
Recent Strategies in Transition-Metal-Catalyzed Sequential C–H Activation/Annulation for One-Step Construction of Functionalized Indazole Derivatives


Abstract
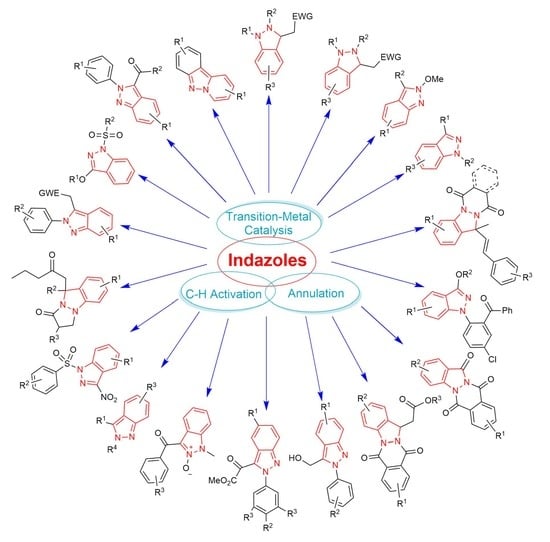
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
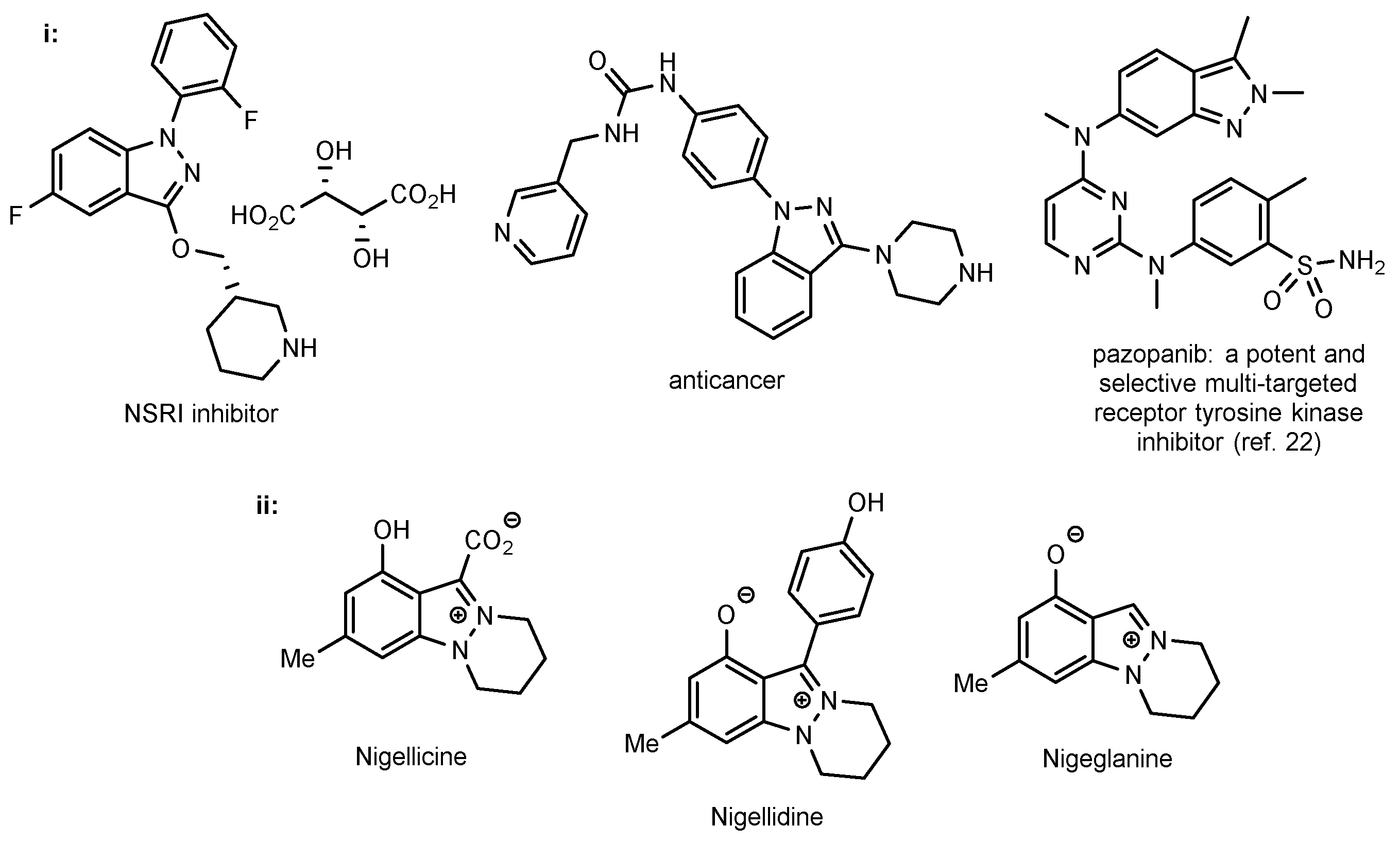
1. Introduction
2. Different Synthesis Routes for the Construction of Indazoles via Transition-Metal-Catalyzed Sequential C–H Activation/Annulation
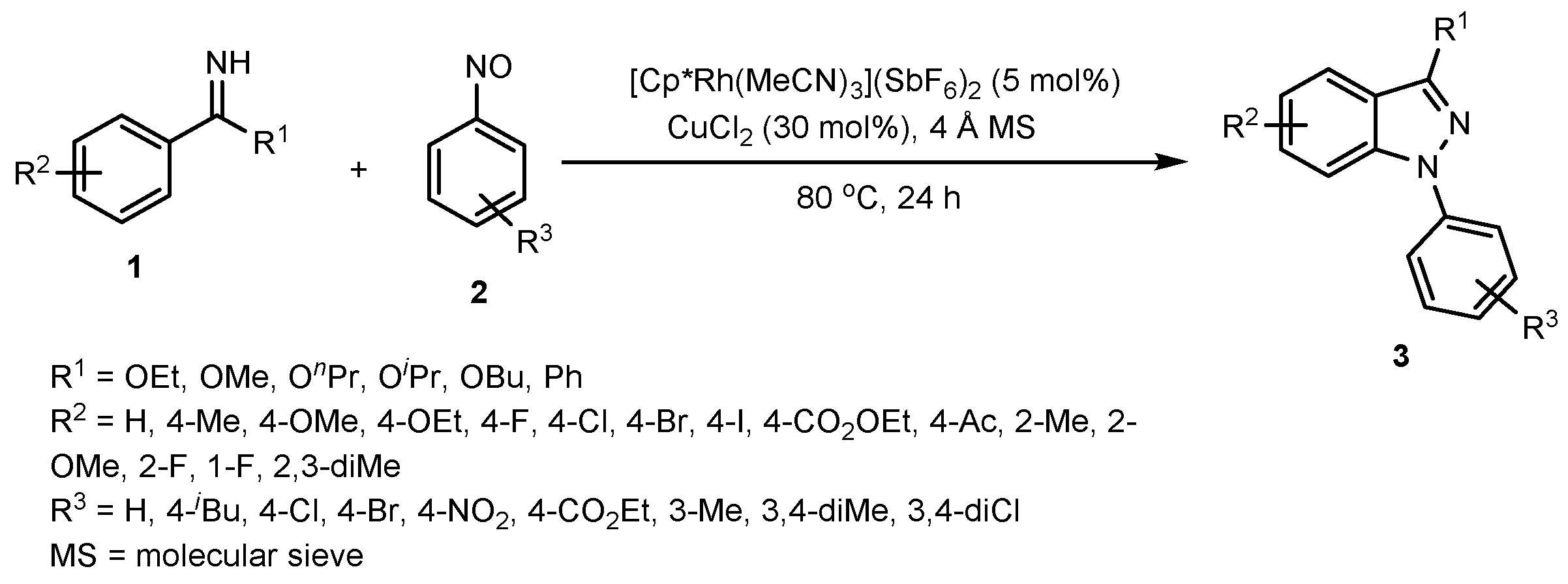
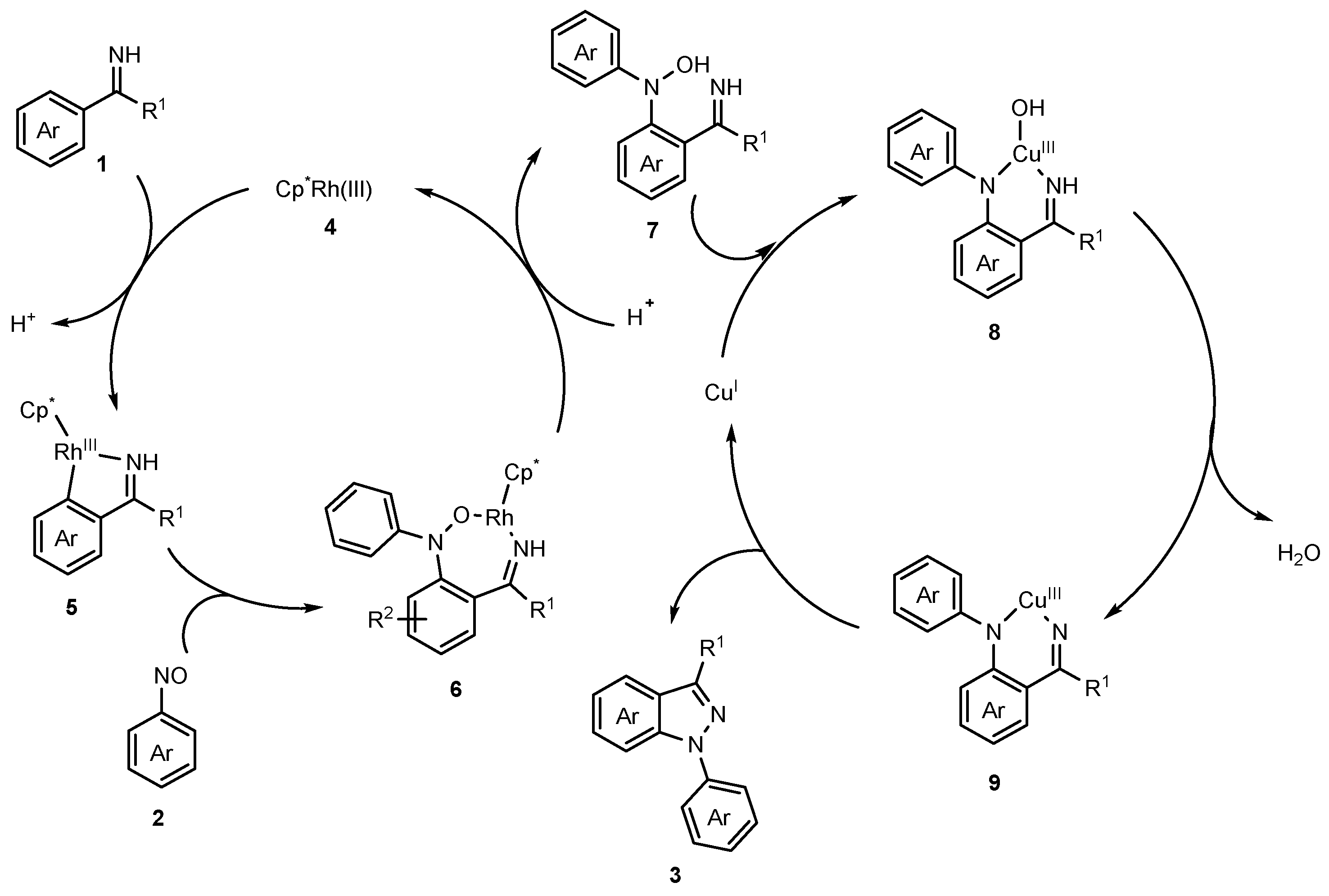
2.1. Synthesis of Indazoles Using Rhodium and Copper Salts
2.2. Synthesis of Indazoles Using Ruthenium Salt in the Presence of Sodium Acetate
2.3. Synthesis of Indazoles Using Rhodium Complexes and Silver Salts
2.4. Synthesis of Indazoles Using Rhodium and Silver Salts in the Presence of Lithium Acetate
2.5. Synthesis of Indazoles Using Rhodium and Silver Salts in the Presence of Sodium Acetate
2.6. Synthesis of Indazoles Using Rhodium and Silver Salts in the Presence of Zinc Acetate
2.7. Synthesis of Indazoles Using Rhodium and Silver Salts in the Presence of Magnesium Sulfate
2.8. Synthesis of Indazoles Using Ruthenium and Copper Salts in the Presence of Potassium Hexafluorophosphate
2.9. Synthesis of Indazoles Using Rhodium, Copper, and Silver Salts
2.10. Synthesis of Indazoles Using Rhodium, Silver, Copper, and Zinc Salts
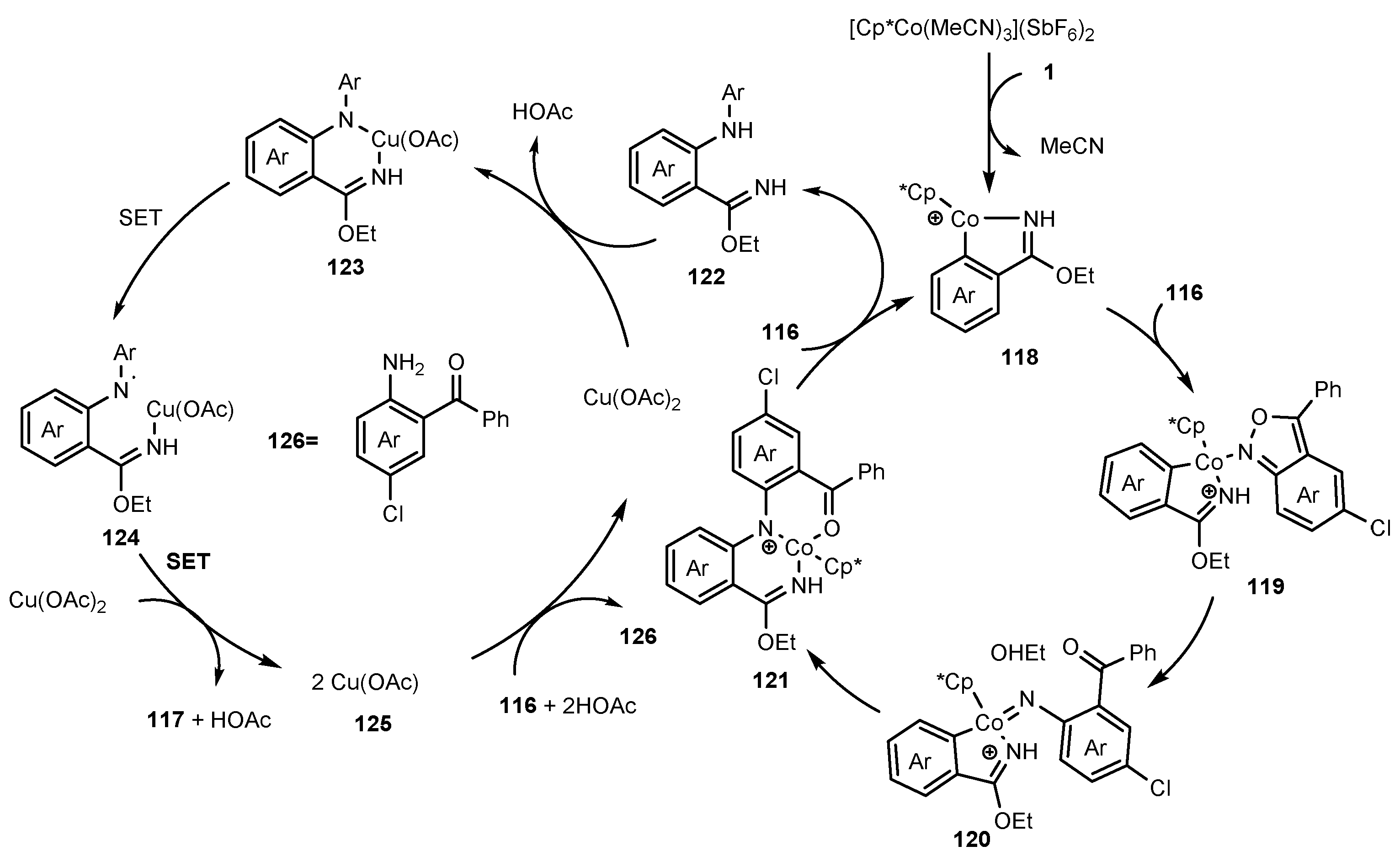
2.11. Synthesis of Indazoles Using Cobalt and Copper Salts
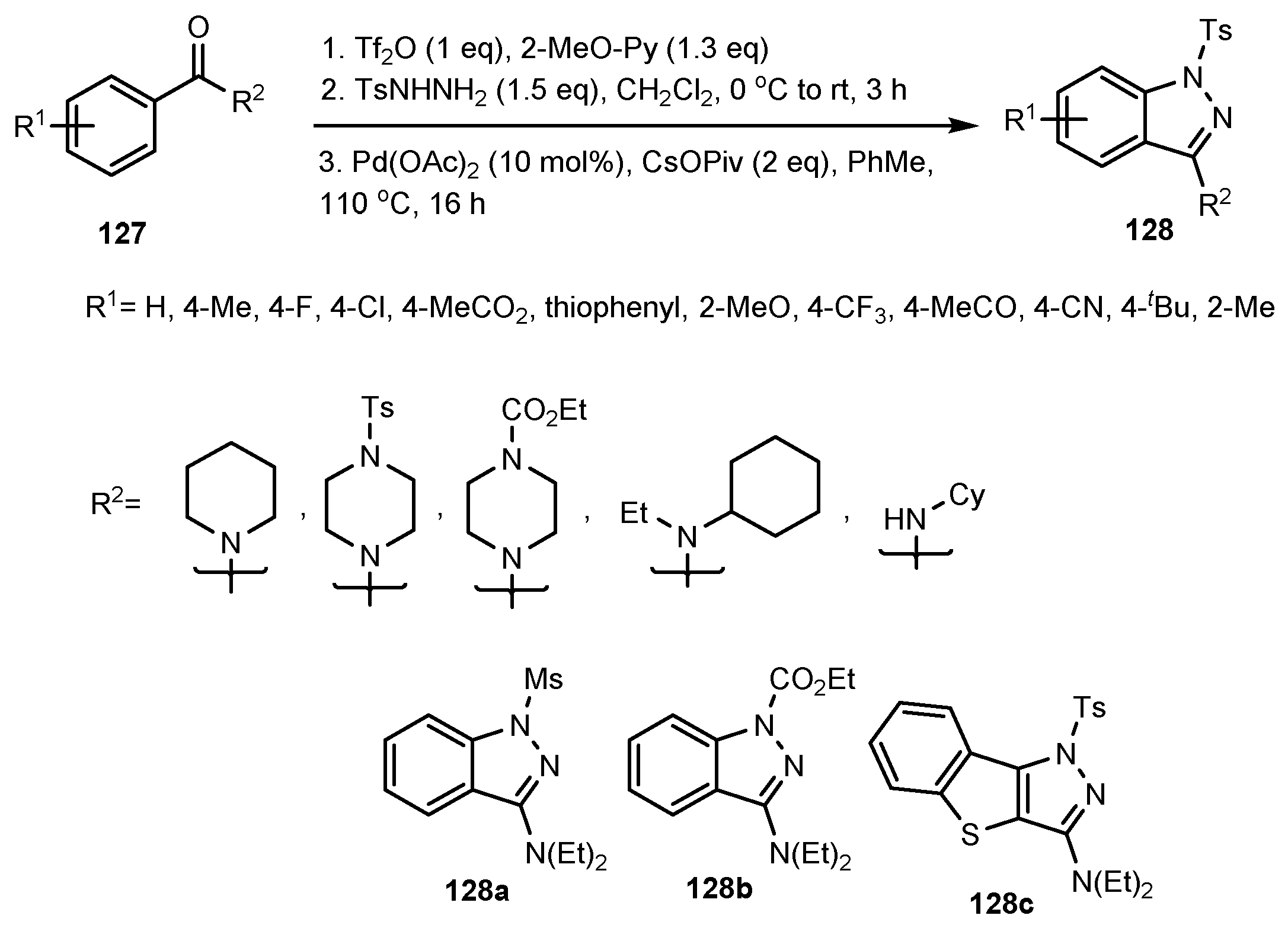
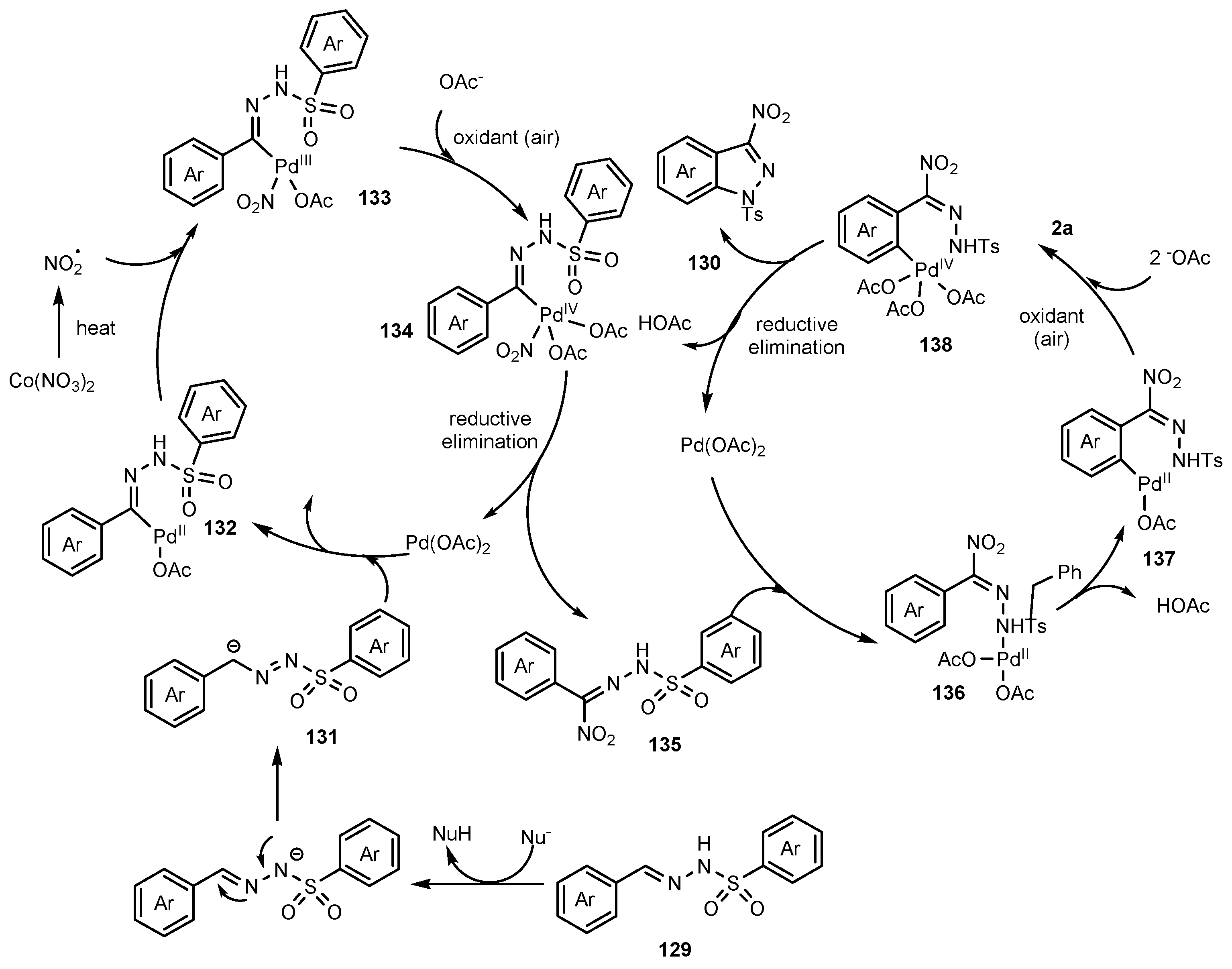
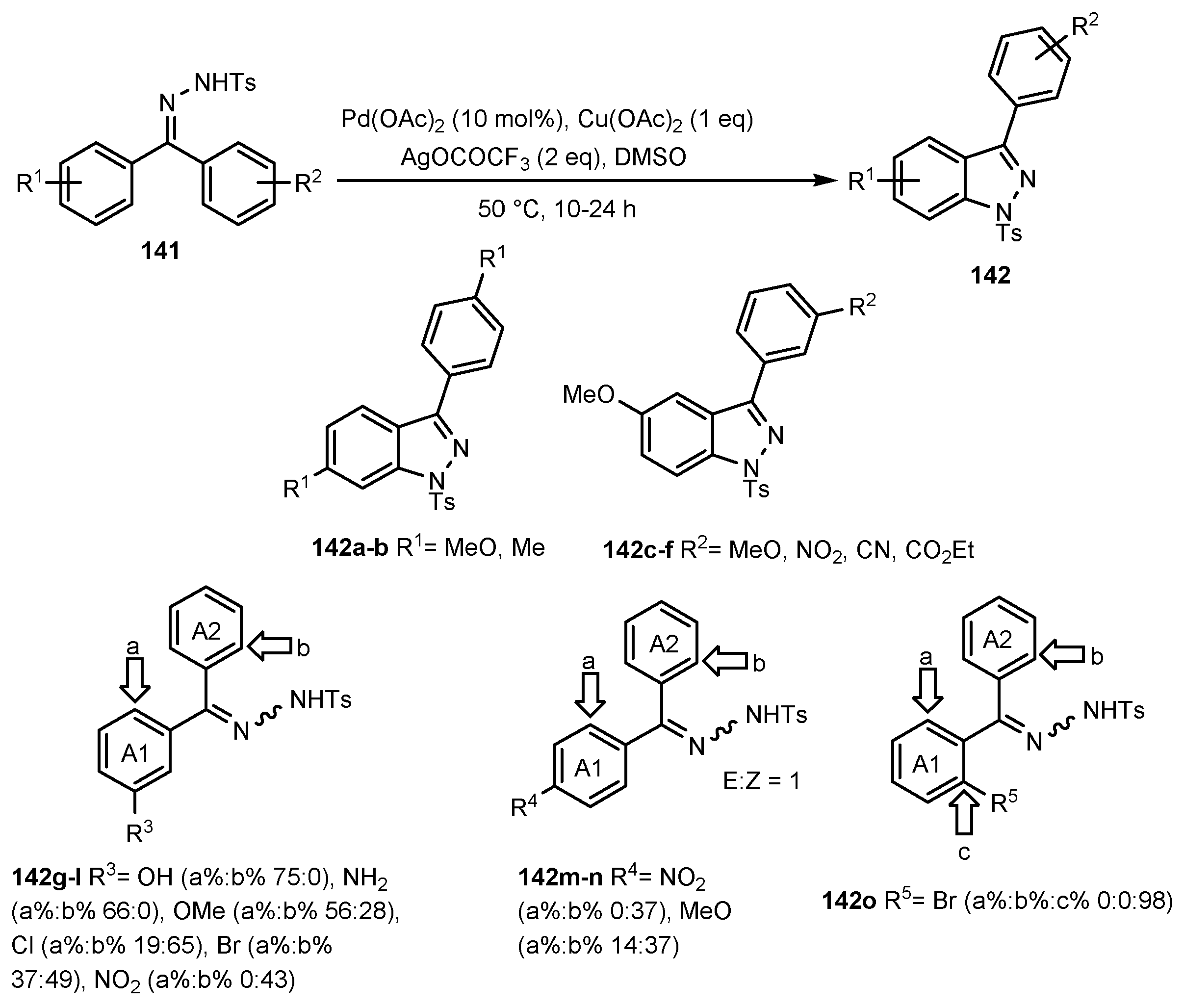
2.12. Synthesis of Indazoles Using Palladium Salts
2.13. Synthesis of Indazoles Using Palladium Salt in the Presence of Cobalt Salts
2.14. Synthesis of Indazoles Using Palladium, Cerium, and Iron Salts
2.15. Synthesis of Indazoles Using Palladium, Copper, and Silver Salts
2.16. Synthesis of Indazoles Using Cobalt Salts
2.17. Synthesis of Indazoles Using Rhenium Salt in the Presence of Sodium Acetate
2.18. Synthesis of Indazoles Using Copper Salts
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Santana, A.C.; Silva Filho, R.C.; Menezes, J.C.; Allonso, D.; Campos, V.R. Nitrogen-based Heterocyclic Compounds: A Promising Class of Antiviral Agents Against Chikungunya Virus. Life 2020, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Shiri, P. An Overview on The Copper-promoted Synthesis of Five-membered Heterocyclic Systems. Appl. Organomet. Chem. 2020, 34, e5600. [Google Scholar] [CrossRef]
- Shiri, P. Novel Hybrid Molecules Based on Triazole-β-lactam as Potential Biological Agents. Mini-Rev. Med. Chem. 2021, 21, 536–553. [Google Scholar] [CrossRef]
- Alizadeh, A.; Roosta, A.; Rezaiyehraad, R. Efficient Access to Pyrido[1, 2-a]pyrimidines and Imidazo[1, 2-a]pyridines Through Knoevenagel Reaction/aza–ene Addition/Intramolecular Cyclization. J. Iran. Chem. Soc. 2020, 17, 1123–1130. [Google Scholar] [CrossRef]
- Alizadeh, A.; Roosta, A. An Efficient Regioselective Access to (11Z)-11-(3-Aryl-5, 6-dihydropyrazin-2 (1H)-ylidene)-11H-indeno[1, 2-b]quinoxaline Derivatives via One-Pot Three-Component Reaction. ChemistrySelect 2019, 4, 13503–13505. [Google Scholar] [CrossRef]
- Khalafi-Nezhad, A.; Panahi, F. Ruthenium-catalyzed Synthesis of Benzoxazoles Using Acceptorless Dehydrogenative Coupling Reaction of Primary Alcohols with 2-Aminophenol under Heterogeneous Conditions. ACS Catal. 2014, 4, 1686–1692. [Google Scholar] [CrossRef]
- Jeong, T.; Han, S.H.; Han, S.; Sharma, S.; Park, J.; Lee, J.S.; Kwak, J.H.; Jung, Y.H.; Kim, I.S. Access to 3-acyl-(2H)-indazoles via Rh (III)-Catalyzed C–H Addition and Cyclization of Azobenzenes with α-Keto Aldehydes. Org. Lett. 2016, 18, 232–235. [Google Scholar] [CrossRef]
- Aboonajmi, J.; Panahi, F.; Sharghi, H. One-Pot Multicomponent Coupling Reaction of Catechols, Benzyl Alcohols/Benzyl Methyl Ethers, and Ammonium Acetate toward Synthesis of Benzoxazoles. ACS Omega 2021, 6, 22395–22399. [Google Scholar] [CrossRef]
- Mohtat, B.; Siadati, S.A.; Khalilzadeh, M.A.; Zareyee, D. The Concern of Emergence of Multi-station Reaction Pathways That Might Make Stepwise the Mechanism of the 1, 3-Dipolar Cycloadditions of Azides and Alkynes. J. Mol. Struct. 2018, 1155, 58–64. [Google Scholar] [CrossRef]
- Schmidt, A.; Beutler, A.; Snovydovych, B. Recent Advances in the Chemistry of Indazoles. Eur. J. Org. Chem. 2008, 2008, 4073–4095. [Google Scholar] [CrossRef]
- Zhang, S.-G.; Liang, C.-G.; Zhang, W.-H. Recent Advances in Indazole-containing Derivatives: Synthesis and Biological Perspectives. Molecules 2018, 23, 2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangadurai, A.; Minu, M.; Wakode, S.; Agrawal, S.; Narasimhan, B. Indazole: A Medicinally Important Heterocyclic Moiety. Med. Chem. Res. 2012, 21, 1509–1523. [Google Scholar] [CrossRef]
- Khalifeh, R.; Karimzadeh, F. Copper Nanoparticles Supported on Charcoal Mediated One-pot Three-component Synthesis of N-Substituted-2H-indazoles via Consecutive Condensation C–N and N–N Bond Formation. Can. J. Chem. 2019, 97, 303–309. [Google Scholar] [CrossRef]
- Khalifeh, R.; Naseri, V.; Rajabzadeh, M. Synthesis of Imidazolium-Based Ionic Liquid on Modified Magnetic Nanoparticles for Application in One-Pot Synthesis of Trisubstituted Imidazoles. ChemistrySelect 2020, 5, 11453–11462. [Google Scholar] [CrossRef]
- Khalili, D.; Evazi, R.; Neshat, A.; Aboonajmi, J. Copper (I) Complex of Dihydro Bis (2-Mercapto Benzimidazolyl) Borate as an Efficient Homogeneous Catalyst for the Synthesis of 2H-Indazoles and 5-Substituted 1H-Tetrazoles. ChemistrySelect 2021, 6, 746–753. [Google Scholar] [CrossRef]
- Sharghi, H.; Aberi, M.; Shiri, P. Silica-supported Cu (II)–quinoline Complex: Efficient and Recyclable Nanocatalyst for One-pot Synthesis of Benzimidazolquinoline Derivatives and 2H-Indazoles. Appl. Organomet. Chem. 2019, 33, e4974. [Google Scholar] [CrossRef]
- Kumar, A.; Verma, S.; Pathak, D.D. Synthesis and Characterization of A Recyclable Graphene Oxide-surface-engineered Copper(II) Schiff Base Complex: Catalytic Application in Synthesis of 1, 2, 3-Triazoles and 2H-Indazoles. J. Environ. Chem. Eng. 2021, 9, 105791. [Google Scholar] [CrossRef]
- Sharghi, H.; Aberi, M.; Shiri, P. Highly Reusable Support-free Copper(II) Complex of Para-hydroxy-substituted Salen: Novel, Efficient and Versatile Catalyst for C─N Bond Forming Reactions. Appl. Organomet. Chem. 2017, 31, e3761. [Google Scholar] [CrossRef]
- Magano, J.; Waldo, M.; Greene, D.; Nord, E. The Synthesis of (S)-5-Fluoro-1-(2-fluorophenyl)-3-(piperidin-3-ylmethoxy)-1H-Indazole, A Norepinephrine/Serotonin Reuptake Inhibitor for the Treatment of Fibromyalgia. Org. Process Res. Dev. 2008, 12, 877–883. [Google Scholar] [CrossRef]
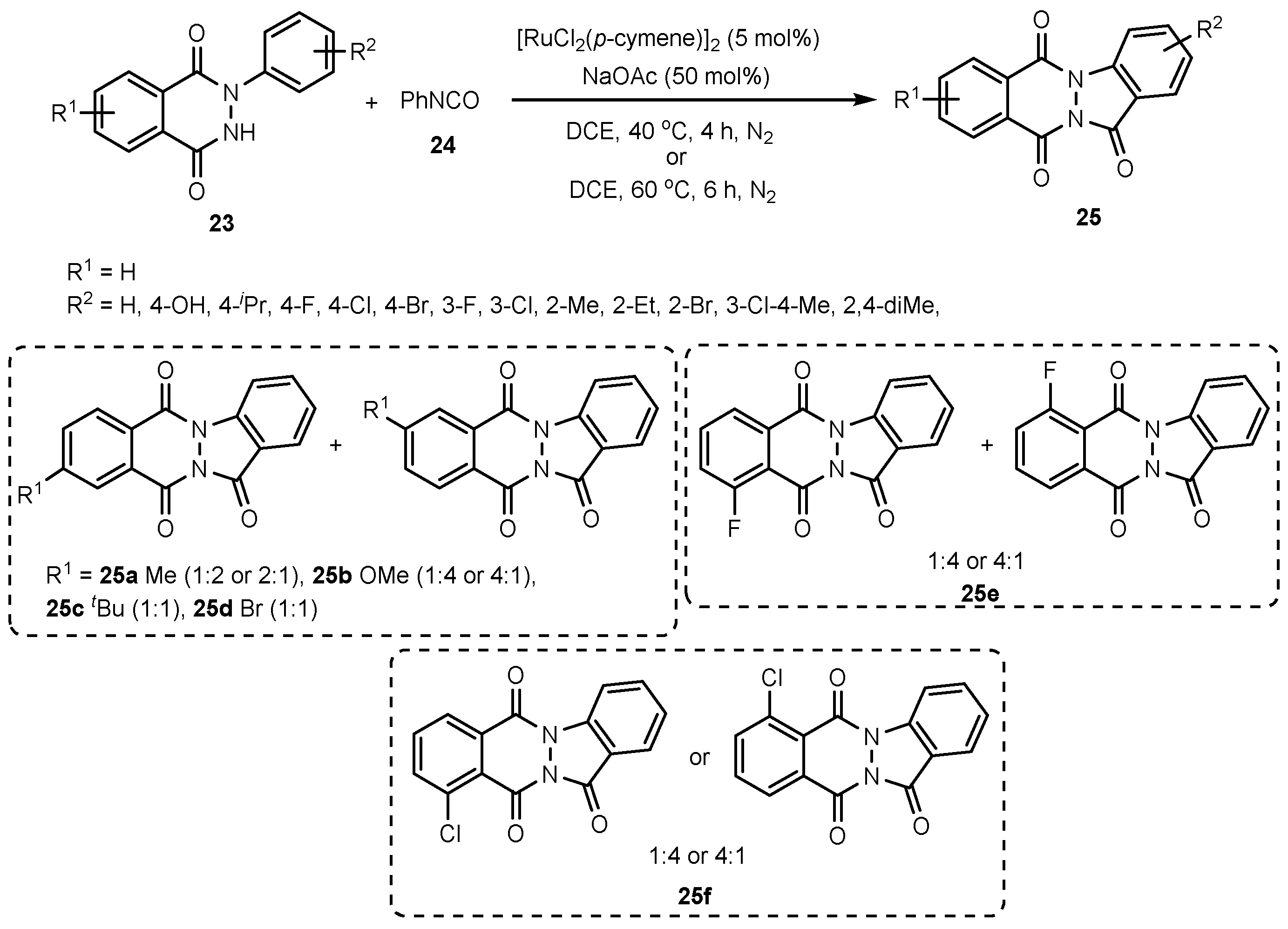
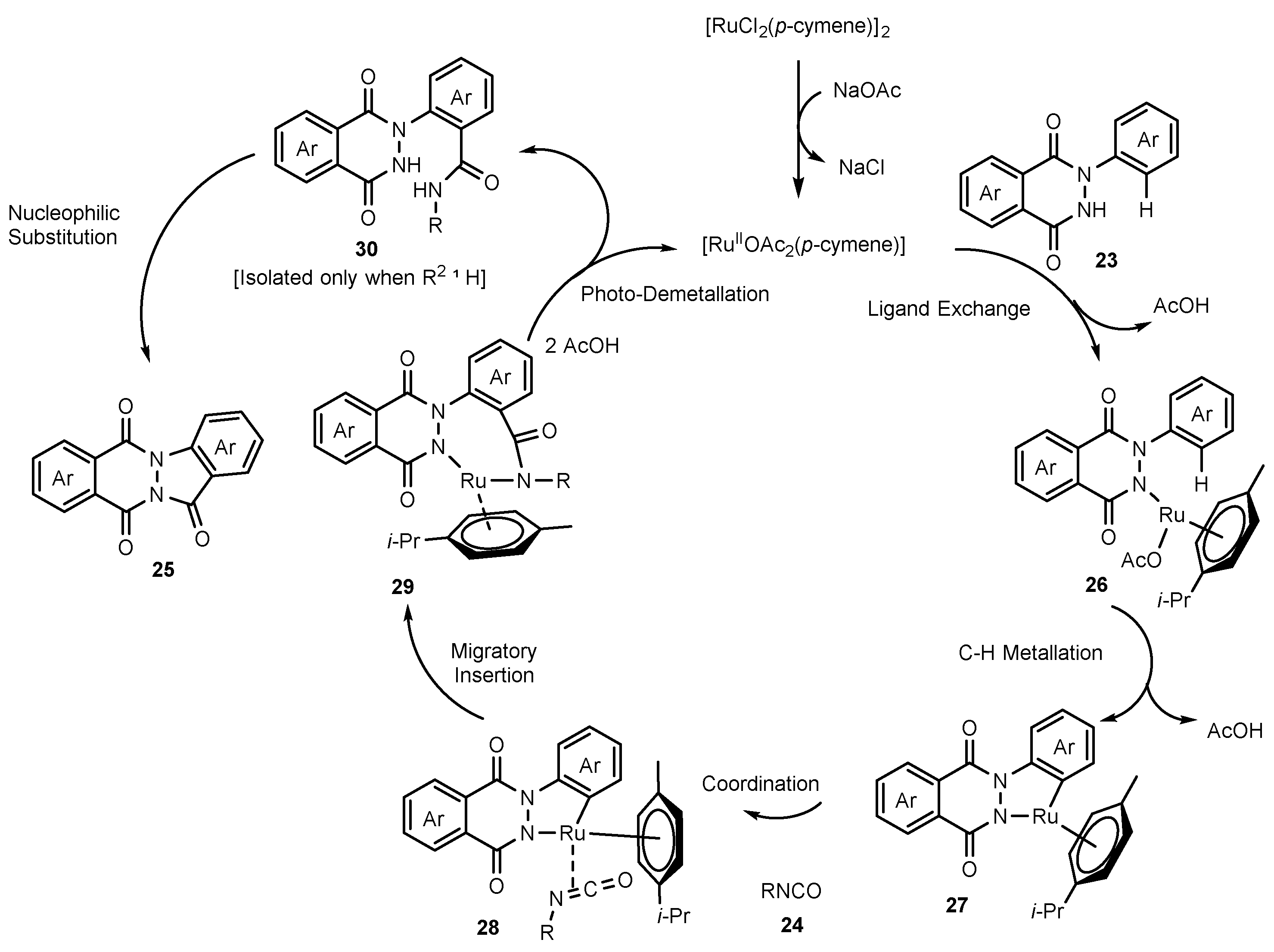
- Karishma, P.; Gogia, A.; Mandal, S.K.; Sakhuja, R. Ruthenium Catalyzed C−H Amidation and Carbocyclization using Isocyanates: An Access to Amidated 2-Phenylphthalazine-1, 4-diones and Indazolo [1, 2-b] Phthalazine-triones. Adv. Synth. Catal. 2021, 363, 762–775. [Google Scholar] [CrossRef]
- Rhee, H.-K.; Yoo, J.H.; Lee, E.; Kwon, Y.J.; Seo, H.-R.; Lee, Y.-S.; Choo, H.-Y.P. Synthesis and Cytotoxicity of 2-Phenylquinazolin-4(3H)-one Derivatives. Eur. J. Med. Chem. 2011, 46, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, L.; Chen, H.; Wu, W.; Jiang, H. Copper-catalyzed Aerobic C (sp2)–H Functionalization for C–N Bond Formation: Synthesis of Pyrazoles and Indazoles. J. Org. Chem. Res. 2013, 78, 3636–3646. [Google Scholar] [CrossRef]
- Cyr, P.; Regnier, S.; Bechara, W.S.; Charette, A.B. Rapid Access to 3-Aminoindazoles from Tertiary Amides. Org. Lett. 2015, 17, 3386–3389. [Google Scholar] [CrossRef] [PubMed]
- Shiri, P.; Ramezanpour, S.; Amani, A.M.; Dehaen, W. A Patent Review on Efficient Strategies for the Total Synthesis of Pazopanib, Regorafenib and Lenvatinib as Novel Anti-angiogenesis Receptor Tyrosine Kinase Inhibitors for Cancer Therapy. Mol. Divers. 2022. [Google Scholar] [CrossRef] [PubMed]
- Cekaviciute, M.; Simokaitiene, J.; Grazulevicius, J.; Buika, G.; Jankauskas, V. New Derivatives of Indazole as Electronically Active Materials. Dye. Pigm. 2012, 92, 654–658. [Google Scholar] [CrossRef]
- Sharghi, H.; Aberi, M. Ligand-free Copper (I) Oxide Nanoparticle Catalyzed Three-component Synthesis of 2H-Indazole Derivatives from 2-Halobenzaldehydes, Amines and Sodium Azide in Polyethylene Glycol as a Green Solvent. Synlett 2014, 25, 1111–1115. [Google Scholar] [CrossRef] [Green Version]
- Gaikwad, D.D.; Chapolikar, A.D.; Devkate, C.G.; Warad, K.D.; Tayade, A.P.; Pawar, R.P.; Domb, A.J. Synthesis of Indazole Motifs and Their Medicinal Importance: An Overview. Eur. J. Med. Chem. 2015, 90, 707–731. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Hasan, S.S.; Choudhary, M.I.; Ni, C.-Z.; Clardy, J. Nigellidine—A New Indazole Alkaloid from The Seeds of Nigella Sativa. Tetrahedron Lett. 1995, 36, 1993–1996. [Google Scholar]
- Boubertakh, B.; Liu, X.-G.; Cheng, X.-L.; Li, P. A Spotlight on Chemical Constituents and Pharmacological Activities of Nigella Glandulifera Freyn et Sint seeds. J. Chem. 2013, 2013, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Inamoto, K.; Katsuno, M.; Yoshino, T.; Arai, Y.; Hiroya, K.; Sakamoto, T. Synthesis of 3-Substituted Indazoles and Benzoisoxazoles via Pd-Catalyzed Cyclization Reactions: Application to the Synthesis of Nigellicine. Tetrahedron 2007, 63, 2695–2711. [Google Scholar] [CrossRef]
- Raut, S.; Hadi, A.; Pathan, M.A. The Efficient Synthesis of 3-[6-(Substituted)-[1, 2, 4] Triazolo [3, 4-b][1, 3, 4] Thiadiazol-3-yl]-1H-indazole. J. Heterocycl. Chem. 2020, 57, 1291–1305. [Google Scholar] [CrossRef]
- Rafique, R.; Khan, K.M.; Chigurupati, S.; Wadood, A.; Rehman, A.U.; Karunanidhi, A.; Hameed, S.; Taha, M.; Al-Rashida, M. Synthesis of New Indazole Based Dual Inhibitors of α-Glucosidase and α-Amylase Enzymes, Their In vitro, In silico and Kinetics Studies. Bioorg. Chem. 2020, 94, 103195. [Google Scholar] [CrossRef]
- Rudavath, D.; Sreenivasulu, R.; Pinapati, S.R.; Raju, R.R. Synthesis and Anticancer Evaluation of Indazole-aryl Hydrazide-hydrazone Derivatives. J. Indian Chem. Soc. 2018, 95, 433–438. [Google Scholar]
- Nie, B.; Wu, W.; Zhang, Y.; Jiang, H.; Zhang, J. Recent Advances in the Synthesis of Bridgehead (or ring-junction) Nitrogen Heterocycles via Transition Metal-catalyzed C–H Bond Activation and Functionalization. Org. Chem. Front. 2020, 7, 3067–3099. [Google Scholar] [CrossRef]
- Kumar, A.; Sherikar, M.S.; Hanchate, V.; Prabhu, K.R. Application of Sulfoxonium Ylide in Transition-metal-catalyzed C−H Bond Activation and Functionalization Reactions. Tetrahedron 2021, 101, 132478. [Google Scholar] [CrossRef]
- Xiang, Y.; Wang, C.; Ding, Q.; Peng, Y. Diazo Compounds: Versatile Synthons for the Synthesis of Nitrogen Heterocycles via Transition Metal-Catalyzed Cascade C–H Activation/Carbene Insertion/Annulation Reactions. Adv. Synth. Catal. 2019, 361, 919–944. [Google Scholar] [CrossRef]
- Zhang, M. Construction of Heterocycle Scaffolds via Transition Metal-Catalyzed sp2 C−H Functionalization. Adv. Synth. Catal. 2009, 351, 2243–2270. [Google Scholar] [CrossRef]
- Kumar, G.R.; Rajesh, M.; Lin, S.; Liu, S. Propargylic Alcohols as Coupling Partners in Transition-Metal-Catalyzed Arene C−H Activation. Adv. Synth. Catal. 2020, 362, 5238–5256. [Google Scholar] [CrossRef]
- Zhang, G.; Fan, Q.; Zhao, Y.; Ding, C. Copper-Promoted Oxidative Intramolecular C–H Amination of Hydrazones to Synthesize 1H-Indazoles and 1H-Pyrazoles Using a Cleavable Directing Group. Eur. J. Org. Chem. 2019, 2019, 5801–5806. [Google Scholar] [CrossRef]
- Verbelen, B.; Leen, V.; Wang, L.; Boens, N.; Dehaen, W. Direct palladium-catalysed C–H arylation of BODIPY dyes at the 3-and 3, 5-positions. Chem. Commun. 2012, 48, 9129–9131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.-L.; Lin, P.-P.; Li, Q. Recent Advances of Allenes in the First-row Transition Metals Catalyzed C−H Activation Reactions. Chin. Chem. Lett. 2019, 30, 1495–1502. [Google Scholar] [CrossRef]
- Karak, P.; Rana, S.S.; Choudhury, J. Cationic π-extended Heteroaromatics via a Catalytic C–H Activation Annulative Alkyne-insertion Sequence. Chem. Commun. 2022, 58, 133–154. [Google Scholar] [CrossRef]
- Mo, J.; Müller, T.; Oliveira, J.C.; Ackermann, L. 1, 4-Iron Migration for Expedient Allene Annulations through Iron-Catalyzed C−H/N−H/C−O/C−H Functionalizations. Angew. Chem. Int. Ed. Engl. 2018, 57, 7719–7723. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Tang, Y.; Wang, X.; Wang, Y.; Huang, X.; Xu, S.; Li, Y. Regioselective Cascade Annulation of Indoles with Alkynediones for Construction of Functionalized Tetrahydrocarbazoles Triggered by Cp*RhIII-catalyzed C–H activation. Org. Chem. Front. 2021, 8, 3809–3814. [Google Scholar] [CrossRef]
- Kalyani, A.; Tulichala, R.P.; Chauhan, S.; Swamy, K.K. Palladium Catalyzed Nitrile Insertion and Cyanation of Biindoles: Synthesis of Indole Fused α-Carboline Scaffolds via Double C–H Activation. Tetrahedron Lett. 2022, 89, 153600. [Google Scholar] [CrossRef]
- Song, X.; Han, X.; Zhang, R.; Liu, H.; Wang, J. Rhodium (III)-catalyzed [4 + 2] Annulation via C−H Activation: Synthesis of Multi-substituted Naphthalenone Sulfoxonium Ylides. Molecules 2019, 24, 1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini-Sarvari, M.; Roosta, A. Synthesis of 2-Amino-4H-Chromen-4-yl phosphonats via C-P Bond Formation Catalyzed by Nano-rods ZnO under Solvent-free Condition. Comb. Chem. High Throughput Screen. 2014, 17, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Van der Eycken, E.V. Transition Metal-Catalyzed Intermolecular Cascade C−H Activation/Annulation Processes for the Synthesis of Polycycles. Chem. Eur. J. 2021, 27, 121–144. [Google Scholar] [CrossRef]
- Duarah, G.; Kaishap, P.; Begum, T.; Gogoi, S. Recent Advances in Ruthenium (II)-Catalyzed C−H Bond Activation and Alkyne Annulation Reactions. Adv. Synth. Catal. 2019, 361, 654–672. [Google Scholar] [CrossRef]
- Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. Recent Advances in the Development of Indazole-based Anticancer Agents. ChemMedChem 2018, 13, 1490–1507. [Google Scholar] [CrossRef] [PubMed]
- Janardhanan, J.C.; Bhaskaran, R.P.; Praveen, V.K.; Manoj, N.; Babu, B.P. Transition-Metal-Catalyzed Syntheses of Indazoles. Asian J. Org. Chem. 2020, 9, 1410–1431. [Google Scholar] [CrossRef]
- Ghosh, D.; Ghosh, S.; Hajra, A. Electrochemical Functionalization of Imidazopyridine and Indazole: An Overview. Adv. Synth. Catal. 2021, 363, 5047–5071. [Google Scholar] [CrossRef]
- Cao, Y.; Luo, C.; Yang, P.; Li, P.; Wu, C. Indazole Scaffold: A Generalist for Marketed and Clinical Drugs. Med. Chem. Res. 2021, 30, 501–518. [Google Scholar] [CrossRef]
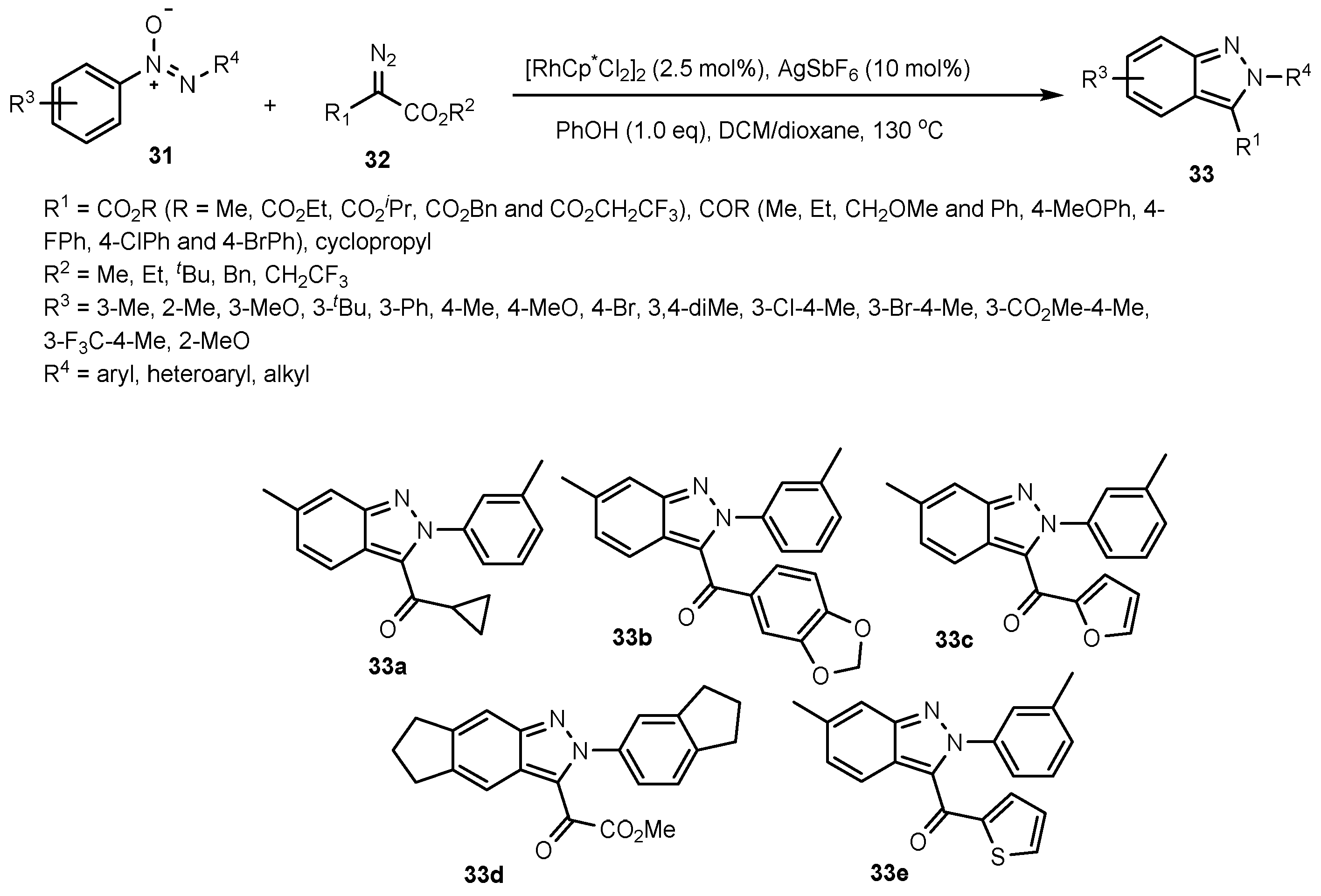
- Wang, Q.; Li, X. Synthesis of 1H-indazoles from Imidates and Nitrosobenzenes via Synergistic Rhodium/copper catalysis. Org. Lett. 2016, 18, 2102–2105. [Google Scholar] [CrossRef]
- Cai, S.; Lin, S.; Yi, X.; Xi, C. Substrate-controlled Transformation of Azobenzenes to Indazoles and Indoles via Rh (III)-catalysis. J. Org. Chem. Res. 2017, 82, 512–520. [Google Scholar] [CrossRef]
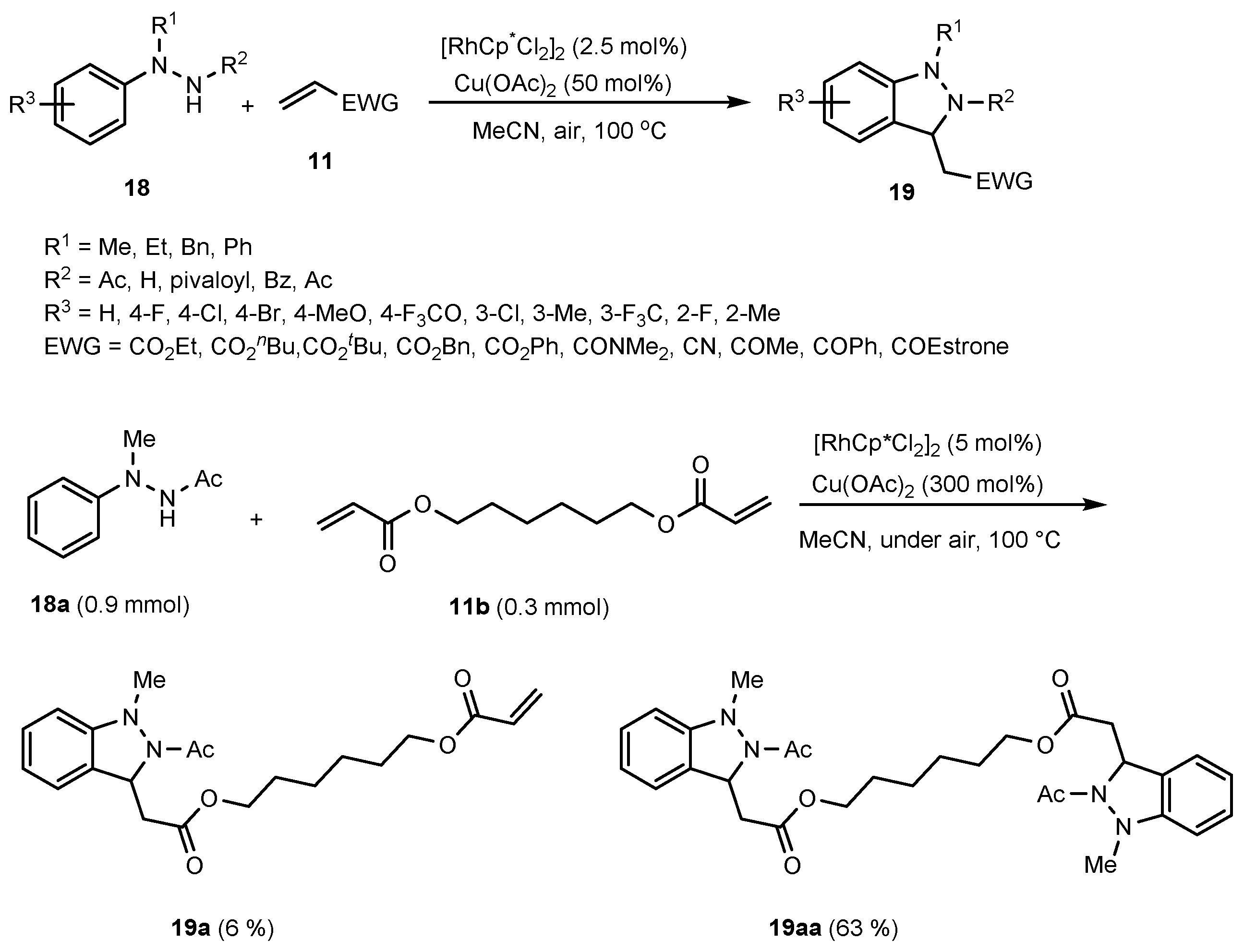
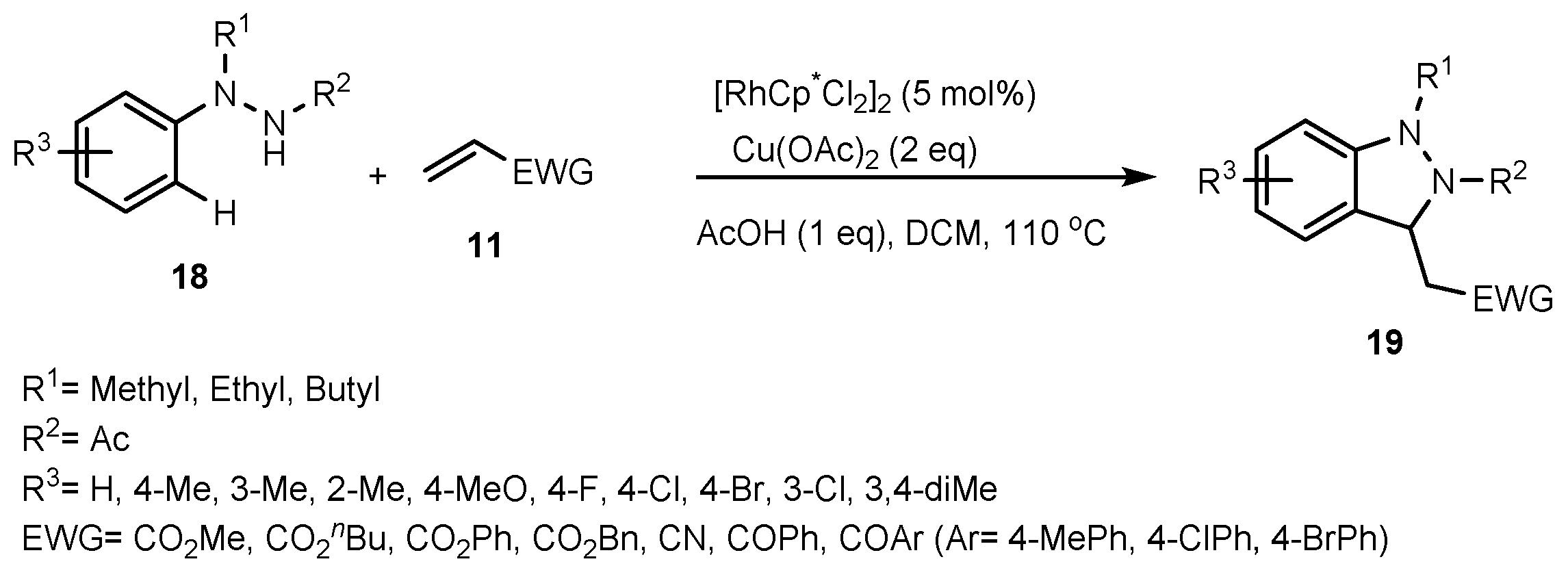
- Han, S.; Shin, Y.; Sharma, S.; Mishra, N.K.; Park, J.; Kim, M.; Kim, M.; Jang, J.; Kim, I.S. Rh (III)-Catalyzed Oxidative Coupling of 1, 2-Disubstituted Arylhydrazines and Olefins: A New Strategy for 2, 3-Dihydro-1H-Indazoles. Org. Lett. 2014, 16, 2494–2497. [Google Scholar] [CrossRef]
- Yao, J.; Feng, R.; Lin, C.; Liu, Z.; Zhang, Y. Synthesis of 2, 3-Dihydro-1H-indazoles by Rh (III)-catalyzed C–H Cleavage of Arylhydrazines. Org. Biomol. Chem. 2014, 12, 5469–5476. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, L.; Chen, J.; Chen, J.; Yin, C.; Yu, C. Rh (III)-Catalyzed [4 + 1] Annulation and Ring Opening for the Synthesis of Pyrazolo [1, 2-a] Indazole Bearing a Quaternary Carbon. Tetrahedron Lett. 2020, 61, 152350. [Google Scholar] [CrossRef]
- Long, Z.; Wang, Z.; Zhou, D.; Wan, D.; You, J. Rh (III)-Catalyzed Regio-and Chemoselective [4 + 1]-Annulation of Azoxy Compounds with Diazoesters for the Synthesis of 2H-Indazoles: Roles of the Azoxy Oxygen Atom. Org. Lett. 2017, 19, 2777–2780. [Google Scholar] [CrossRef] [PubMed]
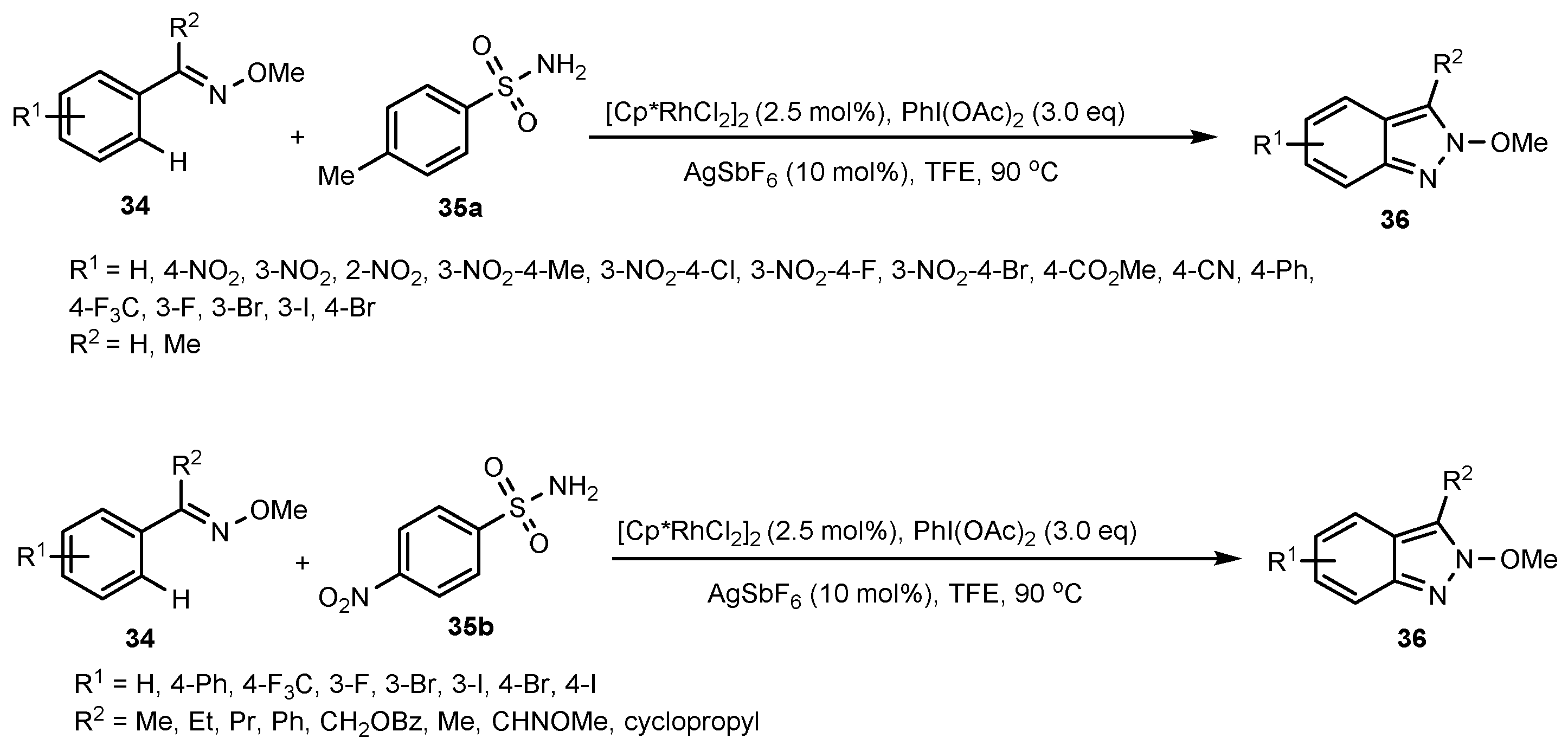
- Wang, N.; Liu, L.; Xu, W.; Zhang, M.; Huang, Z.; Shi, D.; Zhao, Y. Rhodium (III)-Catalyzed Oxidative Annulation of Ketoximes with Sulfonamide: A Direct Approach to Indazoles. Org. Lett. 2019, 21, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Chun, R.; Kim, S.; Han, S.H.; Han, S.; Lee, S.H.; Mishra, N.K.; Jung, Y.H.; Kim, H.S.; Kim, I.S. Synthesis of (2H)-Indazoles from Azobenzenes Using Paraformaldehyde as a One-Carbon Synthon. Adv. Synth. Catal. 2019, 361, 1617–1626. [Google Scholar] [CrossRef]
- Yin, C.; Zhong, T.; Zheng, X.; Li, L.; Zhou, J.; Yu, C. Direct Synthesis of Indazole Derivatives via Rh (III)-catalyzed C–H Activation of Phthalazinones and Allenes. Org. Biomol. Chem. 2021, 19, 7701–7705. [Google Scholar] [CrossRef]
- Park, M.S.; Moon, K.; Oh, H.; Lee, J.Y.; Ghosh, P.; Kang, J.Y.; Park, J.S.; Mishra, N.K.; Kim, I.S. Synthesis of (2H)-Indazoles and Dihydrocinnolinones through Annulation of Azobenzenes with Vinylene Carbonate under Rh (III) Catalysis. Org. Lett. 2021, 23, 5518–5522. [Google Scholar] [CrossRef] [PubMed]
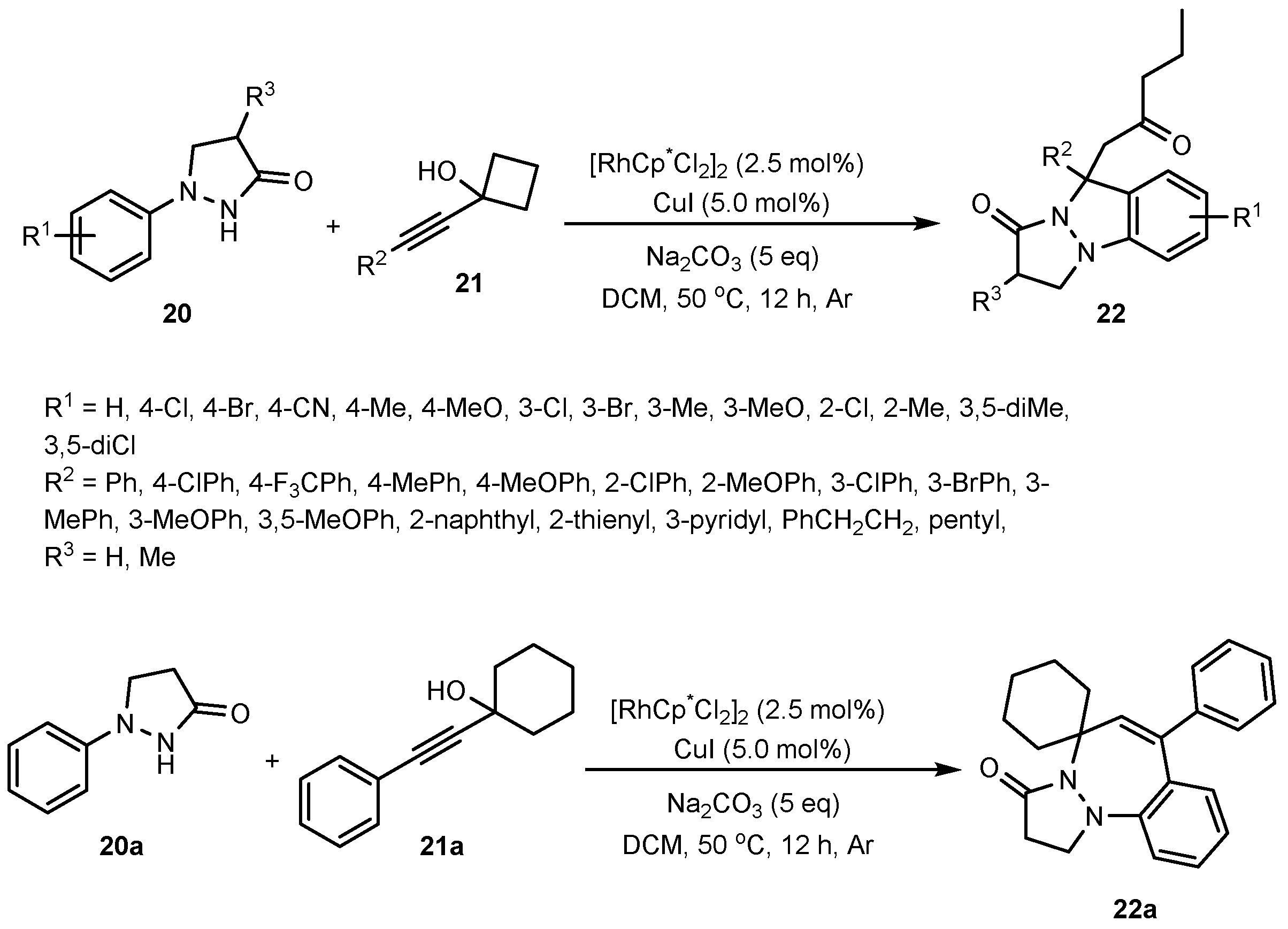
- Zhang, L.; Chen, J.; Chen, X.; Zheng, X.; Zhou, J.; Zhong, T.; Chen, Z.; Yang, Y.-F.; Jiang, X.; She, Y.-B. Rh (III)-catalyzed, Hydrazine-directed C–H Functionalization with 1-Alkynylcyclobutanols: A New strategy for 1H-Indazoles. Chem. Commun. 2020, 56, 7415–7418. [Google Scholar] [CrossRef]
- Lian, Y.; Bergman, R.G.; Lavis, L.D.; Ellman, J.A. Rhodium (III)-catalyzed Indazole Synthesis by C–H Bond Functionalization and Cyclative Capture. J. Am. Chem. Soc. 2013, 135, 7122–7125. [Google Scholar] [CrossRef] [PubMed]
- Gholamhosseyni, M.; Kianmehr, E. A Ruthenium-catalyzed Alkenylation–annulation Approach for the Synthesis of Indazole Derivatives via C–H Bond Activation. Org. Biomol. Chem. 2018, 16, 5973–5978. [Google Scholar] [CrossRef]
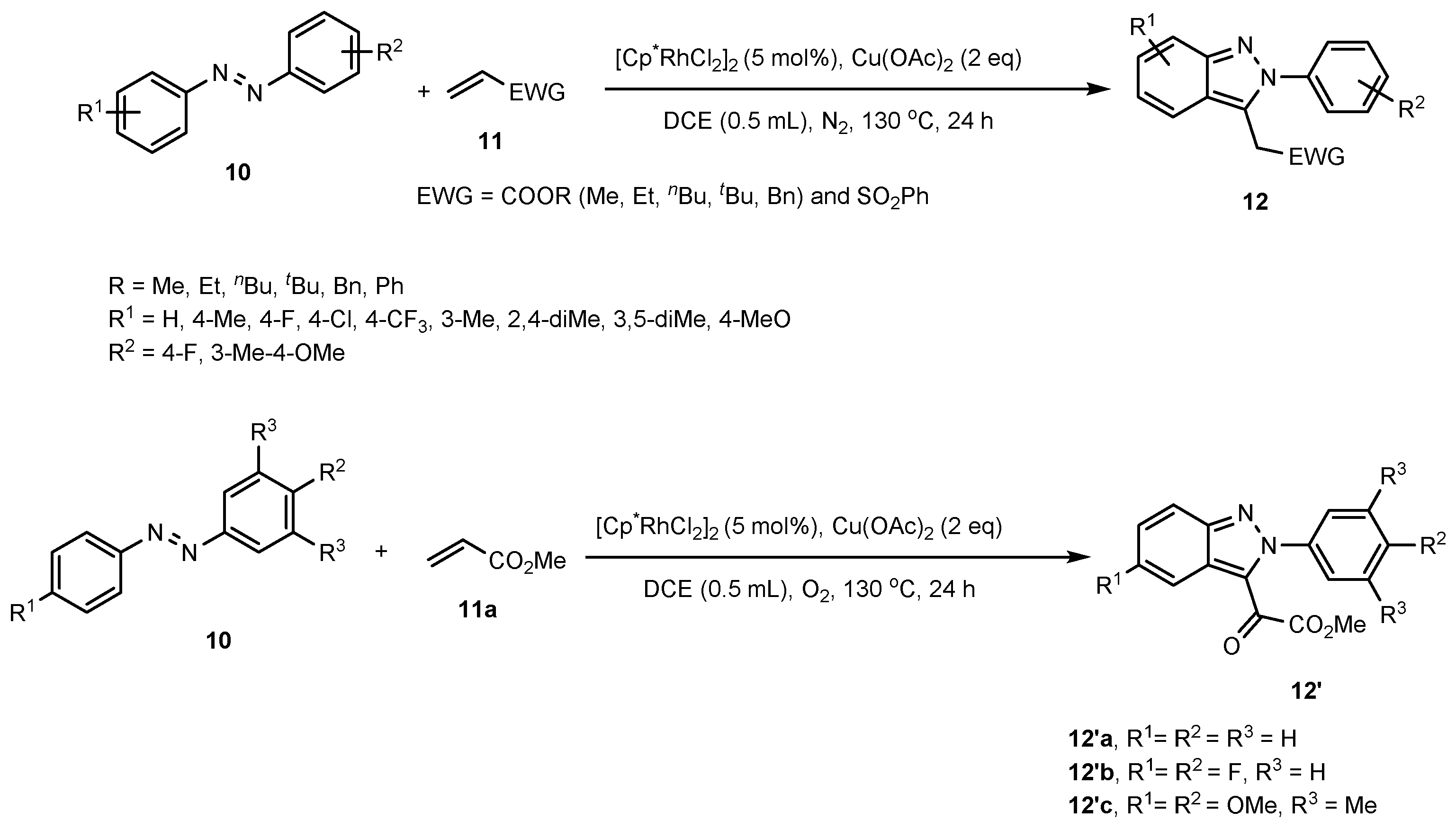
- Yu, D.-G.; Suri, M.; Glorius, F. Rh (III)/Cu (II)-Cocatalyzed Synthesis of 1H-Indazoles Through C–H Amidation and N–N Bond Formation. J. Am. Chem. Soc. 2013, 135, 8802–8805. [Google Scholar] [CrossRef] [PubMed]
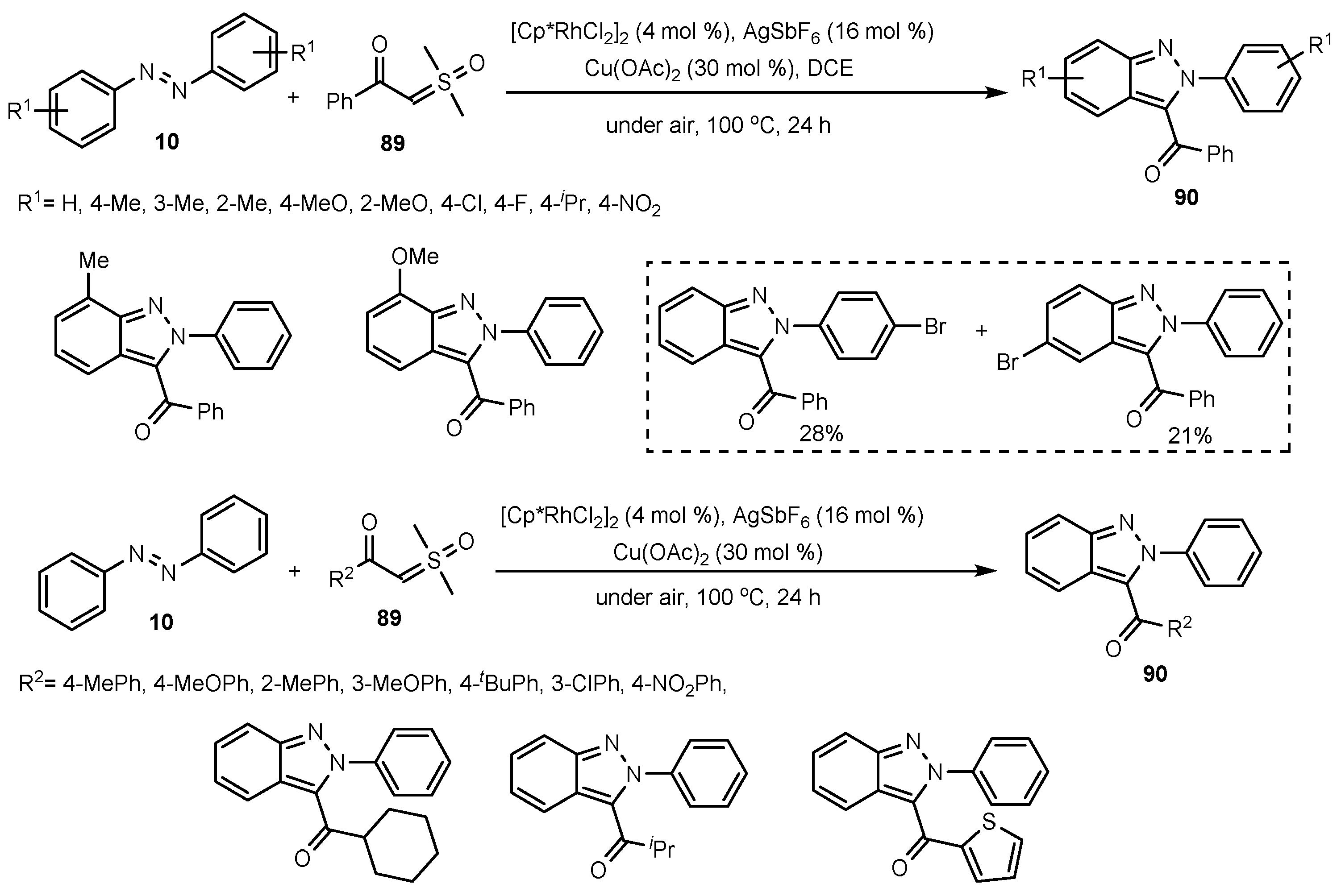
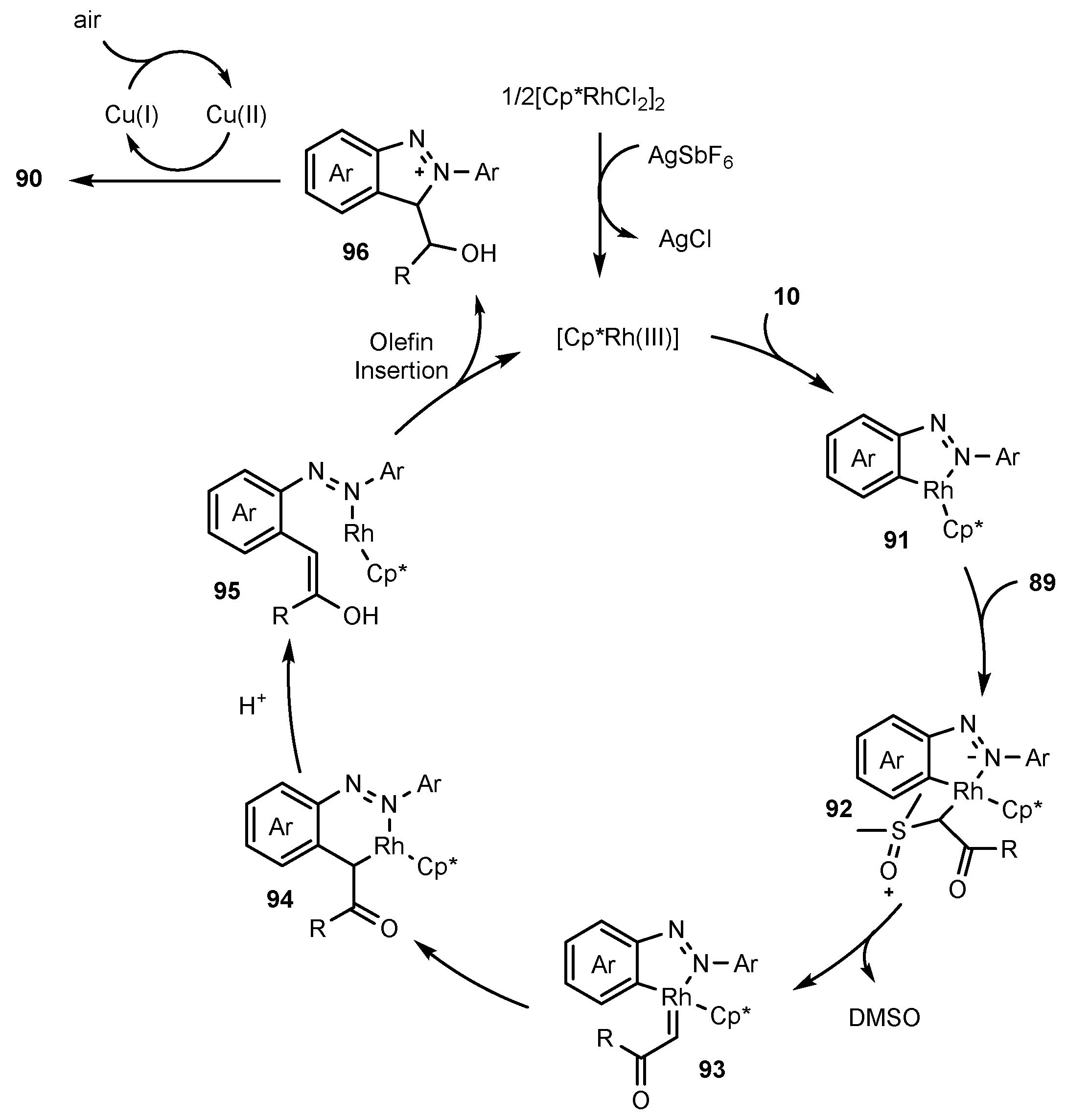
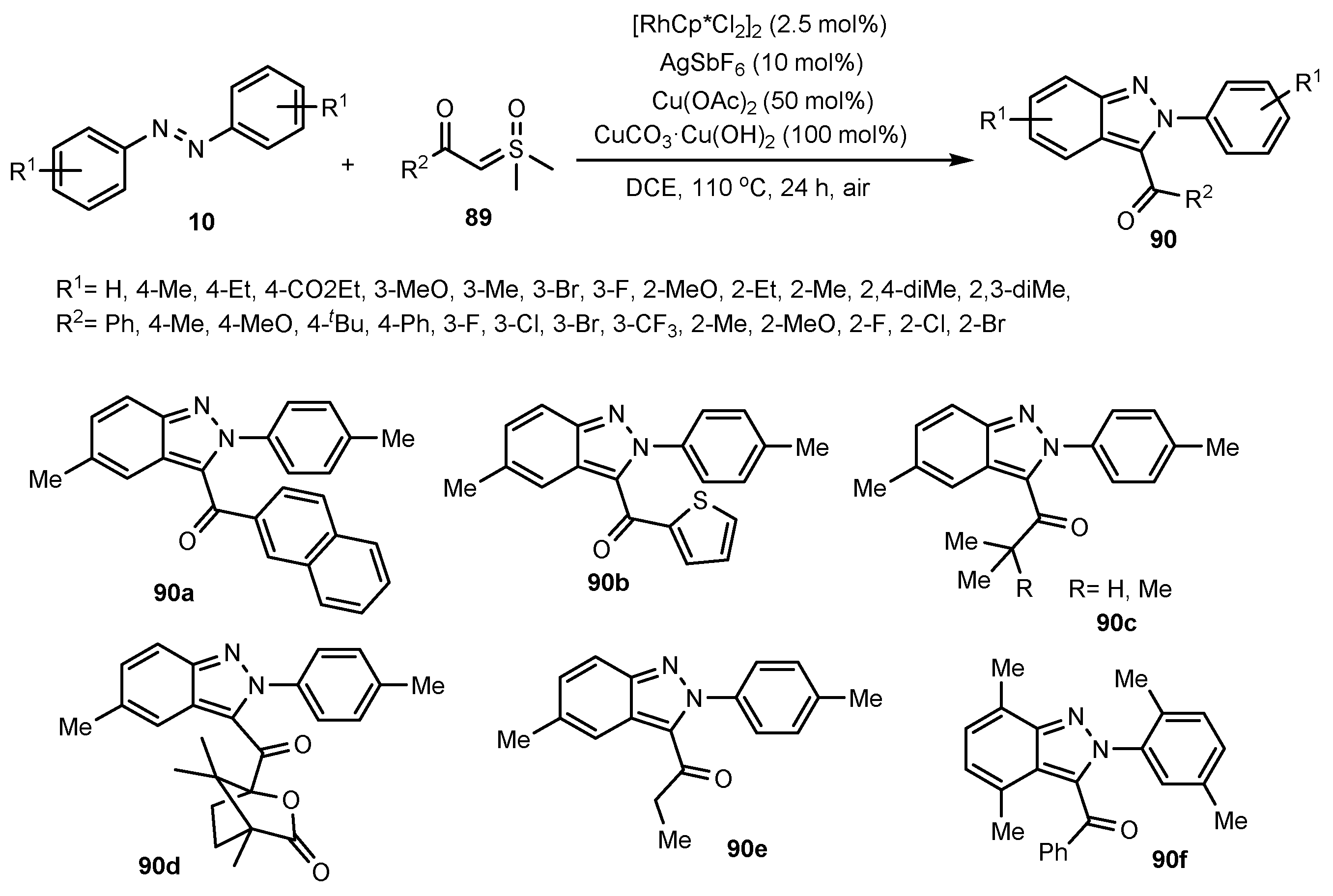
- Zhu, J.; Sun, S.; Cheng, J. Rh (III)-catalyzed [4 + 1]-Annulation of Azobenzenes with α-Carbonyl Sulfoxonium Ylides Toward 3-acyl-(2H)-Indazoles. Tetrahedron Lett. 2018, 59, 2284–2287. [Google Scholar] [CrossRef]
- Oh, H.; Han, S.; Pandey, A.K.; Han, S.H.; Mishra, N.K.; Kim, S.; Chun, R.; Kim, H.S.; Park, J.; Kim, I.S. Synthesis of (2H)-Indazoles through Rh (III)-Catalyzed Annulation Reaction of Azobenzenes with Sulfoxonium Ylides. J. Org. Chem. Res. 2018, 83, 4070–4077. [Google Scholar] [CrossRef] [PubMed]
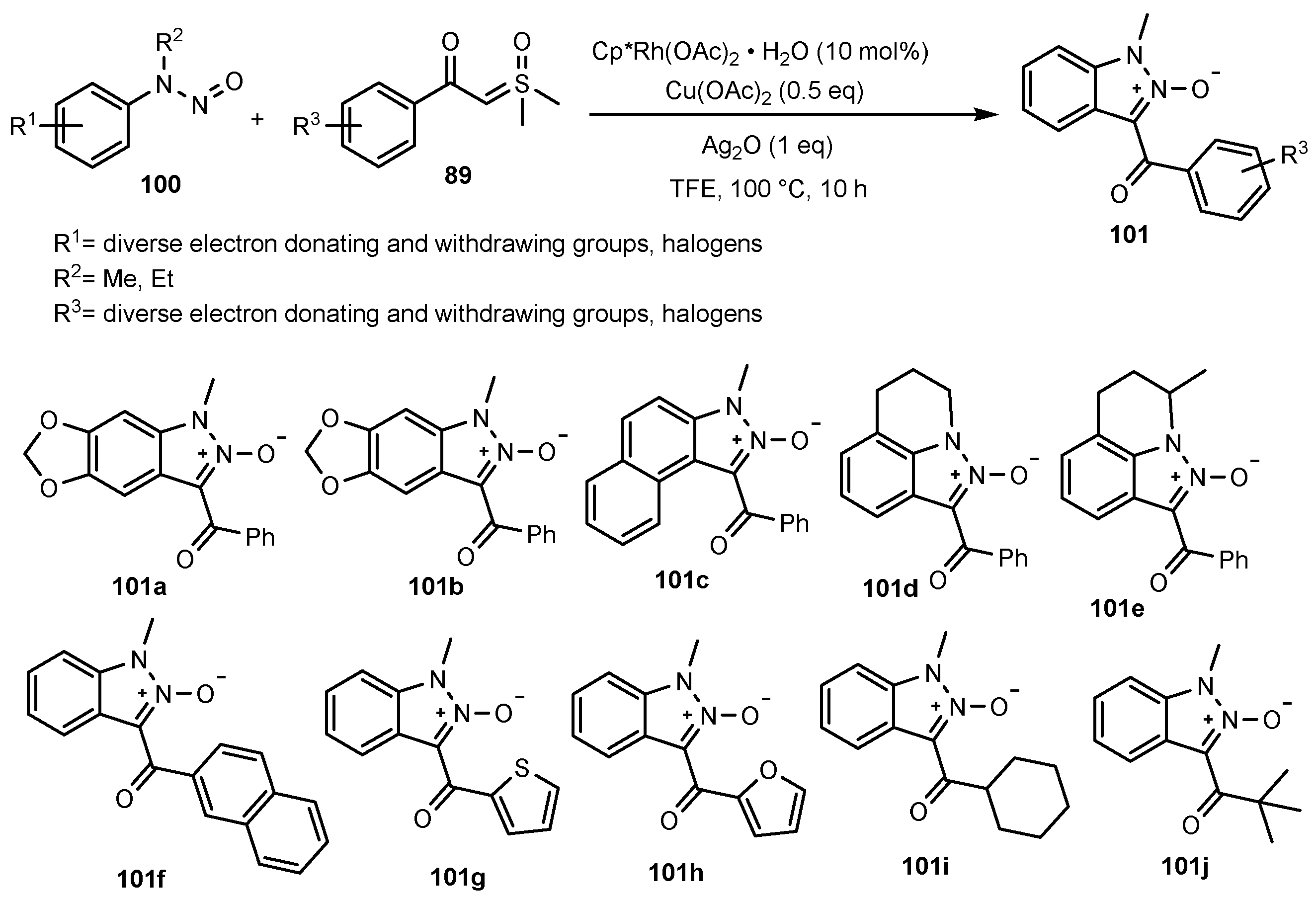
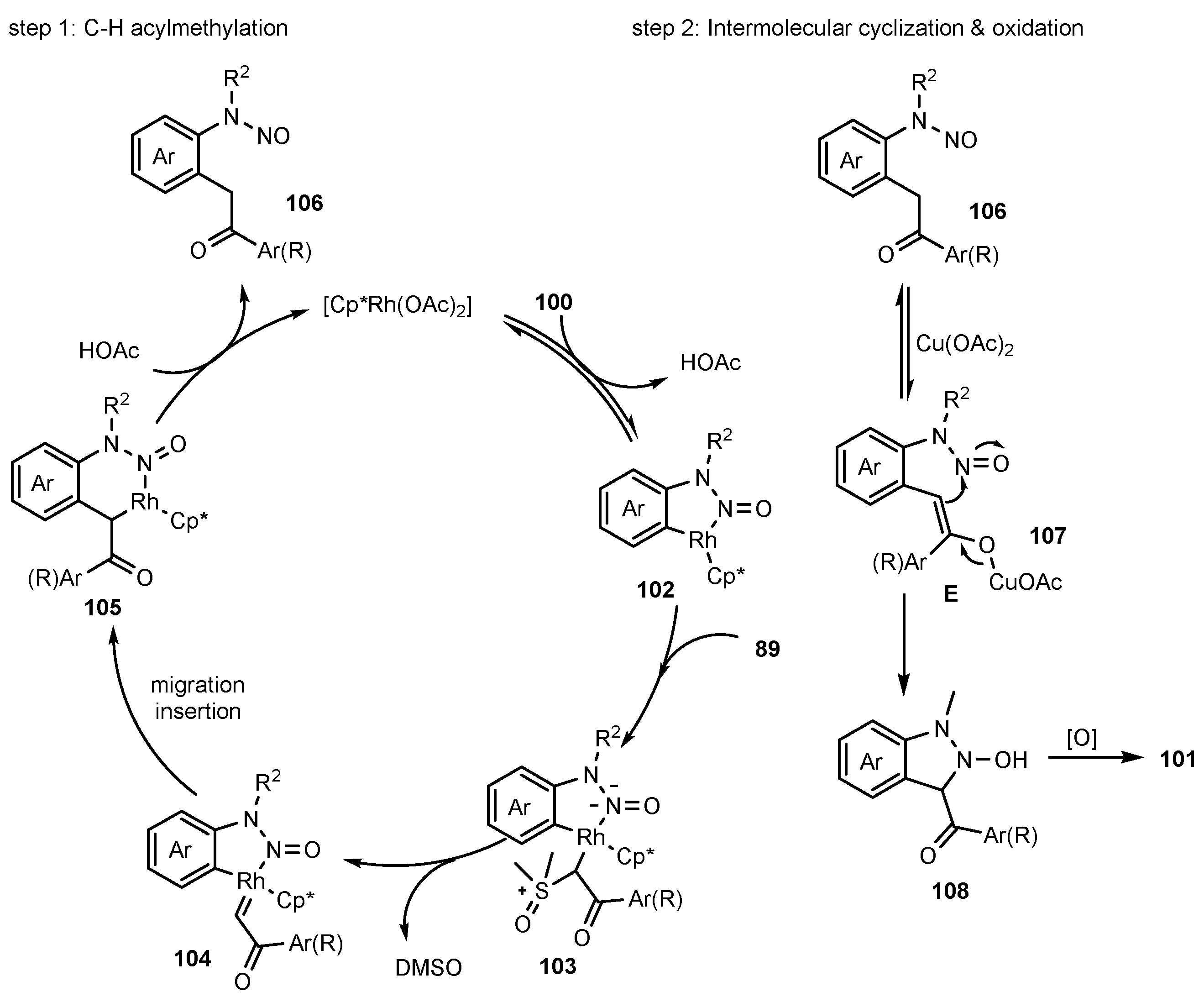
- Cui, X.-F.; Huang, G.-S. Rhodium-catalyzed Tandem Acylmethylation/annulation of N-Nitrosoanilines with Sulfoxonium Ylides for the Synthesis of Substituted Indazole N-Oxides. Org. Biomol. Chem. 2020, 18, 4014–4018. [Google Scholar] [CrossRef] [PubMed]
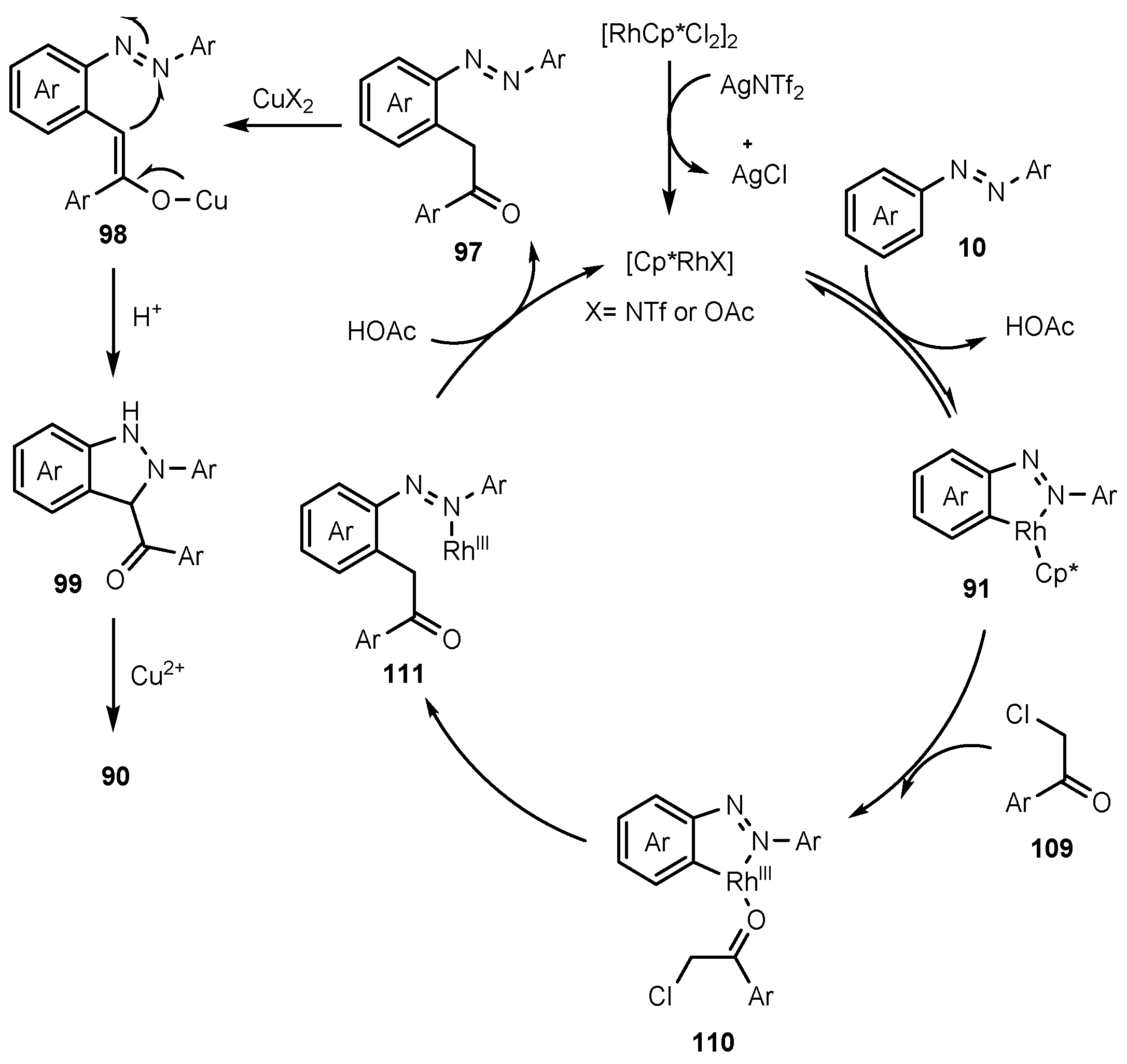
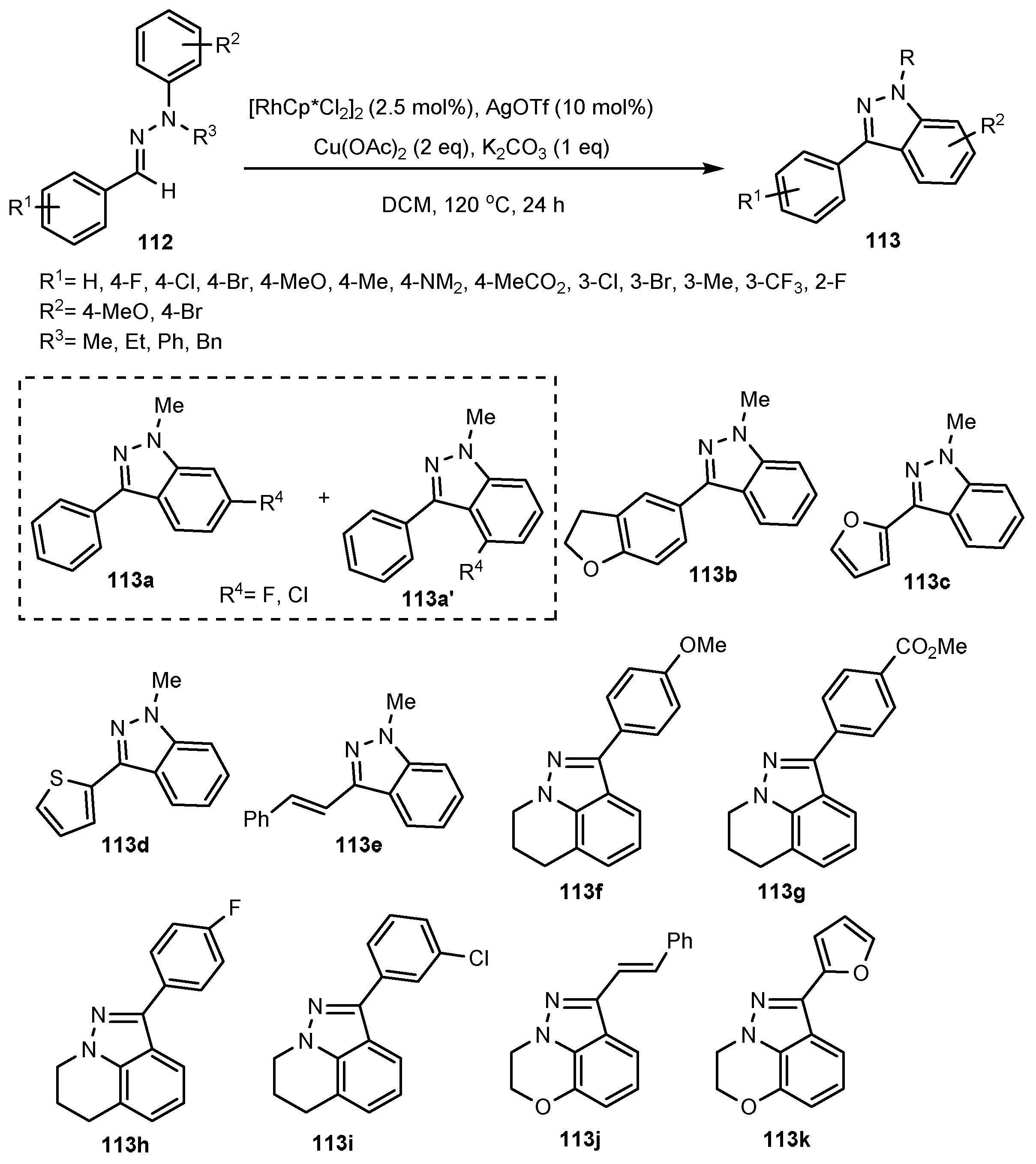
- Li, H.; Han, Y.; Yang, Z.; Yao, Z.; Wang, L.; Cui, X. Rh (III)-catalyzed Annulation of Azobenzenes and α-Cl Ketones toward 3-Acyl-2H-indazoles. Chin. Chem. Lett. 2021, 32, 1709–1712. [Google Scholar] [CrossRef]
- Xu, P.; Wang, G.; Wu, Z.; Zhu, C. Rh (III)-Catalyzed Double C–H Activation of Aldehyde Hydrazones: A Route for Functionalized 1H-Indazole Synthesis. Chem. Sci. 2017, 8, 1303–1308. [Google Scholar] [CrossRef] [Green Version]
- Long, Z.; Yang, Y.; You, J. Rh (III)-Catalyzed [4 + 1]-annulation of Azoxy Compounds with Alkynes: A Regioselective Approach to 2H-Indazoles. Org. Lett. 2017, 19, 2781–2784. [Google Scholar] [CrossRef]
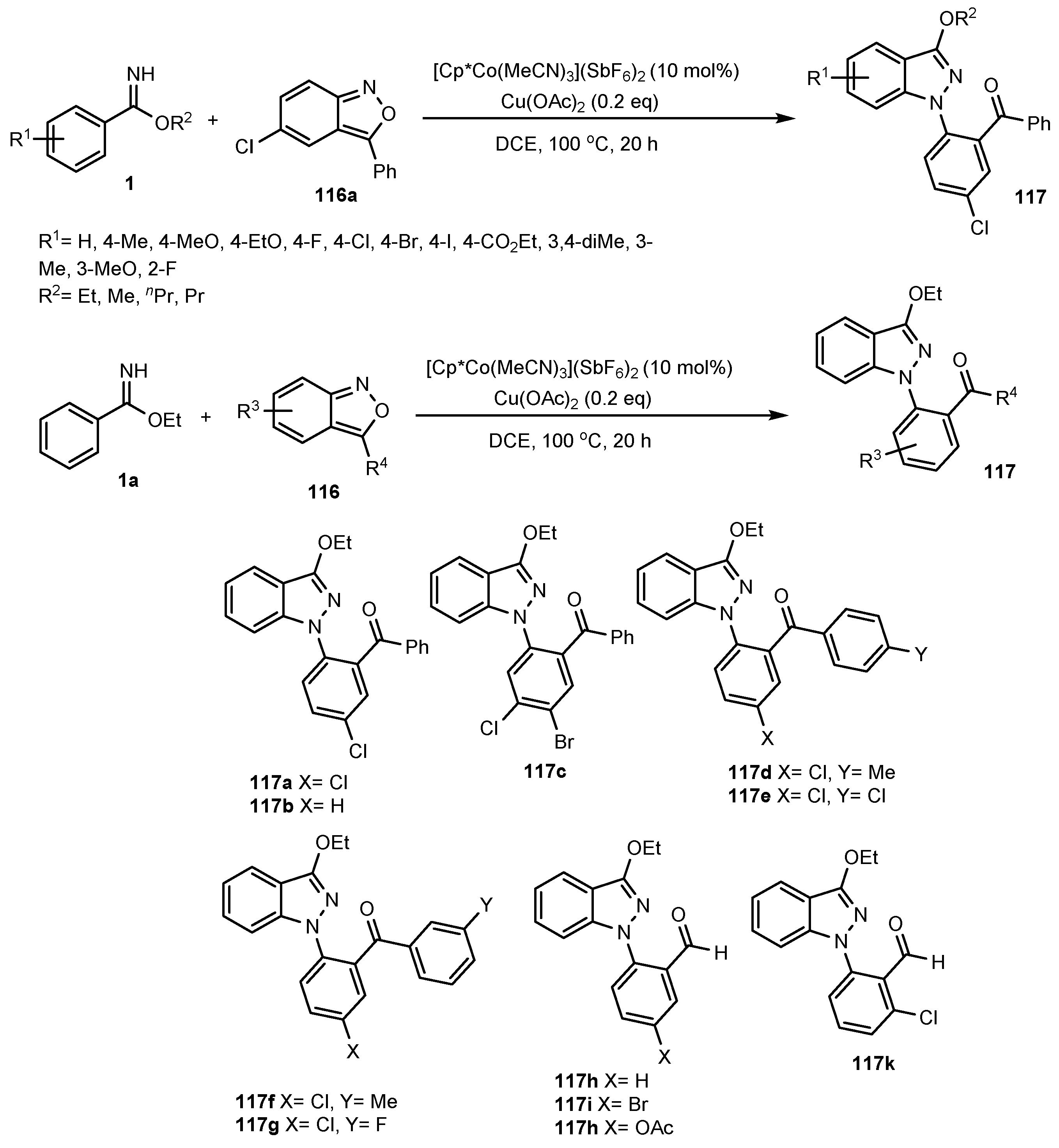
- Li, L.; Wang, H.; Yu, S.; Yang, X.; Li, X. Cooperative Co (III)/Cu (II)-Catalyzed C–N/N–N Coupling of Imidates with Anthranils: Access to 1H-Indazoles via C–H Activation. Org. Lett. 2016, 18, 3662–3665. [Google Scholar] [CrossRef]
- Wang, G.; Sun, J.; Wang, K.; Han, J.; Li, H.; Duan, G.; You, G.; Li, F.; Xia, C. Palladium-catalyzed Direct C–H Nitration and Intramolecular C–H Functionalization for the Synthesis of 3-Nitro-1-(phenylsulfonyl)-1H-indazole Derivatives. Org. Chem. Front. 2019, 6, 1608–1612. [Google Scholar] [CrossRef]
- Zheng, Q.-Z.; Feng, P.; Liang, Y.-F.; Jiao, N. Pd-Catalyzed Tandem C–H Azidation and N–N Bond Formation of Arylpyridines: A Direct Approach to Pyrido [1, 2-b] Indazoles. Org. Lett. 2013, 15, 4262–4265. [Google Scholar] [CrossRef] [PubMed]
- Inamoto, K.; Saito, T.; Katsuno, M.; Sakamoto, T.; Hiroya, K. Palladium-catalyzed C−H Activation/intramolecular Amination Reaction: A New Route to 3-Aryl/alkylindazoles. Org. Lett. 2007, 9, 2931–2934. [Google Scholar] [CrossRef]
- Hummel, J.R.; Ellman, J.A. Cobalt (III)-catalyzed Synthesis of Indazoles and Furans by C–H Bond Functionalization/addition/cyclization Cascades. J. Am. Chem. Soc. 2015, 137, 490–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
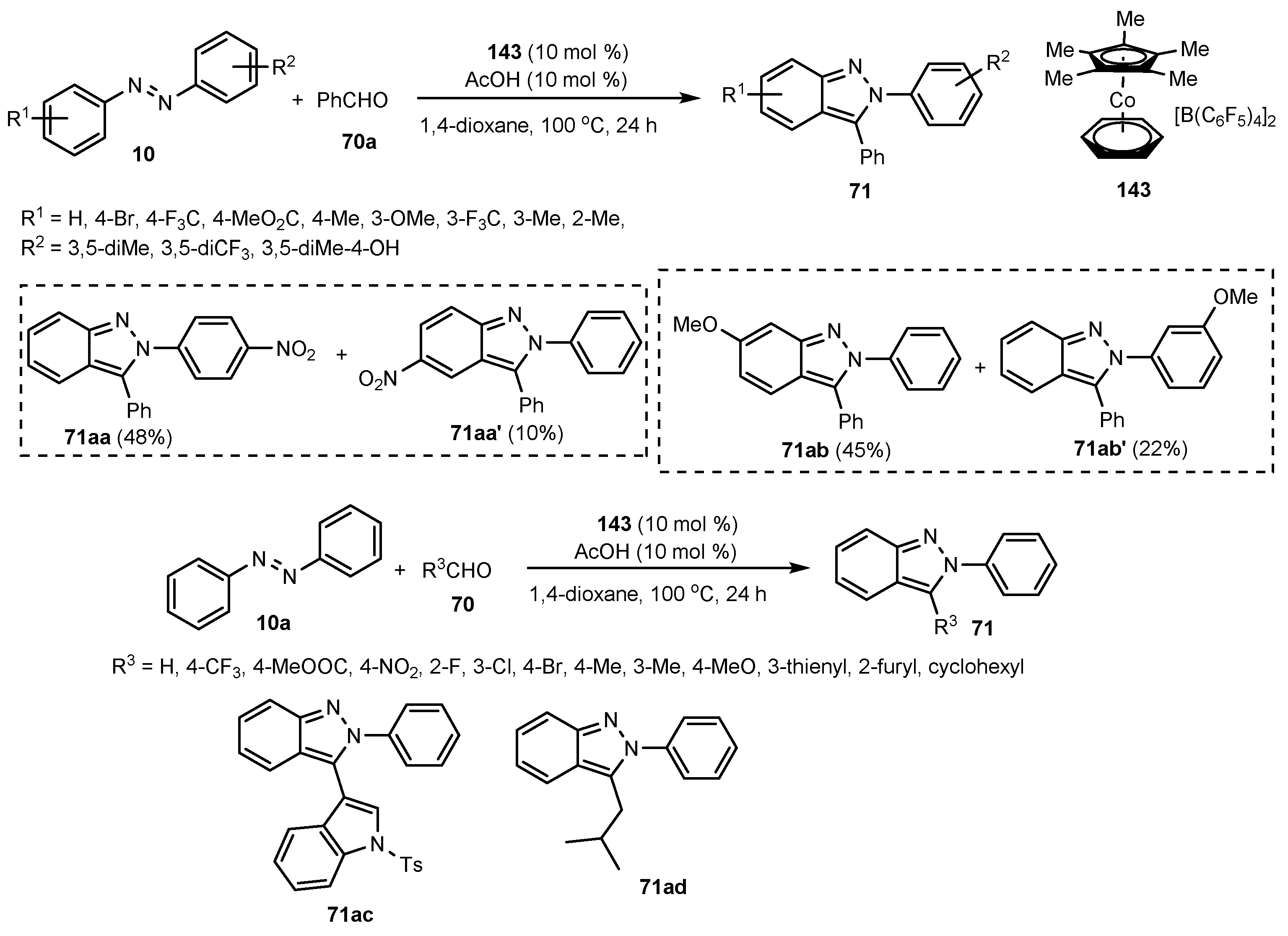
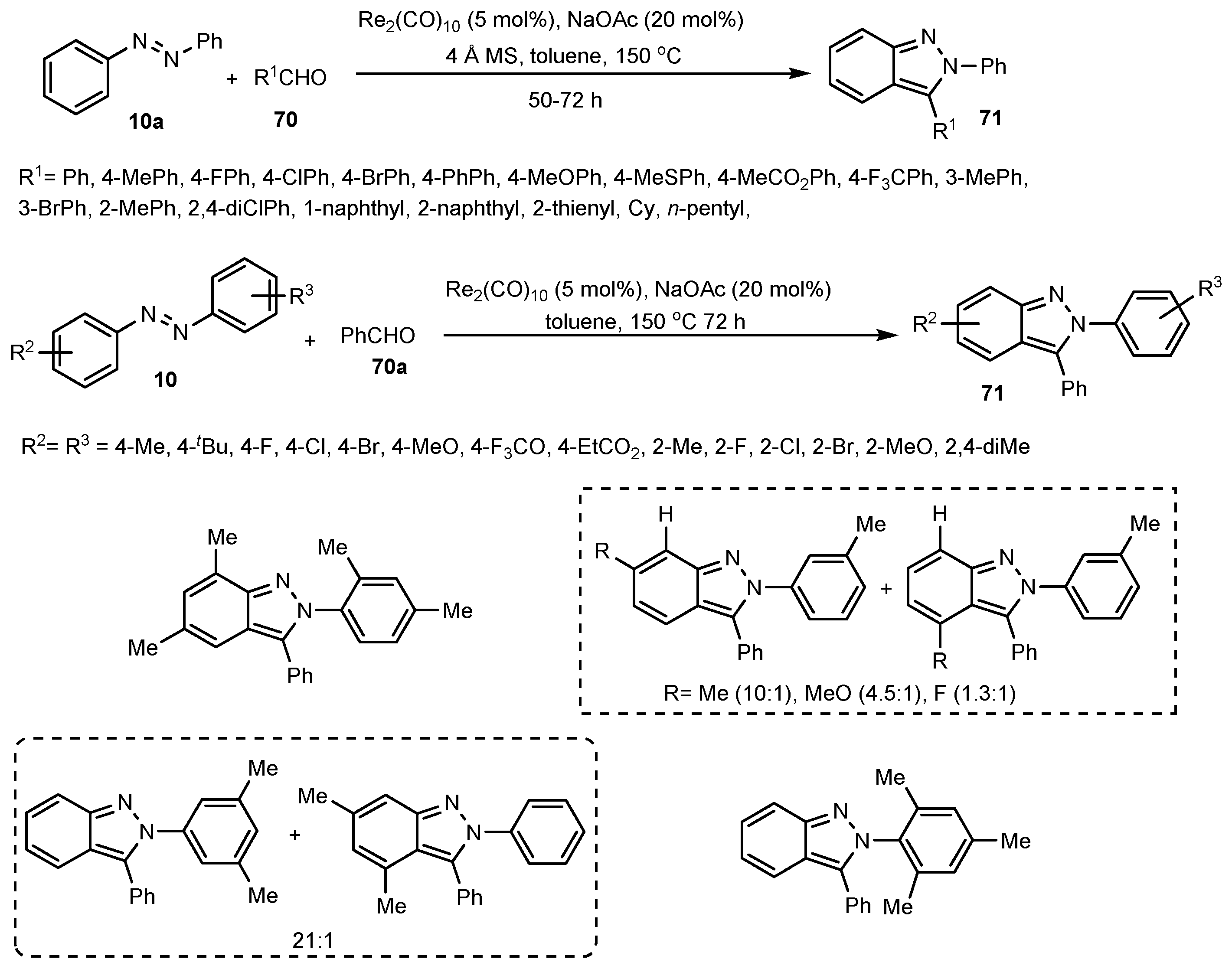
- Geng, X.; Wang, C. Rhenium-catalyzed [4+1] Annulation of Azobenzenes and Aldehydes via Isolable Cyclic Rhenium(I) Complexes. Org. Lett. 2015, 17, 2434–2437. [Google Scholar] [CrossRef]
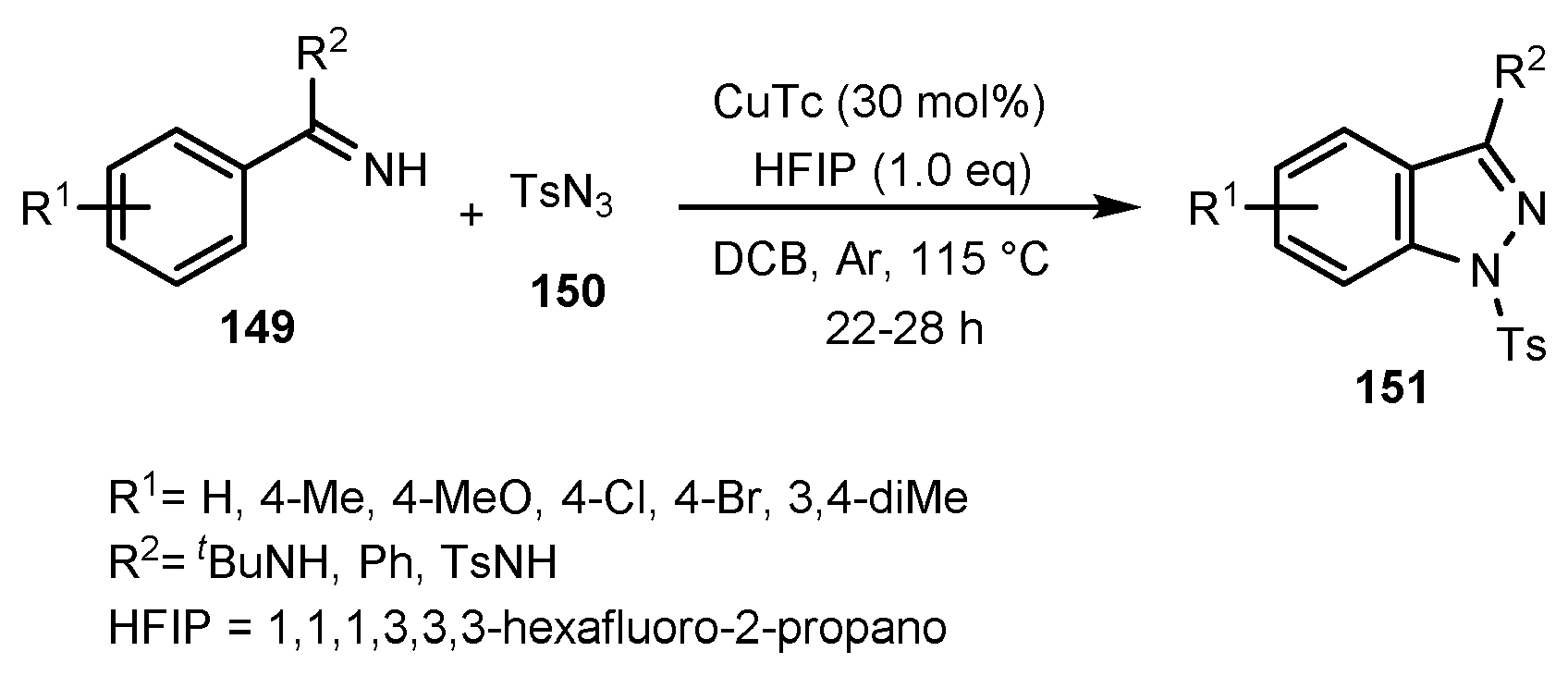
- Peng, J.; Xie, Z.; Chen, M.; Wang, J.; Zhu, Q. Copper-catalyzed C (sp2)–H Amidation with Azides as Amino Sources. Org. Lett. 2014, 16, 4702–4705. [Google Scholar] [CrossRef]

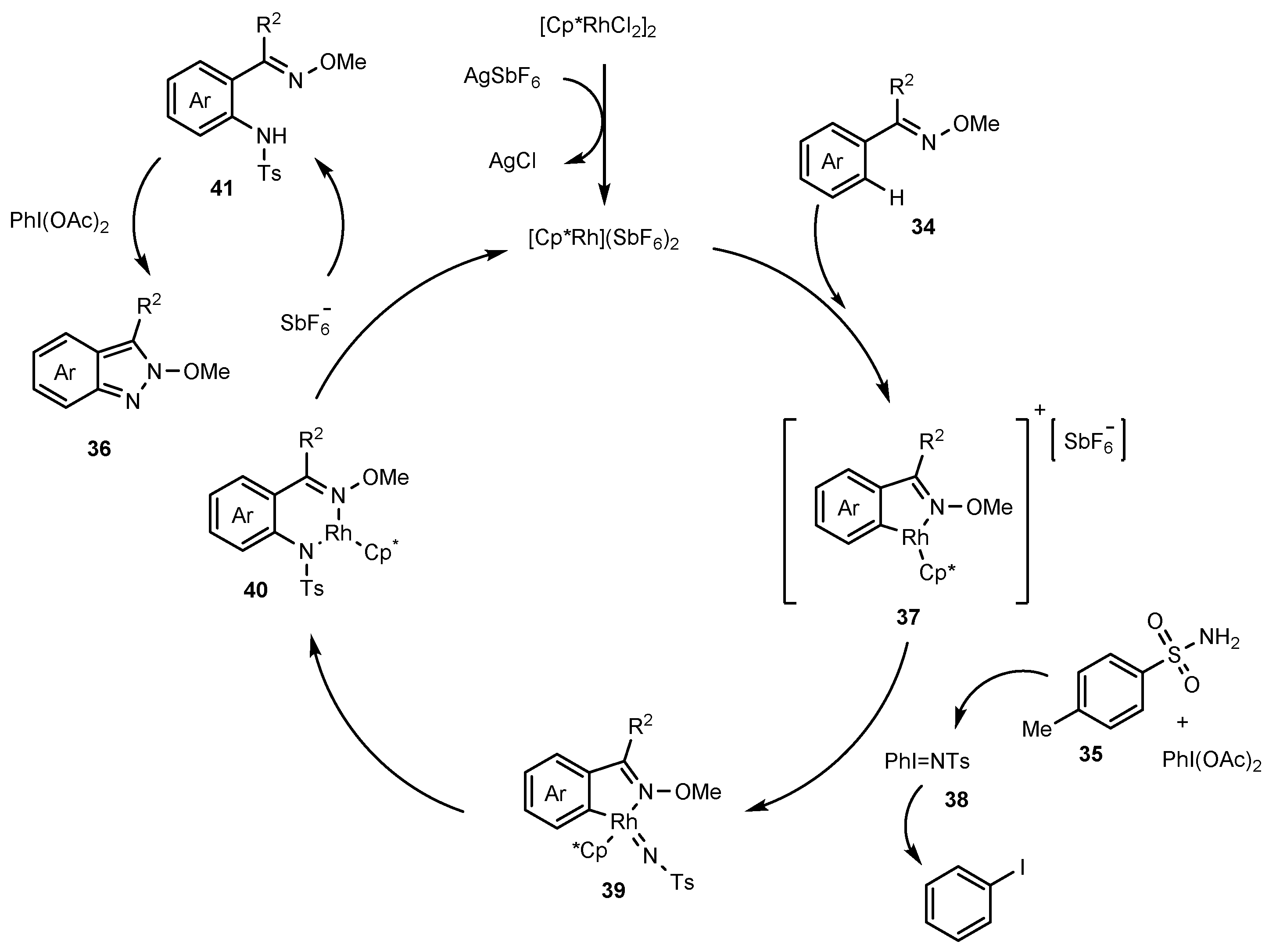
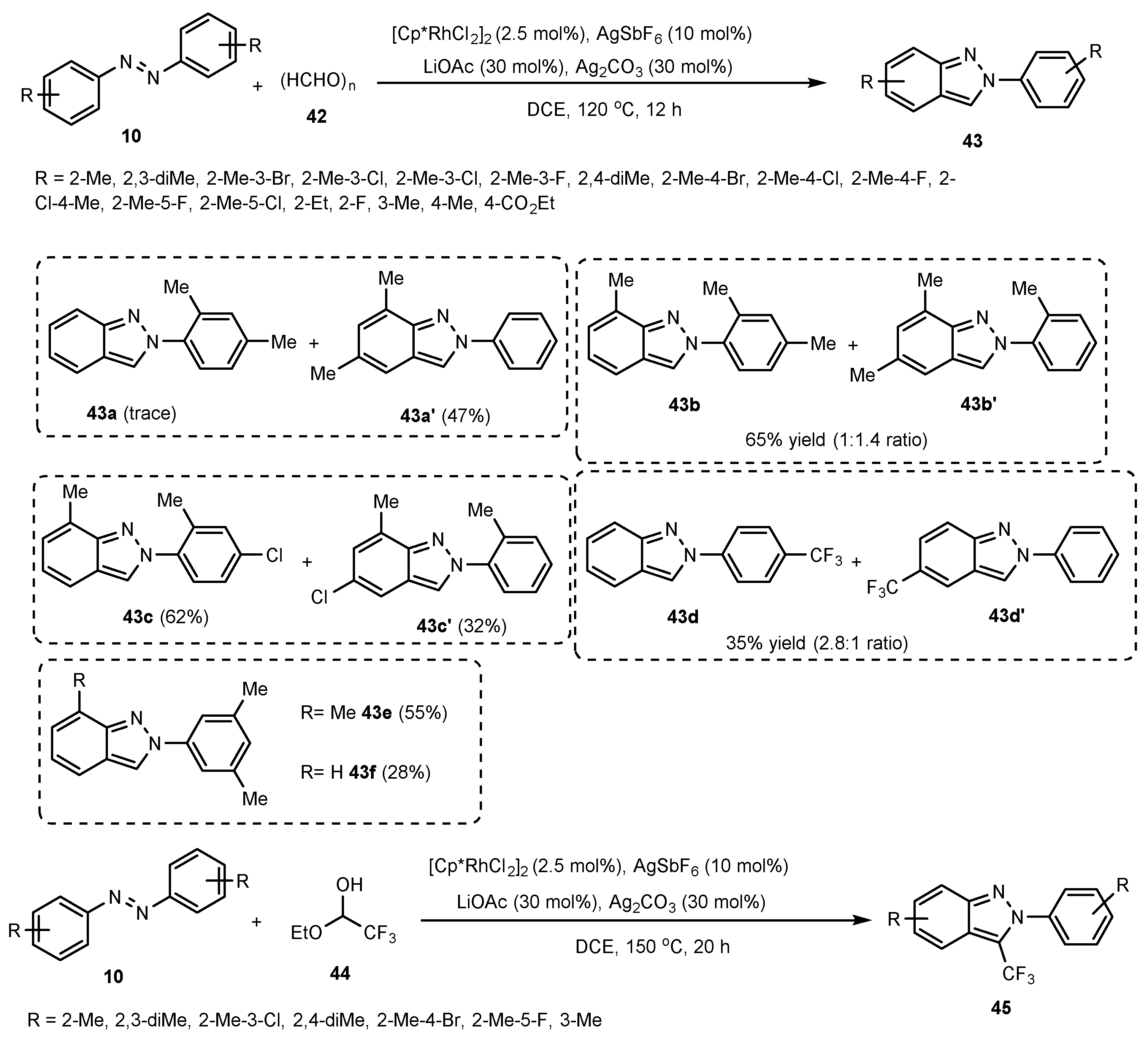

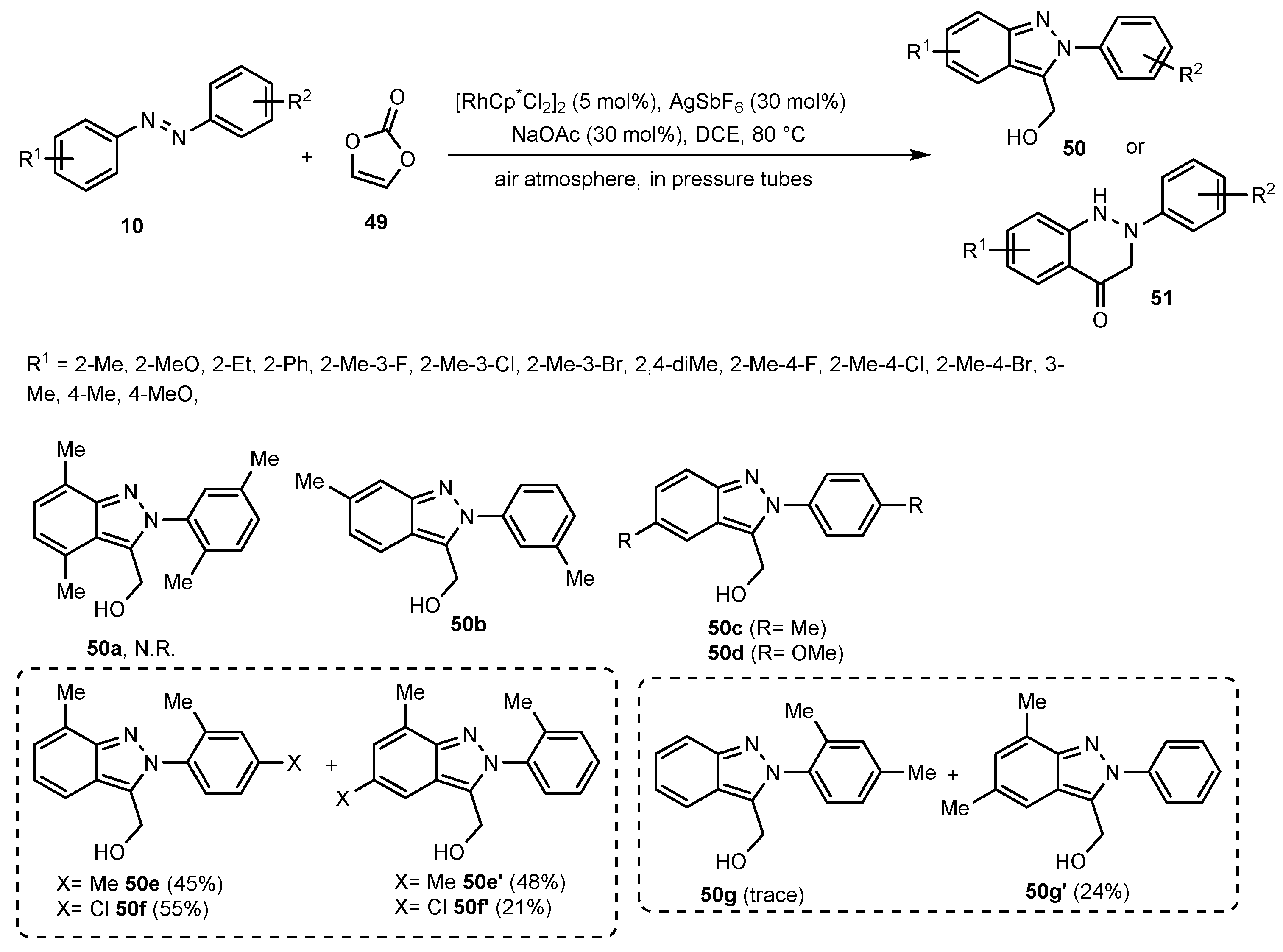


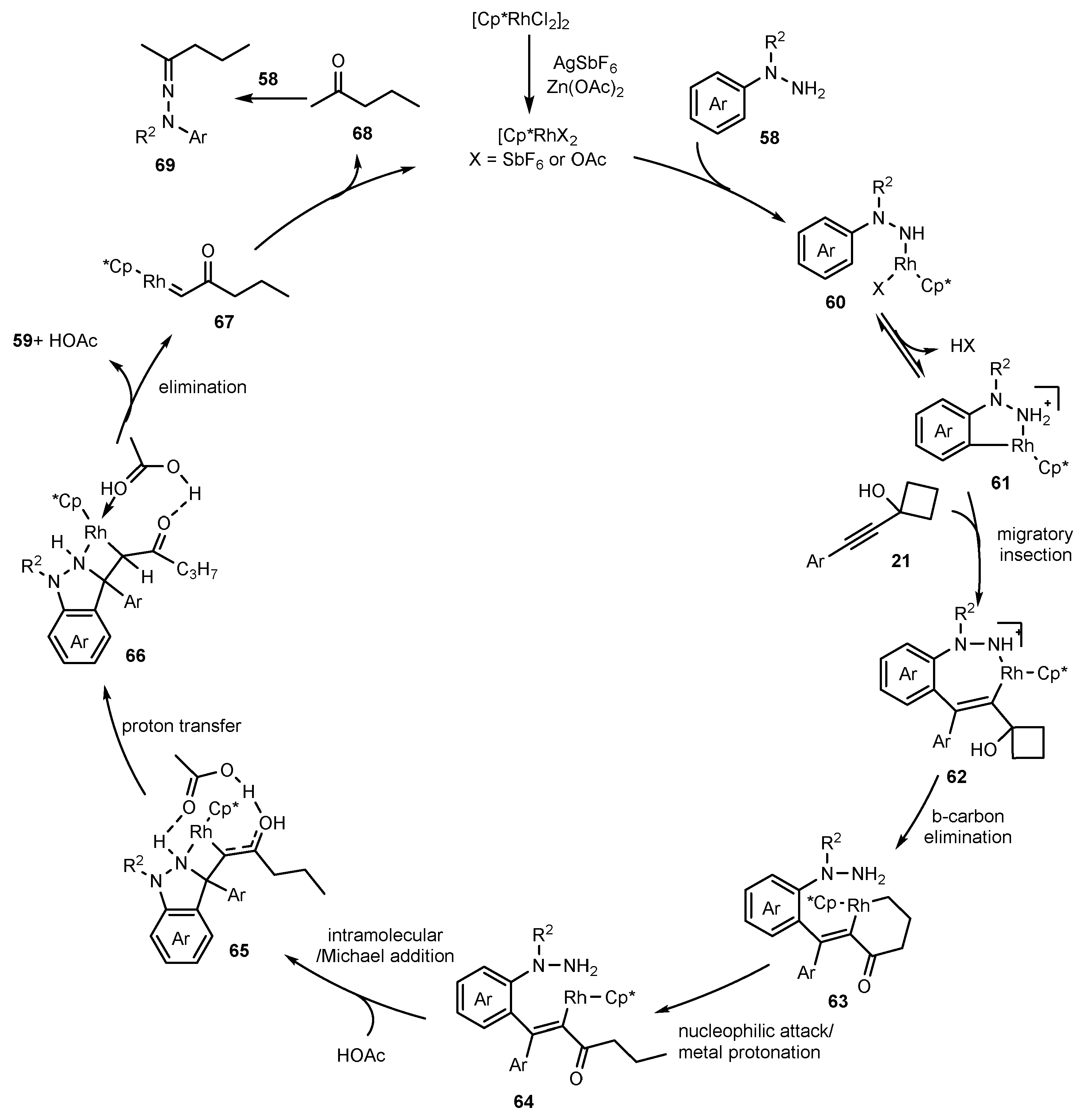
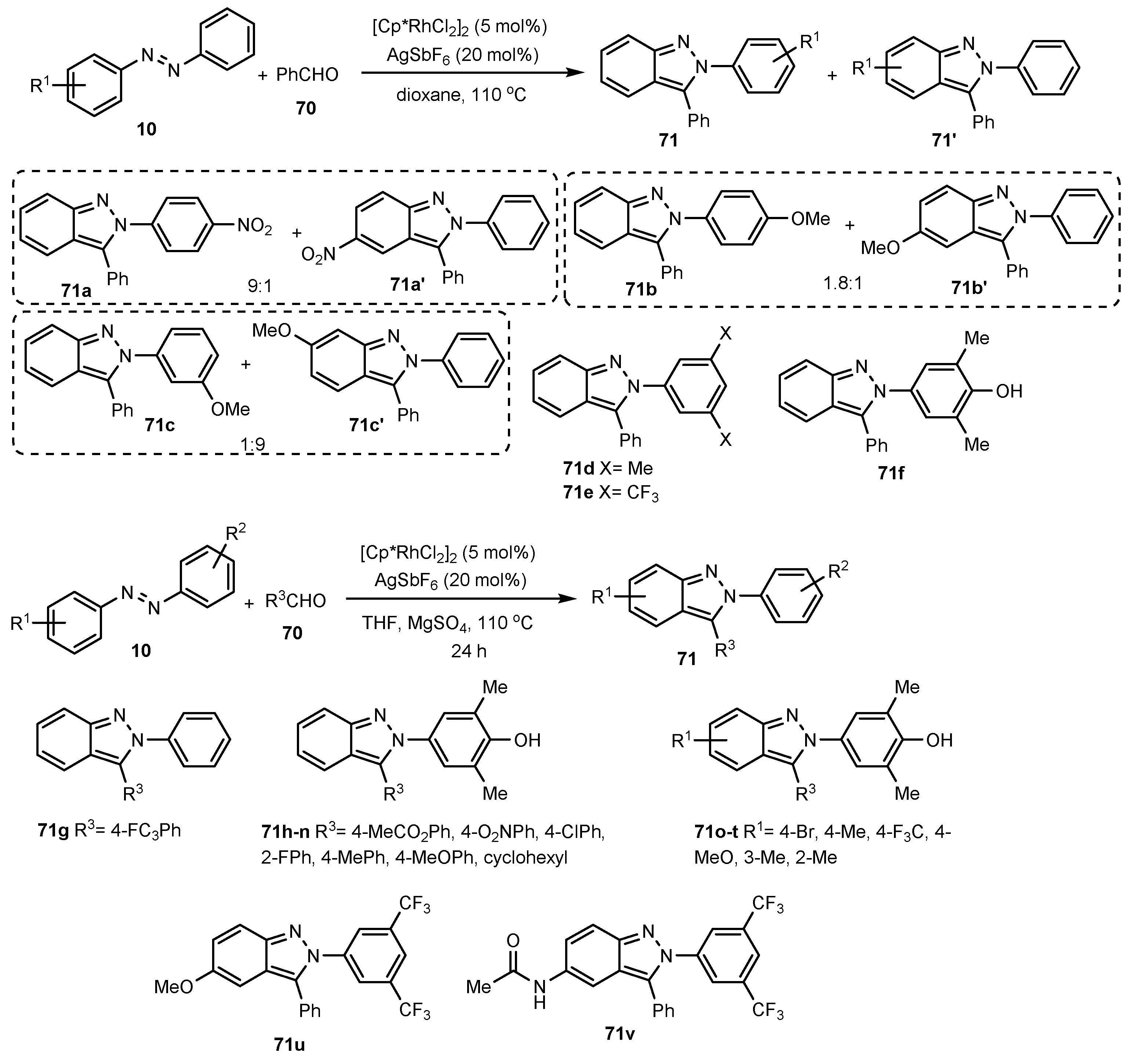
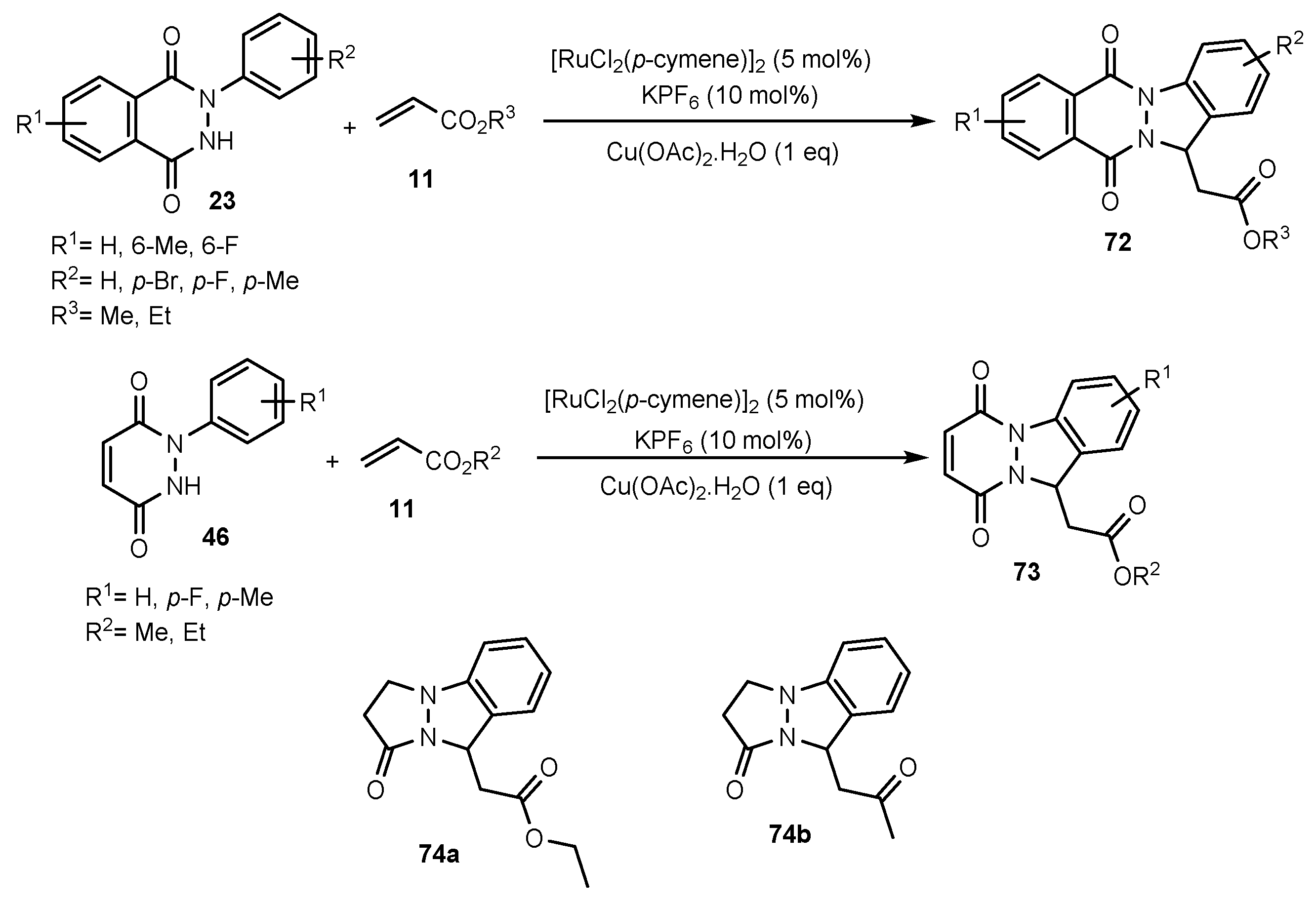
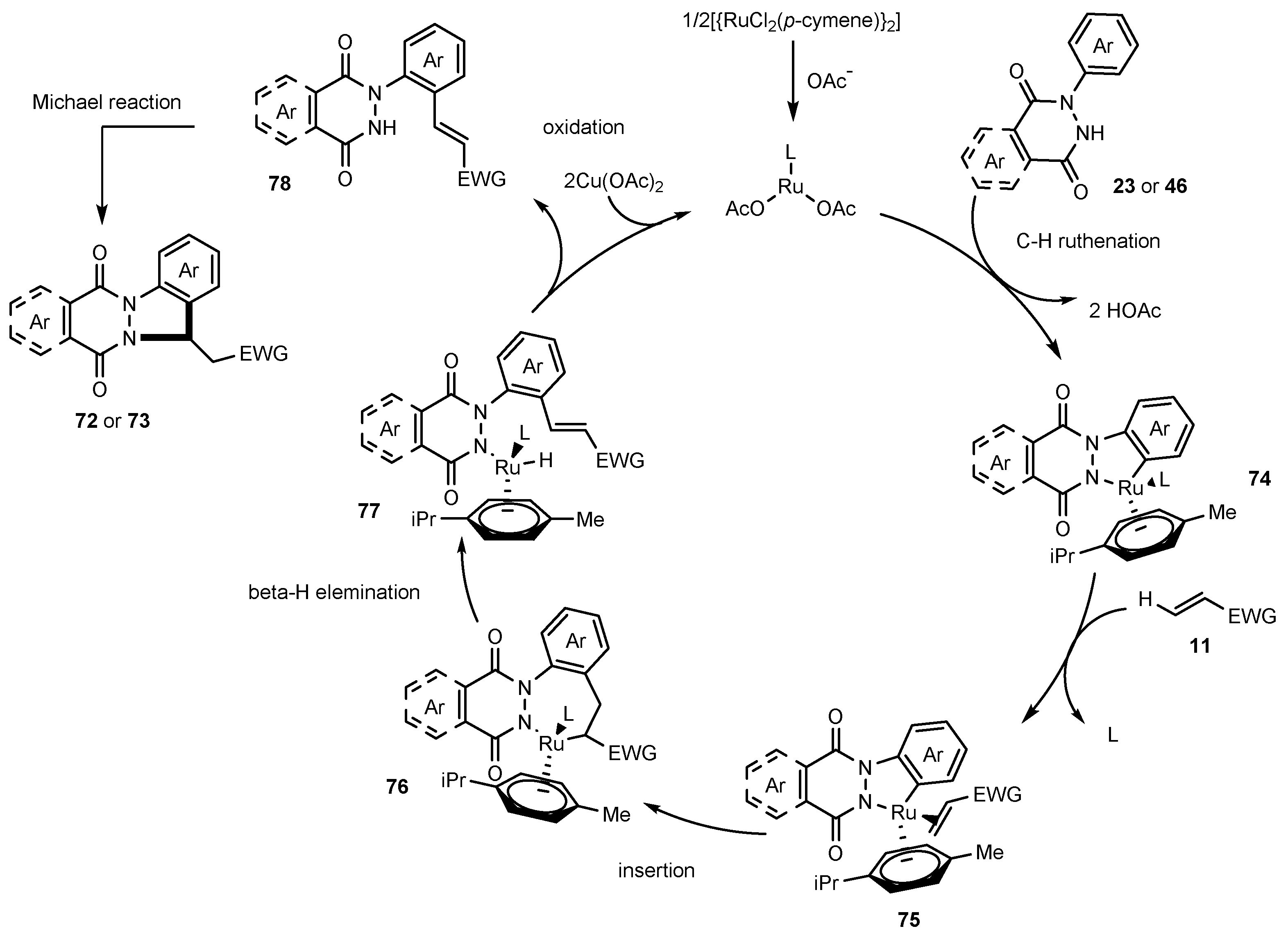

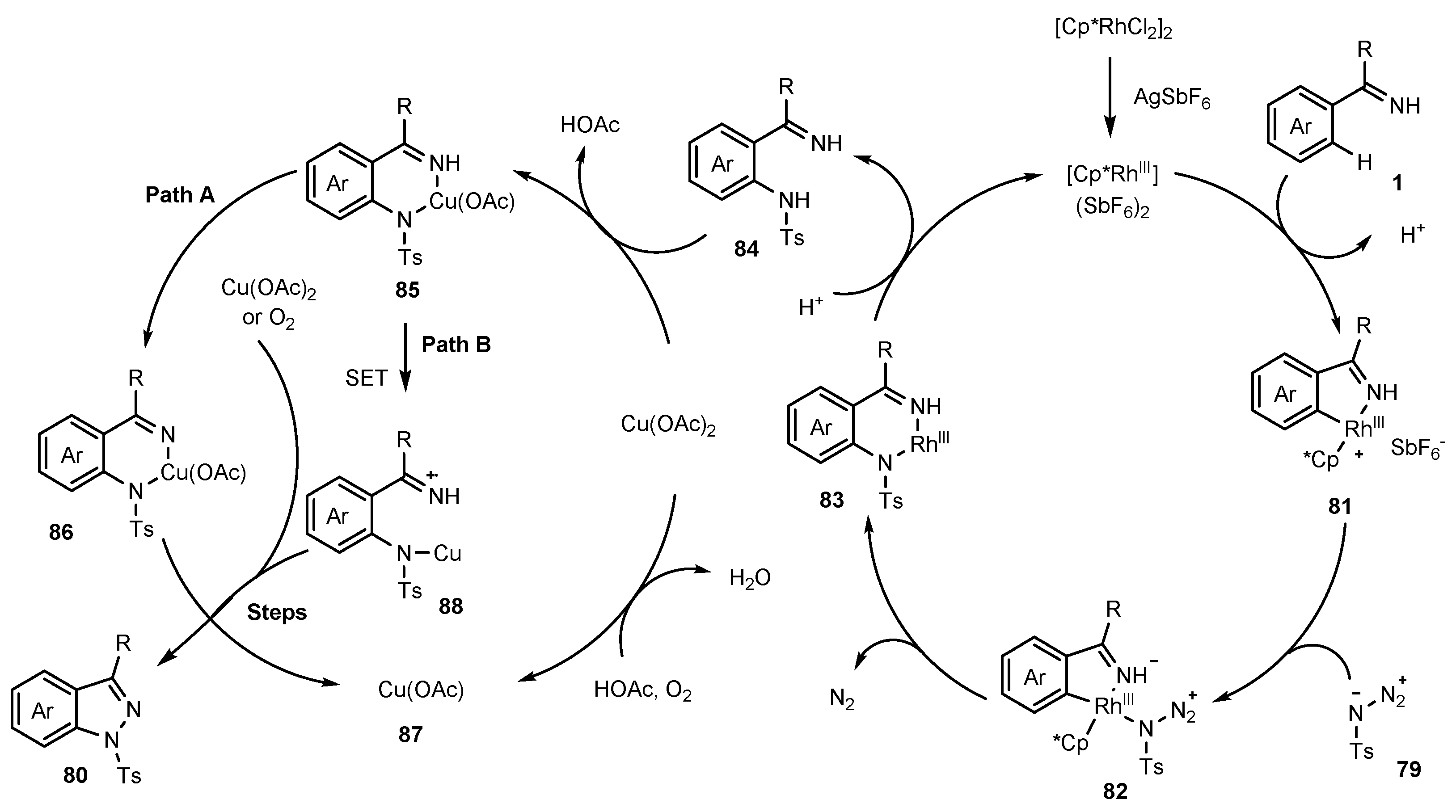








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiri, P.; Roosta, A.; Dehaen, W.; Amani, A.M. Recent Strategies in Transition-Metal-Catalyzed Sequential C–H Activation/Annulation for One-Step Construction of Functionalized Indazole Derivatives. Molecules 2022, 27, 4942. https://doi.org/10.3390/molecules27154942
Shiri P, Roosta A, Dehaen W, Amani AM. Recent Strategies in Transition-Metal-Catalyzed Sequential C–H Activation/Annulation for One-Step Construction of Functionalized Indazole Derivatives. Molecules. 2022; 27(15):4942. https://doi.org/10.3390/molecules27154942
Chicago/Turabian StyleShiri, Pezhman, Atefeh Roosta, Wim Dehaen, and Ali Mohammad Amani. 2022. "Recent Strategies in Transition-Metal-Catalyzed Sequential C–H Activation/Annulation for One-Step Construction of Functionalized Indazole Derivatives" Molecules 27, no. 15: 4942. https://doi.org/10.3390/molecules27154942