; Cabanac, G.; Vásquez-Briones, J.A.; Casavilca-Zambrano, S.; Ponts, N.; Jargeat, P.; Haddad, M.; Bertani, S. Metabotyping of Andean Pseudocereals and Characterization of Emerging Mycotoxins. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen Rearrangement Rules: Computational MS/MS Fragmentation and Structure Elucidation Using MS-FINDER Foftware. Anal. Chem. 2016, 88, 7946–7958. [Google Scholar] [CrossRef] [PubMed]
- Trager, W.; Jensen, J.B. Human Malaria Parasites in Continuous Culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum Erythrocytic Stages in Culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Komlaga, G.; Cojean, S.; Dickson, R.A.; Beniddir, M.A.; Suyyagh-Albouz, S.; Mensah, M.L.K.; Agyare, C.; Champy, P.; Loiseau, P.M. Antiplasmodial Activity of Selected Medicinal Plants Used to Treat Malaria in Ghana. Parasitol. Res. 2016, 115, 3185–3195. [Google Scholar] [CrossRef]
- Bounaadja, L.; Schmitt, M.; Albrecht, S.; Mouray, E.; Tarnus, C.; Florent, I. Selective Inhibition of PfA-M1, over PfA-M17, by an Amino-Benzosuberone Derivative Blocks Malaria Parasites Development in Vitro and in Vivo. Malar. J. 2017, 16, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Li, W.; Liu, G.; Tang, Y. In Silico ADMET Prediction: Recent Advances, Current Challenges and Future Trends. Curr. Top. Med. Chem. 2013, 13, 1273–1289. [Google Scholar] [CrossRef]
- Adityachaudhury, N.; Das, A.K.; Choudhury, A.; Daskanungo, P.L. Aurentiacin, a New Chalcone from Didymocarpus aurentiaca. Phytochemistry 1976, 15, 229–230. [Google Scholar] [CrossRef]
- Resurreccion-Magno, M.H.C.; Villaseñor, I.M.; Harada, N.; Monde, K. Antihyperglycaemic Flavonoids from Syzygium samarangense (Blume) Merr. and Perry. Phytother. Res. 2005, 19, 246–251. [Google Scholar] [CrossRef]
- Wollenweber, E.; Dietz, V.H.; Schilling, G.; Favre-Bonvin, J.; Smith, D.M. Flavonoids from Chemotypes of the Goldback Fern, Pityrogramma triangularis. Phytochemistry 1985, 24, 965–971. [Google Scholar] [CrossRef]
- Basnet, P.; Kadota, S.; Shimizu, M.; Xu, H.-X.; Namba, T. 2’-Hydroxymatteucinol, a New C-Methyl Flavanone Derivative from Matteccia orientalis; Potent Hypoglycemic Activity in Streptozotocin (STZ)-Induced Diabetic Rat. Chem. Pharm. Bull. 1993, 41, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Itokawa, H.; Morita, M.; Mihashi, S. Phenolic Compounds from the Rhizomes of Alpinia speciosa. Phytochemistry 1981, 20, 2503–2506. [Google Scholar] [CrossRef]
- Bick, I.; Brown, R.; Hillis, W. Three Flavanones from Leaves of Eucalyptus sieberi. Aust. J. Chem. 1972, 25, 449–451. [Google Scholar] [CrossRef] [Green Version]
- Mayer, R. A β-Hydroxychalcone from Leptospermum scoparium. Planta Med. 2007, 59, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.M.; Li, S.H.; Peng, L.Y.; Lin, Z.W.; Rao, G.X.; Sun, H.D. Constituents from Limonia Crenulata. J. Asian Nat. Prod. Res. 2001, 3, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Chakrabarty, M.; Kundu, A. Constituents of Pleiospermium alatum: Alatamide and N-Benzoyltyramine Methyl Ether. Aust. J. Chem. 1975, 28, 457–460. [Google Scholar] [CrossRef]
- Di Iorio, N.; Filippini, G.; Mazzanti, A.; Righi, P.; Bencivenni, G. Controlling the C(Sp3)–C(Sp2) Axial Conformation in the Enantioselective Friedel–Crafts-Type Alkylation of β-Naphthols with Inden-1-Ones. Org. Lett. 2017, 19, 6692–6695. [Google Scholar] [CrossRef]
- Szmant, H.H.; Nanjundiah, R. Thiol-Olefin Cooxidation Reaction. 6. A New Convenient Route to 1-Substituted Indenes. Indenone as Dienophile in Diels-Alder Reactions. J. Org. Chem. 1978, 43, 1835–1837. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | RT | m/z [M + H]+ | Formula | Compound Annotated | cLogP | Level Annotation ** | Classyfire (Superclass/ Class) |
|---|---|---|---|---|---|---|---|
| 1 | 26.51 | 299.123 | C18H18O4 | aurentiacin | 4.47 | generic | flavonoid/chalcone |
| 2 | 23.02 | 285.110 | C17H16O4 | 2′, 4′-dihydroxy-6′-methoxy-3′-methylchalcone | 4.08 | generic | flavonoid/chalcone |
| 3 | 21.61 | 271.094 | C16H14O4 | cardamomin | 3.62 | genus | flavonoid/chalcone |
| 4 | 25.25 | 285.111 | C17H16O4 | strobopinin 7-methyl ether | 4.57 | generic | flavonoid/flavanones |
| 5 | 25.98 | 299.127 | C18H18O4 | 5-hydroxy-7-methoxy-6,8-dimethyl flavanone | 5.03 | genus | flavonoid/flavanones |
| 6 | 23.49 | 285.110 | C17H16O4 | desmethoxymatteucinol | 4.85 | generic | flavonoid/flavanones |
| 7 | 16.75 | 271.495 | C16H14O4 | alpinetin | 3.71 | genus | flavonoid/flavanones |
| 8 | 19.74 | 285.109 | C17H16O4 | pinocembrin | 3.40 | generic | flavonoid/flavanones |
| 9 | 21.48 | 299.127 | C18H18O4 | dimethyl cryptostrobin | 3.86 | generic | flavonoid/flavanones |
| 10 | 17.80 | 285.111 | C17H16O4 | NA | 2.44 | NA | Phenylpropanoi/cinnamic acid and derivative |
| 11 | 18.23 | 256.133 | C16H17NO2 | N-Benzoyltyrarnine methyl ether | 2.79 | generic | alkaloid/alkylamide |
| 12 * | 16.01 | 131.048 | C9H6O | 1H-inden-1-one | 2.21 | generic | naphthalenes/naphthoquinone (indanone) |
| Cpd | m/z [M + H]+ | Adduct m/z Expected with heme-Fe (III) | 3D7 Sensitive Strain IC50 (µM) | AB943 Primary Human Fibroblast CC50 (µM) | Selectivity Index CC50/IC50 | DV50 (eV) |
|---|---|---|---|---|---|---|
| 1 | 299 | 915 | 2.25 | 68.5 | 30.4 | 5.15 |
| 2 | 285 | 900 | 51 | >100 | 1.9 | 5.93 |
| 3 | 271 | 886 | 5.5 | 58 | 10.5 | 5.60 |
| 4 | 285 | 900 | 60 | >100 | <1 | NA |
| 5 | 299 | 915 | 33.2 | 23 | <1 | NA |
| 6 | 285 | 900 | 71 | >100 | 1.4 | NA |
| 7 | 271 | 886 | 72 | >100 | 1.4 | 8.40 |
| 8 | 285 | 900 | 78 | >100 | 1.3 | 8.68 |
| 9 | 299 | 915 | 85 | >100 | 1.2 | 7.68 |
| 10 | 285 | 900 | >100 | >100 | 1 | NA |
| 11 | 256 | 871 | >100 | >100 | 1 | 9.45 |
| 12 | 131 | 746 * | >100 | >100 | 1 | NA |
| Crude extract | 1.36 ± 0.06 µg/mL | 37.5 µg/mL | 27.6 | NT | ||
| CQ | 320 | 935 | 0.04 ± 3.25 | NT | NT | 14.27 |
| ART | 283 | 898 (heme-Fe(II) | 0.004 ± 0.1 [69] | NT | NT | 15.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vásquez-Ocmín, P.G.; Gallard, J.-F.; Van Baelen, A.-C.; Leblanc, K.; Cojean, S.; Mouray, E.; Grellier, P.; Guerra, C.A.A.; Beniddir, M.A.; Evanno, L.; et al. Biodereplication of Antiplasmodial Extracts: Application of the Amazonian Medicinal Plant Piper coruscans Kunth. Molecules 2022, 27, 7638. https://doi.org/10.3390/molecules27217638
Vásquez-Ocmín PG, Gallard J-F, Van Baelen A-C, Leblanc K, Cojean S, Mouray E, Grellier P, Guerra CAA, Beniddir MA, Evanno L, et al. Biodereplication of Antiplasmodial Extracts: Application of the Amazonian Medicinal Plant Piper coruscans Kunth. Molecules. 2022; 27(21):7638. https://doi.org/10.3390/molecules27217638
Chicago/Turabian StyleVásquez-Ocmín, Pedro G., Jean-François Gallard, Anne-Cécile Van Baelen, Karine Leblanc, Sandrine Cojean, Elisabeth Mouray, Philippe Grellier, Carlos A. Amasifuén Guerra, Mehdi A. Beniddir, Laurent Evanno, and et al. 2022. "Biodereplication of Antiplasmodial Extracts: Application of the Amazonian Medicinal Plant Piper coruscans Kunth" Molecules 27, no. 21: 7638. https://doi.org/10.3390/molecules27217638