
Synthesis of Functionalized Thiophene Based Pyrazole Amides via Various Catalytic Approaches: Structural Features through Computational Applications and Nonlinear Optical Properties


 ,
,  ,
,  and
and
Abstract
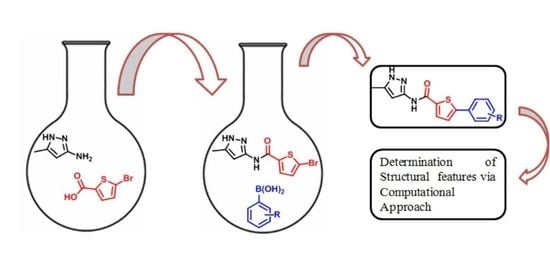
:
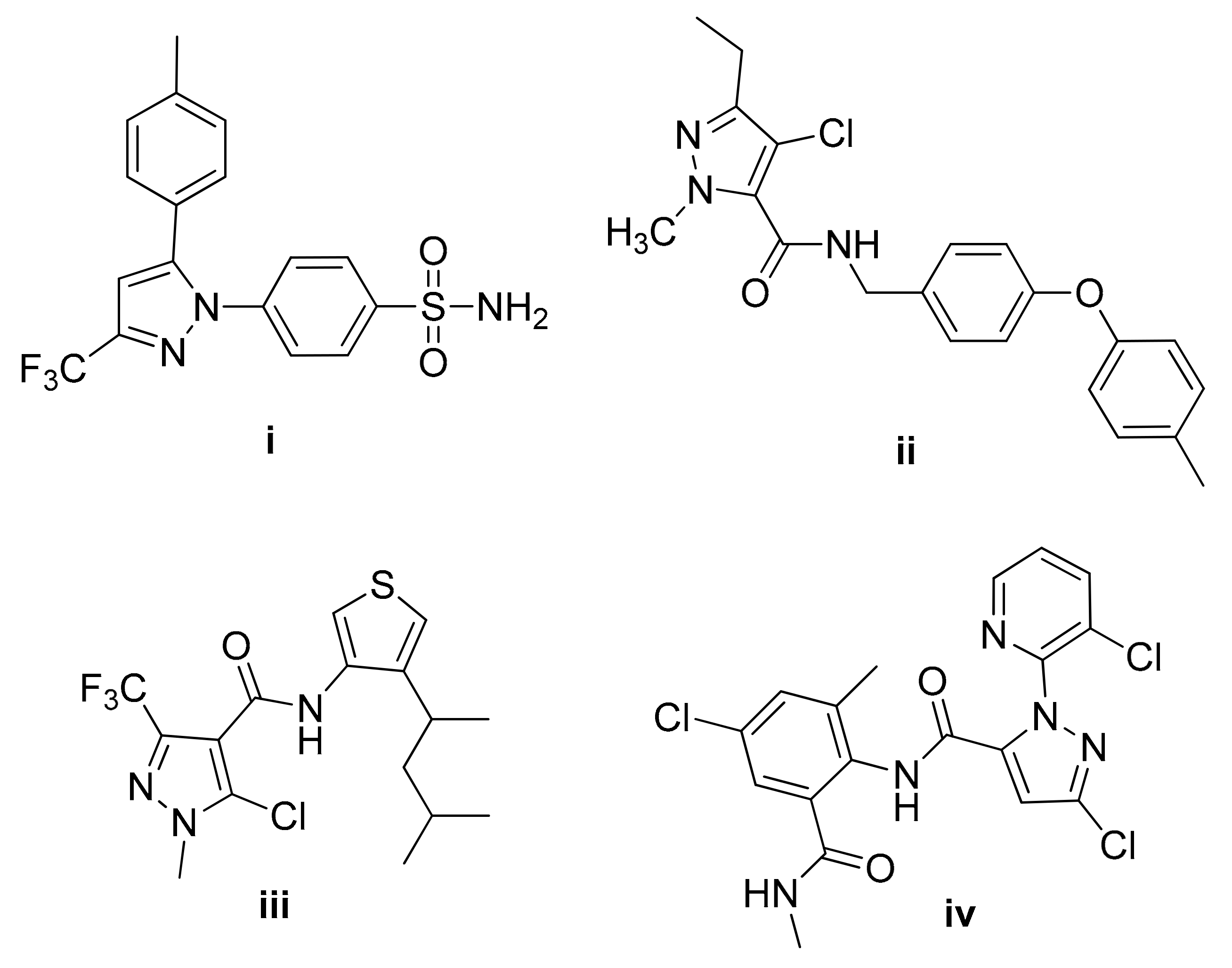
1. Introduction
2. Results and Discussion
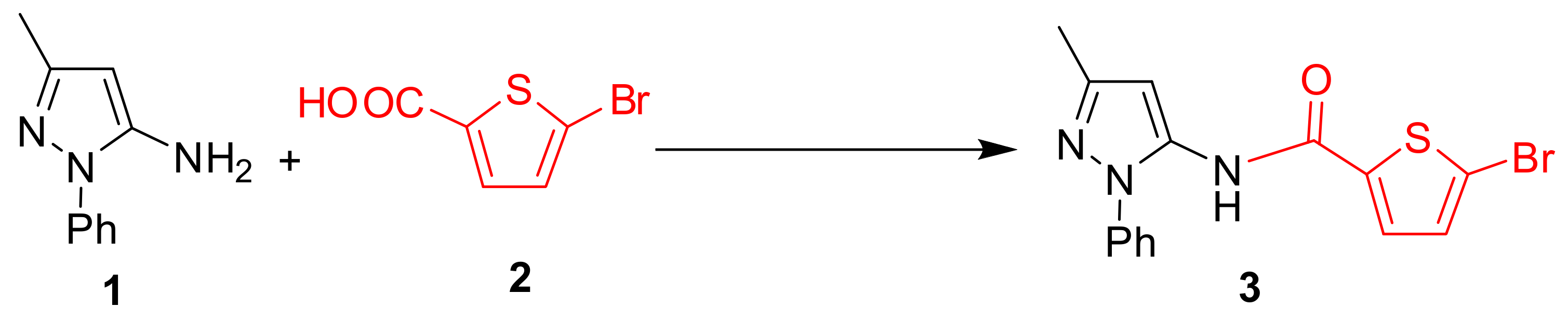
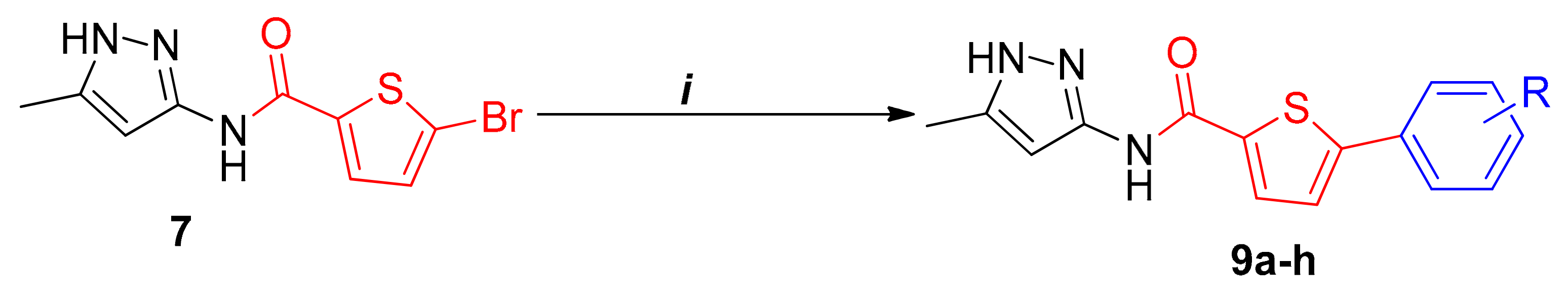
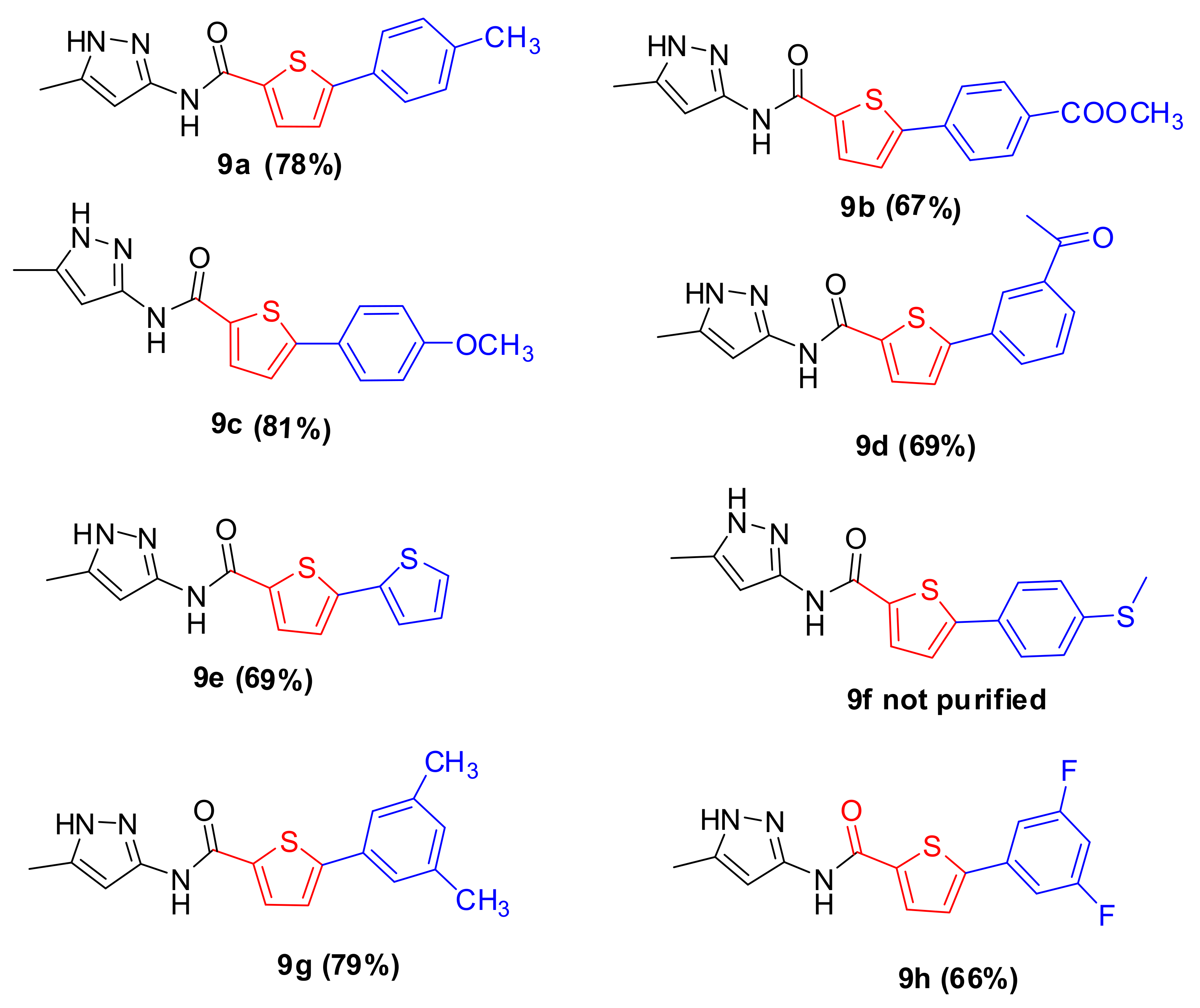
2.1. Chemistry
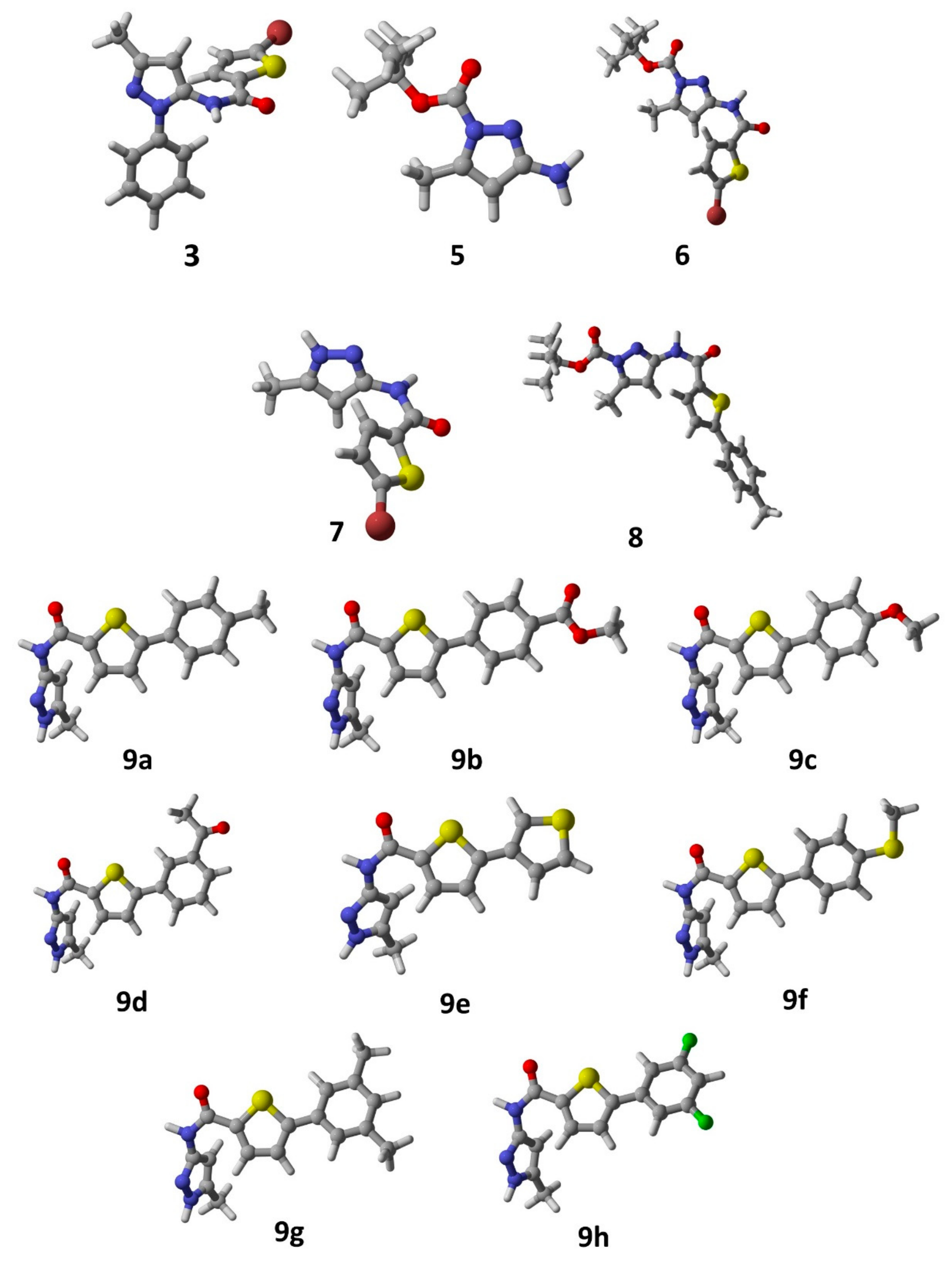
2.2. Computational Studies
2.2.1. Computation of NMR Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 9a | ||||
|---|---|---|---|---|
| Carbon No. | Carbon Type | 1H-NMR (Experimental) δ, ppm | 1H-NMR (Computed) δ, ppm | Δδ, ppm |
| 2 | C | - | - | - |
| 3 | CH | 8.06 | 7.77 | 0.29 |
| 4 | CH | 7.50 | 7.74 | 0.24 |
| 5 | C | - | - | - |
| 1′N | NH | 12.15 | 9.12 | 3.03 |
| 3′ | C | - | - | - |
| 4′ | CH | 6.36 | 6.86 | 0.50 |
| 5′ | C | - | - | - |
| 5′-Me | CH3 | 2.23 | 2.25 | 0.02 |
| 1″ | C | - | - | - |
| 2″ | CH | 7.62 | 8.14 | 0.52 |
| 3″ | CH | 7.26 | 7.66 | 0.40 |
| 4″ | C | - | - | - |
| 4″-Me | CH3 | 2.33 | 2.38 | 0.05 |
| 5″ | CH | 7.26 | 7.56 | 0.30 |
| 6″ | CH | 7.62 | 7.95 | 0.33 |
| Mean Absolute Error (MAE) = 0.24 Root Mean Square Error (RMSE) = 0.65 | ||||
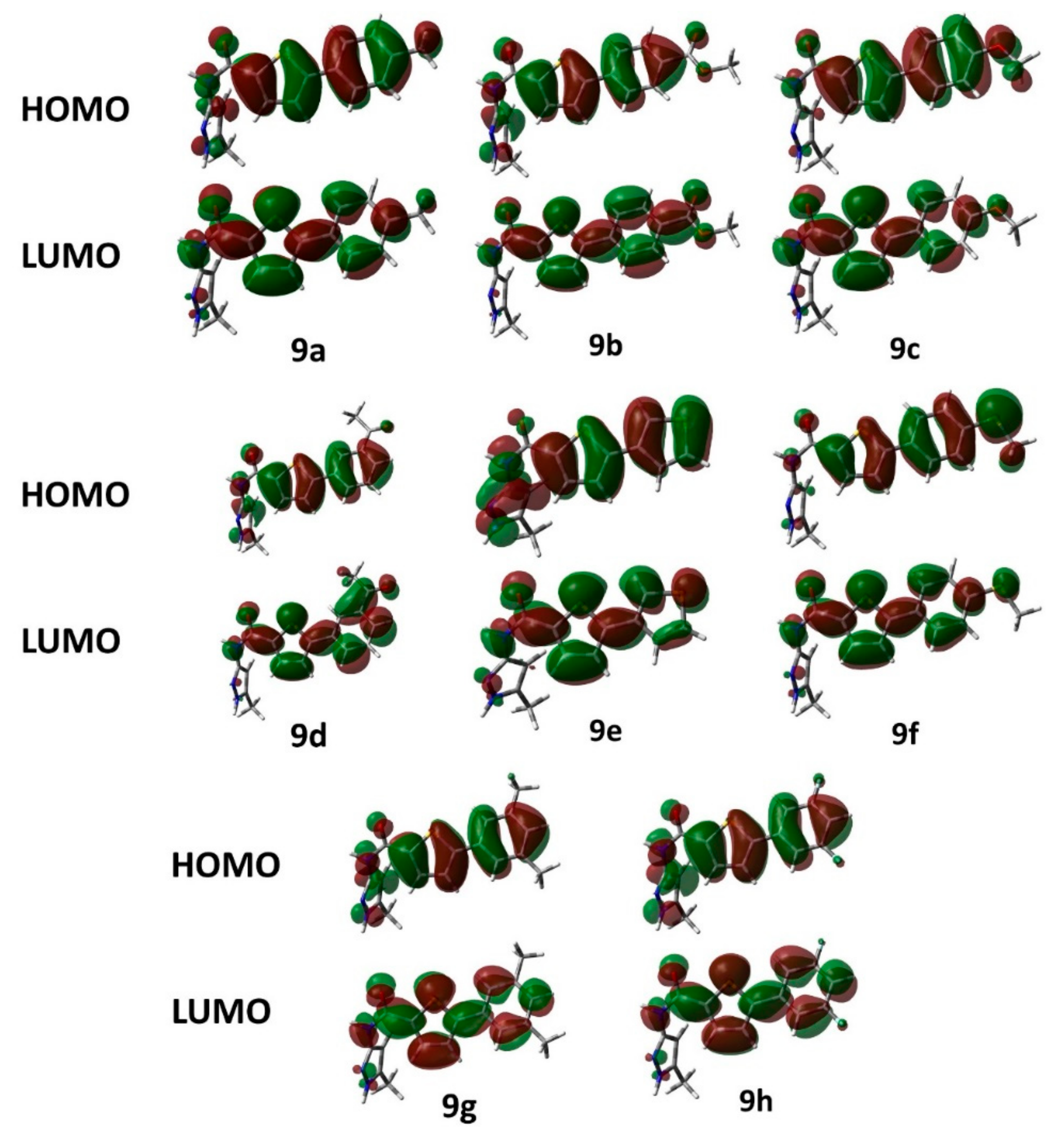
2.2.2. Frontier Molecular Orbital (FMO) Analysis and Hyperpolarizability
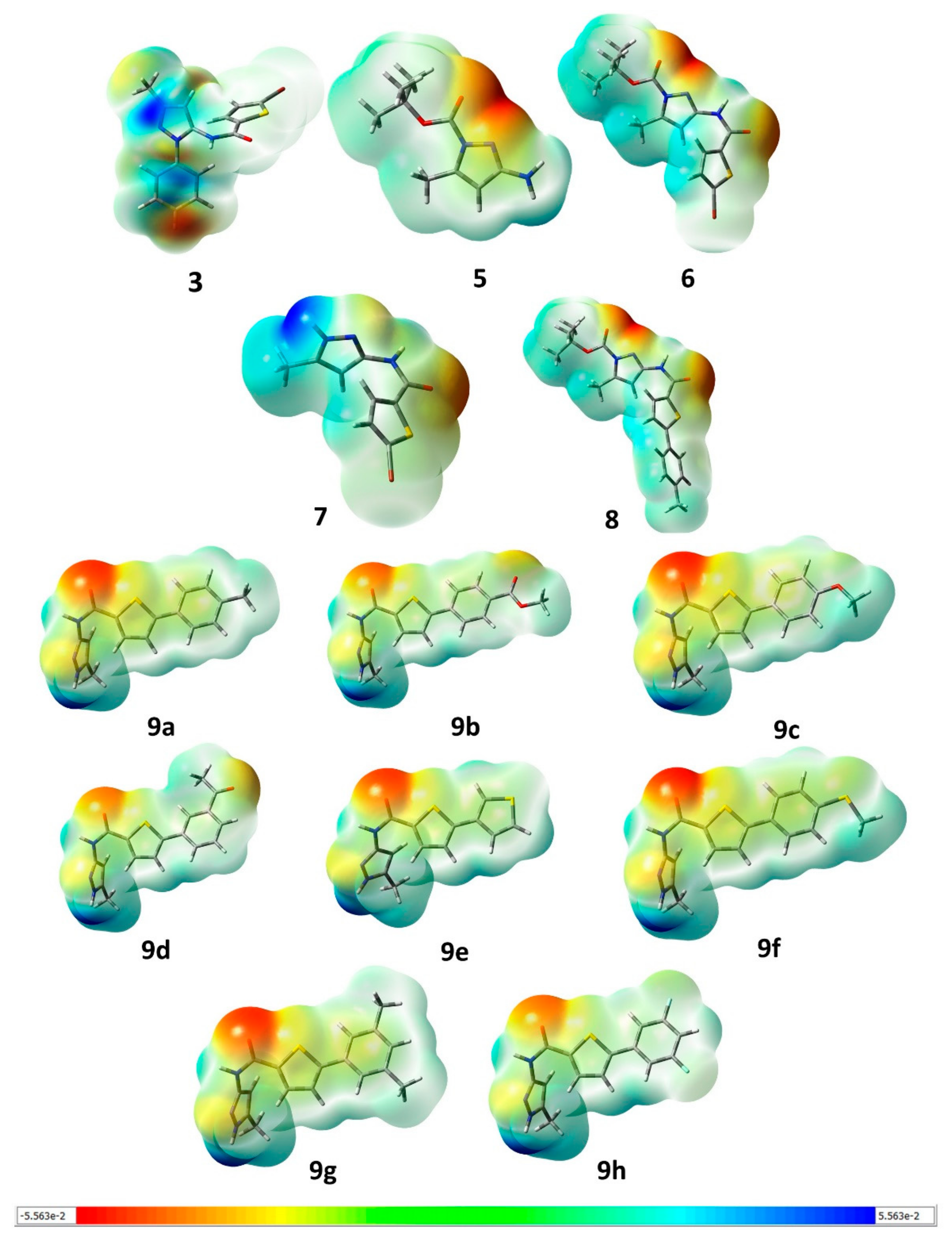
2.2.3. Molecular Electrostatic Potential (MESP)
2.2.4. UV-Vis Spectral Analysis
2.2.5. Conceptual DFT Reactivity Descriptors
3. Materials and Methods
3.1. General Information
3.2. Procedure for the Synthesis of Compound (3)
3.3. Procedure for the Synthesis of Tert-butyl 3-amino-5-methyl-1H-pyrazole-1-carboxylate (5)
3.4. Synthesis of Tertbutyl-3-(5-bromothiophene-2-carboxamido)-5-methyl-pyrazole-1-carboxylate (6)
3.5. Procedure for the Synthesis of 5-Bromo-N-(5-methyl-1H-pyrazol-3-yl)thiophene-2-carboxamide (7)
3.6. Synthesis of N-(5-Methyl-1H-pyrazol-3-yl)-5-(p-tolyl)thiophene-2-carboxamide (8b)
3.7. Arylation of 5-Bromo-N-(5-methyl-1H-pyrazol-3-yl)Thiophene-2-carboxamide (9a–h)
3.8. Computational Method
3.9. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Aly, M.F.; El-Nagger, G.M.; El-Emary, T.I.; Grigg, R.; Metwally, S.A.; Sivagnanam, S. X Y–ZH Compounds as potential 1, 3-Dipoles.: Part 41. Azomethine ylide formation from the reactions of α-amino acids and esters with alloxan (strecker degradation) and with 1-phenyl-3-methylpyrazolin-4, 5-dione. Tetrahedron 1994, 50, 895–906. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M. Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Mert, S.; Kasımoğulları, R.; Iça, T.; Çolak, F.; Altun, A.; Ok, S. Synthesis, structure–activity relationships, and in vitro antibacterial and antifungal activity evaluations of novel pyrazole carboxylic and dicarboxylic acid derivatives. Eur. J. Med. Chem. 2014, 78, 86–96. [Google Scholar] [CrossRef]
- Wu, J.; Wang, J.; Hu, D.; He, M.; Jin, L.; Song, B. Synthesis and antifungal activity of novel pyrazolecarboxamide derivatives containing a hydrazone moiety. Chem. Cent. J. 2012, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Khera, R.A.; Ali, A.; Hussain, M.; Tatar, J.; Villinger, A.; Langer, P. Synthesis of Arylated Pyrazoles by Site-Selective Suzuki-Miyaura Reactions of Tribromopyrazoles. Synlett 2010, 2010, 1923–1926. [Google Scholar]
- Rizwan, K.; Rasool, N.; Rehman, R.; Mahmood, T.; Ayub, K.; Rasheed, T.; Ahmad, G.; Malik, A.; Khan, S.A.; Akhtar, M.N.; et al. Facile synthesis of N- (4-bromophenyl)-1- (3-bromothiophen-2-yl)methanimine derivatives via Suzuki cross-coupling reaction: Their characterization and DFT studies. Chem. Cent. J. 2018, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, K.; Zubair, M.; Rasool, N.; Mahmood, T.; Ayub, K.; Alitheen, N.B.; Aziz, M.N.M.; Akhtar, M.N.; Nasim, F.-U.-H.; Bukhary, S.M.; et al. Palladium(0) catalyzed Suzuki cross-coupling reaction of 2,5-dibromo-3-methylthiophene: Selectivity, characterization, DFT studies and their biological evaluations. Chem. Cent. J. 2018, 12, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, G.; Rasool, N.; Rizwan, K.; Imran, I.; Zahoor, A.F.; Zubair, M.; Sadiq, A.; Rashid, U. Synthesis, in-vitro cholinesterase inhibition, in-vivo anticonvulsant activity and in-silico exploration of N-(4-methylpyridin-2-yl)thiophene-2-carboxamide analogs. Bioorg. Chem. 2019, 92, 103216. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Mei, Y.-J.; Cao, X.-T.; Zhang, P.-F.; Zheng, H. The synthesis of pyrazole derivatives based on glucose. Int. J. Chem. Eng. Appl. 2013, 4, 238. [Google Scholar] [CrossRef] [Green Version]
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; dos Santos, M.S.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903. [Google Scholar] [CrossRef]
- Rzepecki, P.; Wehner, M.; Molt, O.; Zadmard, R.; Harms, K.; Schrader, T. Aminopyrazole Oligomers for β-SheetStabilization of Peptides. Synthesis 2003, 2003, 1815–1826. [Google Scholar]
- Rzepecki, P.; Schrader, T. β-sheet ligands in action: KLVFF recognition by aminopyrazole hybrid receptors in water. J. Am. Chem. Soc. 2005, 127, 3016–3025. [Google Scholar] [CrossRef] [PubMed]
- Rzepecki, P.; Gallmeier, H.; Geib, N.; Cernovska, K.; König, B.; Schrader, T. New heterocyclic β-sheet ligands with peptidic recognition elements. J. Org. Chem. 2004, 69, 5168–5178. [Google Scholar] [CrossRef]
- Prashad, M.; Har, D.; Hu, B.; Kim, H.-Y.; Girgis, M.J.; Chaudhary, A.; Repic, O.; Blacklock, T.J.; Marterer, W. Process development of a large-scale synthesis of TKA731: A tachykinin receptor antagonist. Org. Process. Res. Dev. 2004, 8, 330–340. [Google Scholar] [CrossRef]
- Carmalt, C.J.; Peters, E.S.; Parkin, I.P.; Tocher, D.A. Synthesis and characterisation of titanium pyridine-and pyrimidine-thiolates and their application as precursors to titanium disulfide. Polyhedron 2007, 26, 43–48. [Google Scholar] [CrossRef]
- Kusakiewicz-Dawid, A.; Górecki, Ł.; Masiukiewicz, E.; Rzeszotarska, B. Susceptibility of Methyl 3-Amino-1 H-pyrazole-5-carboxylate to Acylation. Synth. Commun. 2009, 39, 4122–4132. [Google Scholar] [CrossRef]
- Fleckenstein, C.A.; Plenio, H. Highly efficient Suzuki–Miyaura coupling of heterocyclic substrates through rational reaction design. Chem. Eur. J. 2008, 14, 4267–4279. [Google Scholar] [CrossRef]
- Western, E.C.; Daft, J.R.; Johnson, E.M.; Gannett, P.M.; Shaughnessy, K.H. Efficient one-step Suzuki arylation of unprotected halonucleosides, using water-soluble palladium catalysts. J. Org. Chem. 2003, 68, 6767–6774. [Google Scholar] [CrossRef]
- Western, E.C.; Shaughnessy, K.H. Inhibitory effects of the guanine moiety on Suzuki couplings of unprotected halonucleosides in aqueous media. J. Org. Chem. 2005, 70, 6378–6388. [Google Scholar] [CrossRef] [PubMed]
- Čapek, P.; Pohl, R.; Hocek, M. Cross-coupling reactions of unprotected halopurine bases, nucleosides, nucleotides and nucleoside triphosphates with 4-boronophenylalanine in water. Synthesis of (purin-8-yl)-and (purin-6-yl) phenylalanines. Org. Biomol. Chem. 2006, 4, 2278–2284. [Google Scholar] [CrossRef]
- Muhammad, S.; Al-Sehemi, A.G.; Irfan, A.; Chaudhry, A.R.; Gharni, H.; AlFaify, S.; Shkir, M.; Asiri, A.M. The impact of position and number of methoxy group (s) to tune the nonlinear optical properties of chalcone derivatives: A dual substitution strategy. J. Mol. Modeling 2016, 22, 73. [Google Scholar] [CrossRef]
- Sarotti, A.M.; Pellegrinet, S.C. Application of the Multi-standard Methodology for Calculating 1H NMR Chemical Shifts. J. Org. Chem. 2012, 77, 6059–6065. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.N.; Bibi, A.; Mahmood, T.; Asiri, A.M.; Ayub, K. Synthesis, crystal structures and spectroscopic properties of triazine-based hydrazone derivatives; a comparative experimental-theoretical study. Molecules 2015, 20, 5851–5874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israr, H.; Rasool, N.; Rizwan, K.; Hashmi, M.A.; Mahmood, T.; Rashid, U.; Hussein, M.Z.; Akhtar, M.N. Synthesis and Reactivities of Triphenyl Acetamide Analogs for Potential Nonlinear Optical Material Uses. Symmetry 2019, 11, 622. [Google Scholar] [CrossRef] [Green Version]
- Leggio, A.; Bagalà, J.; Belsito, E.L.; Comandè, A.; Greco, M.; Liguori, A. Formation of amides: One-pot condensation of carboxylic acids and amines mediated by TiCl 4. Chem. Cent. J. 2017, 11, 87. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, G.; Rasool, N.; Qamar, M.U.; Alam, M.M.; Kosar, N.; Mahmood, T.; Imran, M. Facile synthesis of 4-aryl-N-(5-methyl-1H-pyrazol-3-yl) benzamides via Suzuki Miyaura reaction: Antibacterial activity against clinically isolated NDM-1-positive bacteria and their Docking Studies. Arab. J. Chem. 2021, 14, 103270. [Google Scholar] [CrossRef]
- Savjani, J.K.; Mulamkattil, S.; Variya, B.; Patel, S. Molecular docking, synthesis and biological screening of mefenamic acid derivatives as anti-inflammatory agents. Eur. J. Pharmacol. 2017, 801, 28–34. [Google Scholar] [CrossRef]
- Sarswat, P.K.; Sathyapalan, A.; Zhu, Y.; Free, M.L. Design, synthesis, and characterization of TPA-thiophene-based amide or imine functionalized molecule for potential optoelectronic devices. J. Theor. Appl. Phys. 2013, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Arslan, M. Synthesis and characterization of novel bio-based benzoxazines from gallic acid with latent catalytic characteristics. React. Funct. Polym. 2019, 139, 9–16. [Google Scholar] [CrossRef]
- Imran, H.M.; Rasool, N.; Kanwal, I.; Hashmi, M.A.; Altaf, A.A.; Ahmed, G.; Malik, A.; Kausar, S.; Khan, S.U.-D.; Ahmad, A. Synthesis of halogenated [1, 1′-biphenyl]-4-yl benzoate and [1, 1′: 3′, 1 ″-terphenyl]-4′-yl benzoate by palladium catalyzed cascade C–C coupling and structural analysis through computational approach. J. Mol. Struct. 2020, 1222, 128839. [Google Scholar] [CrossRef]
- Malik, A.; Rasool, N.; Kanwal, I.; Hashmi, M.A.; Zahoor, A.F.; Ahmad, G.; Altaf, A.A.; Shah, S.A.A.; Sultan, S.; Zakaria, Z.A. Suzuki–miyaura reactions of (4-bromophenyl)-4, 6-dichloropyrimidine through commercially available palladium catalyst: Synthesis, optimization and their structural aspects identification through computational studies. Processes 2020, 8, 1342. [Google Scholar] [CrossRef]
- Tùng, Đ.T.; Tuân, Đ.T.; Rasool, N.; Villinger, A.; Reinke, H.; Fischer, C.; Langer, P. Regioselective Palladium (0)-Catalyzed Cross-Coupling Reactions and Metal-Halide Exchange Reactions of Tetrabromothiophene: Optimization, Scope and Limitations. Adv. Synth. Catal. 2009, 351, 1595–1609. [Google Scholar] [CrossRef]
- Dang, T.T.; Rasool, N.; Dang, T.T.; Reinke, H.; Langer, P. Synthesis of tetraarylthiophenes by regioselective Suzuki cross-coupling reactions of tetrabromothiophene. Tetrahedron Lett. 2007, 48, 845–847. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154122. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Cammi, R.; Mennucci, B.; Tomasi, J. Fast Evaluation of Geometries and Properties of Excited Molecules in Solution: A Tamm-Dancoff Model with Application to 4-Dimethylaminobenzonitrile. J. Phys. Chem. A 2000, 104, 5631–5637. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V. Solvent effect on vertical electronic transitions by the polarizable continuum model. J. Chem. Phys. 2000, 112, 2427–2435. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V. Time-dependent density functional theory for molecules in liquid solutions. J. Chem. Phys. 2001, 115, 4708–4717. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Polarizable dielectric model of solvation with inclusion of charge penetration effects. J. Chem. Phys. 2001, 114, 5691–5701. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]


| Entry | Catalyst | Solvent | Base | % Yield |
|---|---|---|---|---|
| 3 | TiCl4 | Pyridine | Pyridine | 12 |
| DCC/DMAP | DCM | - | 8 | |
| 4-methylphenyl boronic acid | Toluene | - | 7 | |
| - | Xylene | - | 9 |
| Compound | EHOMO | ELUMO | HOMO-LUMO Gap | Hyperpolarizability |
|---|---|---|---|---|
| 3 | −6.67 | −1.68 | 4.99 | 1597.42 |
| 5 | −6.01 | −0.35 | 5.66 | 1568.38 |
| 6 | −6.70 | −1.67 | 5.03 | 1570.87 |
| 7 | −6.76 | −1.55 | 5.21 | 882.79 |
| 8a | −6.20 | −1.79 | 4.41 | 4477.25 |
| 8b | −6.16 | −1.72 | 4.44 | 3174.72 |
| 9a | −6.16 | −1.71 | 4.45 | 3181.71 |
| 9b | −6.44 | −2.10 | 4.33 | 3785.18 |
| 9c | −5.92 | −1.62 | 4.30 | 6825.00 |
| 9d | −6.39 | −1.86 | 4.54 | 1144.23 |
| 9e | −6.14 | −1.75 | 4.40 | 2517.82 |
| 9f | −5.79 | −1.74 | 4.45 | 10,221.04 |
| 9g | −6.23 | −1.70 | 4.33 | 1876.18 |
| 9h | −6.44 | −1.93 | 4.30 | 799.16 |
| Compounds | µ (Debye) | βtot (a.u.) | βvec (a.u.) | βvec/βtot |
|---|---|---|---|---|
| 3 | 3.561 | 176.66 | −80.6 | −0.456 |
| 5 | 3.776 | 135.50 | −15.6 | −0.115 |
| 6 | 7.433 | 352.12 | 113.7 | 0.322 |
| 7 | 9.127 | 241.55 | 130.4 | 0.539 |
| 8 | 9.265 | 376.69 | 227.2 | 0.603 |
| 9a | 7.416 | 206.53 | 188.2 | 0.911 |
| 9b | 7.396 | 233.41 | 229.0 | 0.981 |
| 9c | 5.574 | 229.21 | 111.4 | 0.486 |
| 9d | 9.795 | 612.59 | 449.1 | 0.733 |
| 9e | 8.316 | 424.17 | 210.0 | 0.495 |
| 9f | 10.543 | 452.47 | 347.5 | 0.853 |
| 9g | 8.734 | 237.68 | 231.2 | 0.972 |
| 9h | 9.387 | 259.34 | 259.5 | 1.001 |
| Compound | Ionization Potential, I (eV) | Electron Affinity, A (eV) | Chemical Hardness, ƞ (eV) | Electronic Chemical Potential, μ (eV) | Electrophilicity Index, ω (eV) |
|---|---|---|---|---|---|
| 3 | 6.67 | 1.68 | −2.49 | 4.17 | −3.49 |
| 5 | 6.01 | 0.35 | −2.83 | 3.18 | −1.79 |
| 6 | 6.70 | 1.67 | −2.52 | 4.18 | −3.48 |
| 7 | 6.76 | 1.55 | −2.61 | 4.16 | −3.32 |
| 8a | 6.20 | 1.79 | −2.20 | 4.00 | −3.62 |
| 8b | 6.16 | 1.72 | −2.22 | 3.94 | −3.49 |
| 9a | 6.16 | 1.71 | −2.22 | 3.94 | −3.48 |
| 9b | 6.44 | 2.10 | −2.17 | 4.27 | −4.21 |
| 9c | 5.92 | 1.62 | −2.15 | 3.77 | −3.31 |
| 9d | 6.39 | 1.86 | −2.27 | 4.13 | −3.75 |
| 9e | 6.14 | 1.75 | −2.20 | 3.94 | −3.54 |
| 9f | 6.16 | 1.71 | −2.22 | 3.94 | −3.48 |
| 9g | 6.44 | 2.10 | −2.17 | 4.27 | −4.21 |
| 9h | 5.92 | 1.62 | −2.15 | 3.77 | −3.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanwal, I.; Rasool, N.; Zaidi, S.H.M.; Zakaria, Z.A.; Bilal, M.; Hashmi, M.A.; Mubarik, A.; Ahmad, G.; Shah, S.A.A. Synthesis of Functionalized Thiophene Based Pyrazole Amides via Various Catalytic Approaches: Structural Features through Computational Applications and Nonlinear Optical Properties. Molecules 2022, 27, 360. https://doi.org/10.3390/molecules27020360
Kanwal I, Rasool N, Zaidi SHM, Zakaria ZA, Bilal M, Hashmi MA, Mubarik A, Ahmad G, Shah SAA. Synthesis of Functionalized Thiophene Based Pyrazole Amides via Various Catalytic Approaches: Structural Features through Computational Applications and Nonlinear Optical Properties. Molecules. 2022; 27(2):360. https://doi.org/10.3390/molecules27020360
Chicago/Turabian StyleKanwal, Iram, Nasir Rasool, Syeda Huda Mehdi Zaidi, Zainul Amiruddin Zakaria, Muhammad Bilal, Muhammad Ali Hashmi, Adeel Mubarik, Gulraiz Ahmad, and Syed Adnan Ali Shah. 2022. "Synthesis of Functionalized Thiophene Based Pyrazole Amides via Various Catalytic Approaches: Structural Features through Computational Applications and Nonlinear Optical Properties" Molecules 27, no. 2: 360. https://doi.org/10.3390/molecules27020360