
Competing Metal–Ligand Interactions in Tris(cyclopentadienyl)-cyclohexylisonitrile Complexes of Trivalent Actinides and Lanthanides


Abstract
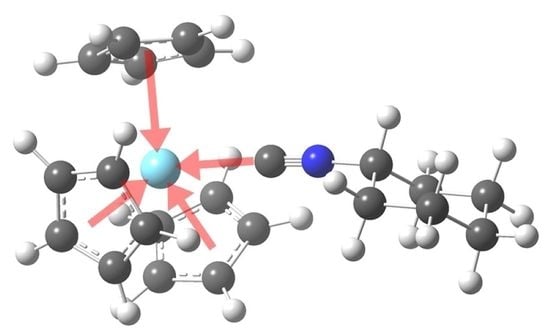
:
1. Introduction
2. Results and Discussion
2.1. Crystal and Molecular Structure

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | M-CC≡N | C≡N | M-CCp,av | νC≡N |
|---|---|---|---|---|
| La | 2.686(16) | 1.147(6) | 2.830 | 2180 3 |
| La’ | 2.818(6) | 1.149(8) | 2.854 | - |
| Ce | 2.641(15) | 1.145(18) | 2.795 | 2197 3 |
| Pr 2 | 2.65(1) | 1.11(1) | 2.78 | 2203 3 |
| Nd | 2.618(4) | 1.143(5) | 2.779 | 2207 4 |
| Sm | 2.576(4) | 1.149(5) | 2.758 | 2202 3 |
| Eu | 2.567(5) | 1.149(5) | 2.739 | 2200 3 |
| Gd | 2.535(2) | 1.147(3) | 2.737 | 2196 3 |
| Tb | 2.513(3) | 1.150(3) | 2.724 | 2205 4 |
| Dy | 2.497(2) | 1.149(2) | 2.710 | 2204 3 |
| Ho | 2.475(2) | 1.150(2) | 2.705 | 2205 4 |
| Er | 2.460(2) | 1.147(2) | 2.699 | 2206 3 |
| Tm | 2.447(2) | 1.148(2) | 2.692 | 2204 3 |
| Yb | 2.443(3) | 1.155(3) | 2.685 | 2203 4 |
| Lu | 2.415(2) | 1.149(3) | 2.680 | 2210 3 |
| U | - | - | - | 2160 5 |
| Np | - | - | - | 2166 3 |
| Pu | 2.58(3) | 1.10(3) | 2.762 | 2190 3 |

2.2. Structure of the (Cp)3La(C≡NCy)2 Bis-Adduct
2.3. 1H and 13C NMR Characteristics of (Cp)3Pu(C≡NCy)
2.4. Bonding Analysis
2.5. Competing Metal–Ligand Interactions: Cp3 vs. C≡NCy
3. Materials and Methods
3.1. Syntheses
3.2. Single Crystal XRD
3.3. NMR Spectroscopy
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilkinson, G.; Birmingham, J.M. Cyclopentadienyl Compounds of Sc, Y, La, Ce and Some Lanthanide Elements. J. Am. Chem. Soc. 1954, 76, 6210. [Google Scholar] [CrossRef]
- Birmingham, J.M.; Wilkinson, G. The Cyclopentadienides of Scandium, Yttrium and Some Rare Earth Elements. J. Am. Chem. Soc. 1956, 78, 42–44. [Google Scholar] [CrossRef]
- Evans, W.J. Tutorial on the role of cyclopentadienyl ligands in the discovery of molecular complexes of the rare-earth and actinide metals in new oxidation states. Organometallics 2016, 35, 3088–3100. [Google Scholar] [CrossRef]
- Reynolds, L.T.; Wilkinson, G. π-Cyclopentadienyl compounds of uranium-IV and thorium-IV. J. Inorg. Nucl. Chem. 1956, 2, 246–253. [Google Scholar] [CrossRef]
- Baumgärtner, F.; Fischer, E.O.; Kanellakopulos, B.; Laubereau, P. Triscyclopentadienylplutonium. Angew. Chem. Int. Ed. 1965, 4, 878. [Google Scholar] [CrossRef]
- Baumgärtner, F.; Fischer, E.O.; Kanellakopulos, B.; Laubereau, P. Tri (cyclopentadienyl) americium (III). Angew. Chem. Int. Ed. 1966, 5, 134–135. [Google Scholar] [CrossRef]
- Kanellakopulos, B.; Fischer, E.O.; Dornberger, E.; Baumgärtner, F. Über tricyclopentadienyluran (III) und seine addukte mit tetrahydrofuran, cyclohexylisonitril und l-nicotin. J. Organomet. Chem. 1970, 24, 507–514. [Google Scholar] [CrossRef]
- Baumgärtner, F.; Fischer, E.O.; Billich, H.; Dornberger, E.; Kanellakopulos, B.; Roth, W.; Stieglitz, L. Tricyclopentadienylcurium (244)-(III). J. Organomet. Chem. 1970, 22, C17–C18. [Google Scholar] [CrossRef]
- Laubereau, P.G.; Burns, J.H. Tricyclopentadienyl-curium. Inorg. Nucl. Chem. Lett. 1970, 6, 59–63. [Google Scholar] [CrossRef]
- Laubereau, P.G.; Burns, J.H. Microchemical preparation of tricyclopentadienyl compounds of berkelium, californium, and some lanthanide elements. Inorg. Chem. 1970, 9, 1091–1095. [Google Scholar] [CrossRef]
- Crisler, L.R.; Eggerman, W.G. A novel synthesis of triscyclopentadienyl plutonium (III). J. Inorg. Nucl. Chem. 1974, 36, 1424–1426. [Google Scholar] [CrossRef]
- Kanellakopulos, B.; Dornberger, E.; Baumgärtner, F. Das erste dreiwertige thorium in einem aromatenkomplex: Tris (cyclopentadienyl) thorium (III). Inorg. Nucl. Chem. Lett. 1974, 10, 155–160. [Google Scholar] [CrossRef]
- Kalina, D.G.; Marks, T.J.; Wachter, W.A. Photochemical synthesis of low-valent organothorium complexes. Evidence for photoinduced. beta.-hydride elimination. J. Am. Chem. Soc. 1977, 99, 3877–3879. [Google Scholar] [CrossRef]
- Albrecht-Schmitt, T.E. (Ed.) Organometallic and Coordination Chemistry of the Actinides; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar] [CrossRef]
- Ephritikhine, M. Recent advances in organoactinide chemistry as exemplified by cyclopentadienyl compounds. Organometallics 2013, 32, 2464–2488. [Google Scholar] [CrossRef]
- Arnold, P.L.; Dutkiewicz, M.S.; Walter, O. Organometallic Neptunium Chemistry. Chem. Rev. 2017, 117, 11460–11475. [Google Scholar] [CrossRef]
- Walter, O. Actinide Organometallic Complexes with π-Ligands. Chem. Eur. J. 2019, 25, 2927–2934. [Google Scholar] [CrossRef] [Green Version]
- Apostolidis, C.; Dutkiewicz, M.S.; Kovács, A.; Walter, O. Solid-State Structure of Tris-Cyclopentadienide Uranium (III) and Plutonium (III). Chem. Eur. J. 2018, 24, 2841–2844. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Batista, E.R.; Yang, P. Electronic Structure and Spectroscopy of f-Element Tris (cyclopentadienyl) Complexes. In Rare Earth Elements and Actinides: Progress in Computational Science Applications, ch. 14; American Chemical Society: Washington, DC, USA, 2021; Volume 1388, pp. 285–327. [Google Scholar]
- Bursten, B.E.; Rhodes, L.F.; Strittmatter, R.J. Bonding in tris (.eta.5-cyclopentadienyl) actinide complexes. 2. The ground electronic configurations of “base-free” Cp3An complexes (An = thorium, protactinium, uranium, neptunium, plutonium). J. Am. Chem. Soc. 1989, 111, 2756–2758. [Google Scholar] [CrossRef]
- Bursten, B.E.; Rhodes, L.F.; Strittmatter, R.J. The bonding in tris (η5-cyclopentadienyl) actinide complexes IV: Electronic structural effects in AnCl3 and (η5-C5H5)3An (An = Th—Cf) complexes. J. Less Common Metals 1989, 149, 207–211. [Google Scholar] [CrossRef]
- Strittmatter, R.J.; Bursten, B.E. Bonding in tris (η5-cyclopentadienyl) actinide complexes. 5. A comparison of the bonding in Np, Pu, and transplutonium compounds with that in lanthanide compounds and a transition-metal analogue. J. Am. Chem. Soc. 1991, 113, 552–559. [Google Scholar] [CrossRef]
- Kaltsoyannis, N.; Bursten, B.E. Electronic structure of f1 lanthanide and actinide complexes. Part 2. Non-relativistic and relativistic calculations of the ground state electronic structures and optical transition energies of [Ce(η-C5H5)3], [Th(η-C5H5)3] and [Pa(η-C8H8)2]. J. Organomet. Chem. 1997, 528, 19–33. [Google Scholar] [CrossRef]
- Benyahia, M.; Belkhiri, L.; Boucekkine, A. A relativistic DFT study of cyclopentadienyl actinide complexes with no transition-metal analogues. J. Mol. Struct. (Theochem) 2006, 777, 61–73. [Google Scholar] [CrossRef]
- Kirker, I.; Kaltsoyannis, N. Does covalency really increase across the 5f series? A comparison of molecular orbital, natural population, spin and electron density analyses of AnCp3 (An = Th- Cm, Cp = η5-C5H5). Dalton Trans. 2011, 40, 124–131. [Google Scholar] [CrossRef]
- Kaltsoyannis, N. Does Covalency Increase or Decrease across the Actinide Series? Implications for Minor Actinide Partitioning. Inorg. Chem. 2013, 52, 3407–3413. [Google Scholar] [CrossRef]
- Neidig, M.L.; Clark, D.L.; Martin, R.L. Covalency in f-element complexes. Coord. Chem. Rev. 2013, 257, 394–406. [Google Scholar] [CrossRef]
- Tassell, M.J.; Kaltsoyannis, N. Covalency in AnCp4 (An = Th–Cm): A comparison of molecular orbital, natural population and atoms-in-molecules analyses. Dalton Trans. 2010, 39, 6719–6725. [Google Scholar] [CrossRef]
- Ferraro, F.; Arratia-Pérez, R. Bonding, energetic, electronic delocalization and optical properties of MCp3 complexes, where M=Sc, Y, La, Ac, Lu, Ce, Yb and Th. Chem. Phys. Lett. 2012, 554, 219–224. [Google Scholar] [CrossRef]
- Fischer, E.O.; Fischer, H. Komplexe von lanthaniden-tricyclopentadienylen mit basen. J. Organomet. Chem. 1966, 6, 141–148. [Google Scholar] [CrossRef]
- Burns, J.H.; Baldwin, W.H. Molecular and crystal structure of the adduct of cyclohexylisonitrile and praseodymium tricyclopentadienide. J. Organomet. Chem. 1976, 120, 361–368. [Google Scholar] [CrossRef]
- Evans, W.J.; Mueller, T.J.; Ziller, J.W. Lanthanide versus Actinide Reactivity in the Formation of Sterically Crowded [(C5Me5)3MLn] Nitrile and Isocyanide Complexes. Chem. Eur. J. 2010, 16, 964–975. [Google Scholar] [CrossRef]
- Stults, S.D.; Andersen, R.A.; Zalkin, A. Structural studies on cyclopentadienyl compounds of trivalent cerium: Tetrameric (MeC5H4)3Ce and monomeric (Me3SiC5H4)3Ce and [(Me3Si)2C5H3]3Ce and their coordination chemistry. Organometallics 1990, 9, 115–122. [Google Scholar] [CrossRef]
- Mehdoui, T.; Berthet, J.C.; Thuery, P.; Ephritikhine, M. CSD Communication. Personal communication, 2013. [Google Scholar]
- Brennan, J.G.; Andersen, R.A.; Robbins, J.L. Preparation of the first molecular carbon monoxide complex of uranium, (Me3SiC5H4)3UCO. J. Am. Chem. Soc. 1986, 108, 335–336. [Google Scholar] [CrossRef]
- del Mar Conejo, M.; Parry, J.S.; Carmona, E.; Schultz, M.; Brennann, J.G.; Beshouri, S.M.; Andersen, R.A.; Rogers, R.D.; Coles, S.; Hursthouse, M.B. Carbon Monoxide and Isocyanide Complexes of Trivalent Uranium Metallocenes. Chem. Eur. J. 1999, 5, 3000–3009. [Google Scholar] [CrossRef]
- Berthet, J.C.; Ephritikhine, M.; Lance, M.; Nierlich, M. First uranium (IV) triflates. Chem. Commun. 1998, 1373–1374. [Google Scholar] [CrossRef]
- Aderhold, C.M. Zur Elektronenstruktur metallorganischer Lanthaniden- und Actinidenverbindungen. Ph.D. Thesis, Ruprecht-Karl-Universität Heidelberg, Heidelberg, Germany, 1975. [Google Scholar]
- Maier, R. Zusammenhänge Zwischen der Molekülstruktur und Ladungsverteilung an Metallorganischen Komplexen der Lanthanoide und Actinoide; KfK 4623; Kernforschungszentrum Karlsruhe: Karlsruhe, Germany, 1989. [Google Scholar]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Seitz, M.; Oliver, A.G.; Raymond, K.N. The Lanthanide Contraction Revisited. J. Am. Chem. Soc. 2007, 129, 11153–11160. [Google Scholar] [CrossRef] [Green Version]
- Kuta, J.; Clark, A.E. Trends in Aqueous Hydration Across the 4f Period Assessed by Reliable Computational Methods. Inorg. Chem. 2010, 49, 7808–7817. [Google Scholar] [CrossRef]
- Regueiro-Figueroa, M.; Esteban-Gómez, D.; de Blas, A.; Rodríguez-Blas, T.; Platas-Iglesias, C. Understanding Stability Trends along the Lanthanide Series. Chem. Eur. J. 2014, 20, 3974–3981. [Google Scholar] [CrossRef]
- Kovács, A.; Apostolidis, C.; Walter, O.; Lindqvist-Reis, P. ‘Lanthanide contraction’ in [Ln(BTP)3](CF3SO3)3 complexes. Struct. Chem. 2015, 26, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Fryer-Kanssen, I.; Austin, J.; Kerridge, A. Topological Study of Bonding in Aquo and Bis (triazinyl) pyridine Complexes of Trivalent Lanthanides and Actinides: Does Covalency Imply Stability? Inorg. Chem. 2016, 55, 10034–10042. [Google Scholar] [CrossRef]
- Apostolidis, C.; Kovács, A.; Morgenstern, A.; Rebizant, J.; Walter, O. Tris-{Hydridotris (1-pyrazolyl) borato}lanthanide Complexes: Synthesis, Spectroscopy, Crystal Structure and Bonding Properties. Inorganics 2021, 9, 44. [Google Scholar] [CrossRef]
- Evans, W.J.; Foster, S.E. Structural trends in bis (pentamethylcyclopentadienyl) lanthanide and yttrium complexes. J. Organomet. Chem. 1992, 433, 79–94. [Google Scholar] [CrossRef]
- Evans, W.J.; Davis, B.L. Chemistry of Tris (pentamethylcyclopentadienyl) f-Element Complexes, (C5Me5)3M. Chem. Rev. 2002, 102, 2119–2136. [Google Scholar] [CrossRef]
- Windorff, C.J.; MacDonald, M.R.; Meihaus, K.R.; Ziller, J.W.; Long, J.R.; Evans, W.J. Expanding the Chemistry of Molecular U2+ Complexes: Synthesis, Characterization, and Reactivity of the {[C5H3(SiMe3)2]3U}− Anion. Chem. Eur. J. 2016, 22, 772–782. [Google Scholar] [CrossRef] [Green Version]
- Windorff, C.J.; Chen, G.P.; Cross, J.N.; Evans, W.J.; Furche, F.; Gaunt, A.J.; Janicke, M.T.; Kozimor, S.A.; Scott, B.L. Identification of the Formal +2 Oxidation State of Plutonium: Synthesis and Characterization of {PuII[C5H3(SiMe3)2]3}−. J. Am. Chem. Soc. 2017, 139, 3970–3973. [Google Scholar] [CrossRef]
- Apostolidis, C.; Walter, O.; Vogt, J.; Liebing, P.; Maron, L.; Edelmann, F.T. A Structurally Characterized Organometallic Plutonium (IV) Complex. Angew. Chem. Int. Ed. 2017, 56, 5066–5070. [Google Scholar] [CrossRef] [Green Version]
- Ammon, R.V.; Fischer, R.D.; Kanellakopulos, B. 1H-NMR-Untersuchungen der konformativen Beweglichkeit von monosubstituiertem Cyclohexan mit Hilfe eines paramagnetischen Seltenerd-Ions im Substituenten: Triscyclopentadienyl-(cyclo-hexylisonitril)-praseodym (III). Chem. Ber. 1971, 104, 1072–1087. [Google Scholar] [CrossRef] [Green Version]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density—Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Matta, C.F.; Boyd, R.J. An Introduction to the Quantum Theory of Atoms in Molecules. In The Quantum Theory of Atoms in Molecules; Matta, C.F., Boyd, R.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007; Chapter 1; pp. 1–34. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theor. Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- SMART, SAINT, SADABS, Programs for Data Collection, Integration and Absorption Correction; Siemens Analytical X-ray Instruments Inc.: Karlsruhe, Germany, 1997.
- APEX2, SAINT-Plus, SADABS, Programs for Data Collection, Integration and Absorption Correction; Bruker AXS Inc.: Madison, WI, USA, 2007.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Soltek, R.; Huttner, G. Winray-32; University of Heidelberg: Heidelberg, Germany, 1998. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2010.
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the rare earth elements. J. Chem. Phys. 1989, 90, 1730–1734. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M. Segmented contraction scheme for small-core lanthanide pseudopotential basis sets. J. Mol. Struct. (Theochem) 2002, 581, 139–147. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-Adjusted Pseudopotentials for the Actinides. Parameter Sets and Test Calculations for Thorium and Thorium Monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M.; Stoll, H. Valence basis sets for relativistic energy-consistent small-core actinide pseudopotentials. J. Chem. Phys. 2003, 118, 487–496. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. AIMAll, Version 17.11.14; TK Gristmill Software: Overland Park, KS, USA, 2017.
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural Bond Orbital Analysis Program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]







| δ(1H) | Intensity | δ(13C) | Assignment 1 |
|---|---|---|---|
| 12.99 | 15H | 75.0 | Cp |
| 3.56 | 1H | 45.6 | H1a |
| 0.51 | 2H | 21.4 | H3e |
| 0.23 | 2H | 21.4 | H3a |
| 0.46 | 2H | 23.2 | H4a, H4e |
| 0.01 | 2H | 35.4 | H2e |
| −0.29 | 2H | 35.4 | H2a |
| M | Atomic Charge | Net CT from | Pop | EPT2 2 from | |||||
|---|---|---|---|---|---|---|---|---|---|
| M | CC≡N | N | 3Cp | C≡N | π*C≡N | CC≡N | πC≡N | M | |
| La | 1.33 | 0.34 | −0.45 | 1.47 | 0.20 | 0.05 | 759 | 20 | 0 |
| Lu | 1.48 | 0.28 | −0.44 | 1.37 | 0.15 | 0.08 | 583 | 60 | 0 |
| U | 0.85 | 0.38 | −0.45 | 1.93 | 0.22 | 0.19 | 1441 | 42 | 35 |
| Np | 0.95 | 0.37 | −0.44 | 1.81 | 0.24 | 0.13 | 1070 | 34 | 28 |
| Pu | 1.04 | 0.37 | −0.45 | 1.73 | 0.22 | 0.12 | 839 | 36 | 24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, A.; Apostolidis, C.; Walter, O. Competing Metal–Ligand Interactions in Tris(cyclopentadienyl)-cyclohexylisonitrile Complexes of Trivalent Actinides and Lanthanides. Molecules 2022, 27, 3811. https://doi.org/10.3390/molecules27123811
Kovács A, Apostolidis C, Walter O. Competing Metal–Ligand Interactions in Tris(cyclopentadienyl)-cyclohexylisonitrile Complexes of Trivalent Actinides and Lanthanides. Molecules. 2022; 27(12):3811. https://doi.org/10.3390/molecules27123811
Chicago/Turabian StyleKovács, Attila, Christos Apostolidis, and Olaf Walter. 2022. "Competing Metal–Ligand Interactions in Tris(cyclopentadienyl)-cyclohexylisonitrile Complexes of Trivalent Actinides and Lanthanides" Molecules 27, no. 12: 3811. https://doi.org/10.3390/molecules27123811