
Hydrazones of 4-(Trifluoromethyl)benzohydrazide as New Inhibitors of Acetyl- and Butyrylcholinesterase




1
Department of Organic and Bioorganic Chemistry, Faculty of Pharmacy in Hradec Králové, Charles University, Akademika Heyrovského 1203, 500 05 Hradec Králové, Czech Republic
2
Department of Biological and Biochemical Sciences, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(4), 989; https://doi.org/10.3390/molecules26040989
Submission received: 10 January 2021
/
Revised: 8 February 2021
/
Accepted: 11 February 2021
/
Published: 13 February 2021
(This article belongs to the Special Issue Recent Advances in the Modulation of Cholinergic Signaling)
Abstract
:Based on the broad spectrum of biological activity of hydrazide–hydrazones, trifluoromethyl compounds, and clinical usage of cholinesterase inhibitors, we investigated hydrazones obtained from 4-(trifluoromethyl)benzohydrazide and various benzaldehydes or aliphatic ketones as potential inhibitors of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). They were evaluated using Ellman’s spectrophotometric method. The hydrazide–hydrazones produced a dual inhibition of both cholinesterase enzymes with IC50 values of 46.8–137.7 µM and 19.1–881.1 µM for AChE and BuChE, respectively. The majority of the compounds were stronger inhibitors of AChE; four of them (2-bromobenzaldehyde, 3-(trifluoromethyl)benzaldehyde, cyclohexanone, and camphor-based 2o, 2p, 3c, and 3d, respectively) produced a balanced inhibition of the enzymes and only 2-chloro/trifluoromethyl benzylidene derivatives 2d and 2q were found to be more potent inhibitors of BuChE. 4-(Trifluoromethyl)-N’-[4-(trifluoromethyl)benzylidene]benzohydrazide 2l produced the strongest inhibition of AChE via mixed-type inhibition determined experimentally. Structure–activity relationships were identified. The compounds fit physicochemical space for targeting central nervous systems with no apparent cytotoxicity for eukaryotic cell line together. The study provides new insights into this CF3-hydrazide–hydrazone scaffold.
1. Introduction
An increasing interest in fluorinated compounds is due to the favorable effect of fluorine on pharmacological properties. It can be conservatively estimated that globally about 20–25% of drugs in the pharmaceutical pipeline contain at least one fluorine atom [1]. Fluorine is making an impact in pharmaceuticals not only in a fast-growing number of fluorinated drugs but also in the development of many of the best health care products [2]. The special properties that make fluorinated motifs very attractive in medicinal chemistry and chemical biology include small atomic radius, high electronegativity, nuclear spin of one-half, and low polarizability of the C–F bond [3,4]. Substituting C–H by C–F bonds clearly benefits from the fact that the size of organofluorine is only slightly larger than the size and volume of the hydrogen substituent and that consequently no particular steric hindrance is encountered in most H→F replacements [5]. Among the fluorinated groups such as F, CF3, OCF3, CHF2 etc., CF3 is a very familiar pharmacophore [6]. This abiotically synthesized group—usually replacing chlorine, methyl, ethyl, or isopropyl group—is a frequently used bioisostere in medicinal chemistry, dating from 1929 [7]. This electron withdrawing functionality increases the biological half-life by impeding the oxidative metabolism, and its lipophilicity improves cellular membrane permeability, thus increasing absorption [8,9].
Given together, the CF3 group belongs to common and useful motifs in central nervous system (CNS)-acting drugs—for example, antipsychotics trifluoperazine (I) and flupentixol (II), aprepitant (III) used against nausea and vomitus; central anesthetics isoflurane (IV) and sevoflurane (V); fluoxetine (VI) and fluvoxamine (VII) prescribed as antidepressants; centrally acting nonsteroidal anti-inflammatory drug celecoxib (VIII); anorectic drug fenfluramine (IX); and benzodiazepine anxiolytics, hypnotics, and anticonvulsants such as triflunordazepam (X) and quazepam (XI) (Figure 1).
Compounds bearing hydrazide–hydrazone scaffold (−CO–NH–N═C(R)−; i.e., fused hydrazide and hydrazone moieties) have a substantial beneficial value in the development of novel antibacterial, mainly antitubercular, antiplatelet, anticancer, anti-inflammatory, and, with regard to targeting the CNS, also of anticonvulsant, analgesic, and antidepressant agents [10,11].
4-(Trifluoromethyl)benzohydrazide (1)-based hydrazones have been reported as potential antimicrobial agents active against Gram-positive and Gram-negative bacteria, mycobacteria, yeasts, and molds [12,13,14]. Moreover, hydrazones of 1 have exhibited inhibition of various enzymes, i.e., bacterial DNA-gyrase and FabH (β-ketoacyl-acyl carrier protein synthase III) [15], urease [14], cyanobacterial fructose-1,6-/sedoheptulose-1,7-bisphosphatase [16] and, more importantly, also acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) [17,18]. The enzyme inhibiting properties are not unique for the CF3 group bearing hydrazide–hydrazones, of course. Illustratively, hydrazones derived from 4-hydroxybenzohydrazide have been studied as a new class of dual inhibitors of thymidine phosphorylase and cancer cell proliferation, which deserves to be further investigated for anti-cancer drug development [19], as well as indole-based acetohydrazide inhibitors of this enzyme [20]. Also, xanthinoxidase can be inhibited by hydrazide–hydrazones in free form as well as after their coordination to copper (II) [21]. Laccase is a copper-containing polyphenol oxidase that catalyzes the radical reduction of atmospheric oxygen to water with simultaneous oxidation of electron-rich aromatic compounds (polyphenols, anilines). Inhibition of this enzyme might weaken activity of the plant pathogens that utilize it in various biochemical processes. The relationship between 4-hydroxybenzohydrazide-hydrazones and laccase from Trametes versicolor was studied and a competitive type of inhibition was identified [22]. In our previous study [23], N-alkyl-2-[4-(trifluoromethyl)benzoyl]hydrazine-1-carboxamides were identified as inhibitors of both AChE and BuChE. Based on here presented findings, we investigated our trifluoromethylated hydrazide–hydrazones 2–3 (Table 1) [12] as potential inhibitors of AChE a BuChE together with their newly prepared analogues 2n–2u.
2. Results and Discussion
2.1. Chemistry
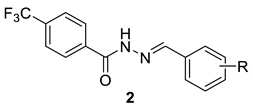
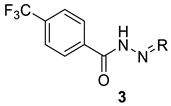
The title hydrazide 1 was prepared from 4-(trifluoromethyl)benzoic acid by a two-step process. First, this acid was esterified by methanol in the presence of a catalytic amount of sulfuric acid under heating in an excess of the alcohol. Then, the methyl ester was converted to hydrazide by hydrazinolysis using hydrazine hydrate in boiling ethanol (Scheme 1). The overall yield was almost quantitative. This procedure was described previously by our group for 4-iodobenzohydrazide [24]. The hydrazone–hydrazides were synthesized by the treatment of 1 (1 mmol) with various aldehydes and ketones (1.1 mmol) in boiling methanol for 2 h (Scheme 2); for ketones with acidic catalyst (concentrated sulfuric acid). The yields of the hydrazide–hydrazones 2 and 3 ranged from 85% to 99% and from 68% to 87%, respectively. The characterization of the compounds 2a–2m and 3 was reported in depth by Krátký et al. [12]; the remaining ones were prepared newly on the basis of the results of biological evaluation for a deeper insight into structure–activity relationships. The group of novel compounds covers positional isomers of the most effective AChE inhibitors (2k–2m), i.e., the derivatives 2n–2s, as well as hydrazide–hydrazones without 4-CF3-benzohydrazide moiety (trifluoromethyl was removed, 2t, or replaced by methyl, 2u).
2.2. Inhibition of Acetyl- and Butyrylcholinesterase
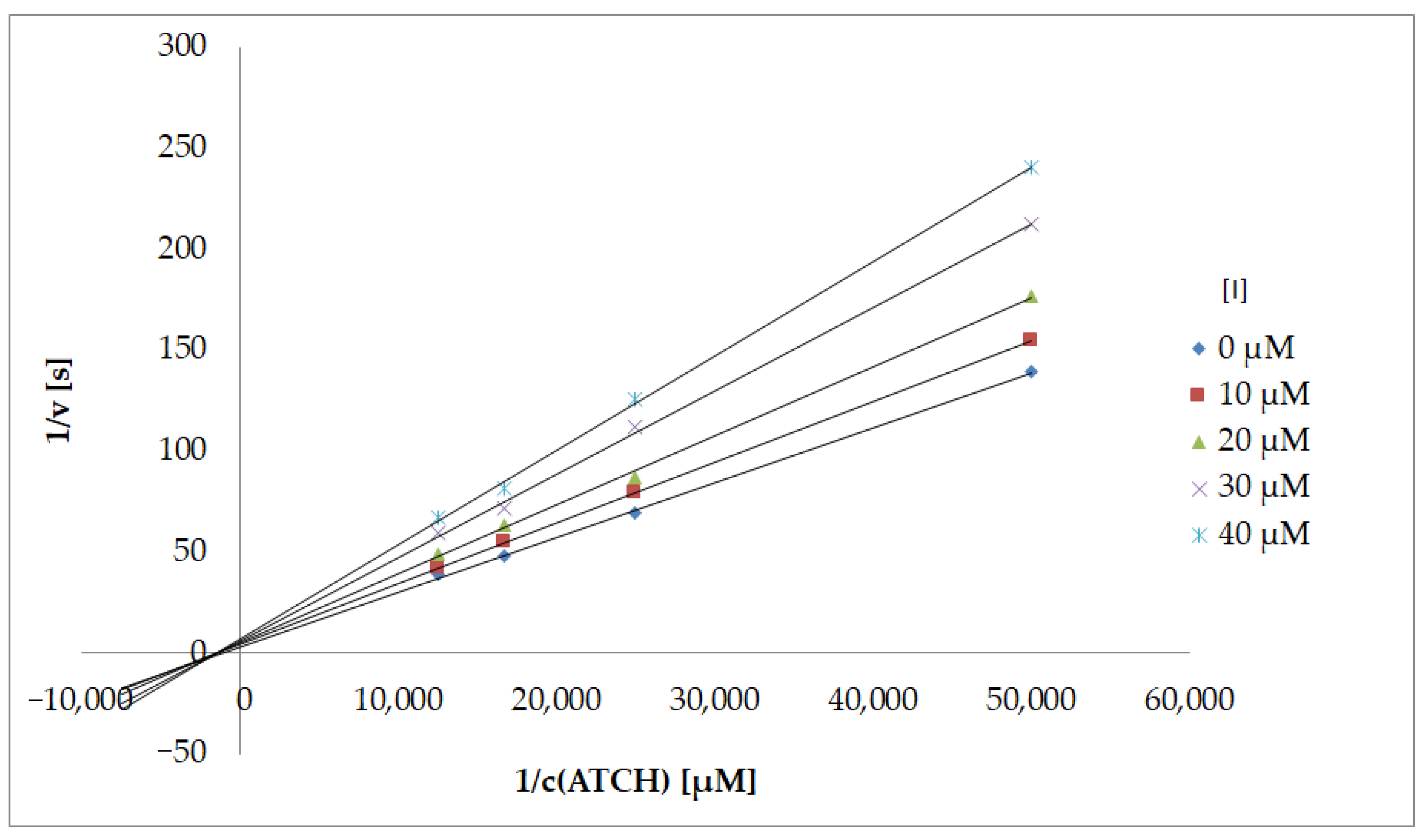
The hydrazide–hydrazones 2 and 3 as well as the parent compound 1 were screened for their potential to affect the function of AChE isolated from electric eel (EeAChE) and BuChE obtained from equine serum (EqBuChE) using Ellman’s spectrophotometric method (Table 1). These enzymes of nonhuman origin were used to screen inhibitory activity and to compare the results with our previous compounds sharing a certain degree of a chemical similarity [23,24]. Their efficacy is expressed as the concentration leading to 50% inhibition of the enzymatic activity (IC50). To quantify the preference to particular acetylcholine decomposing enzymes, selectivity indexes (SI) were calculated. SI is defined as a ratio of IC50 for BuChE/IC50 for AChE (Table 1; illustrated also in Figures S1–S6). Values higher than 1 mean stronger inhibition of AChE and vice versa. Based on analogy with therapeutic index, SI over the borderline of 10 indicates selective AChE inhibition. In addition to 1, galantamine was employed as a standard for comparison of IC50 values. Moreover, we investigated the type of inhibition of the most active AChE inhibitor identified (2l; Figure 2, Table S1).
The original [12] hydrazide–hydrazones 2a–2m and 3 exhibited a dual inhibition of both enzymes with IC50 values of 46.8–137.7 µM and 63.6–881.1 µM for AChE and BuChE, respectively. Focusing on the benzaldehyde derivatives 2 and AChE, 4-(trifluoromethyl)benzylidene hydrazone 2l was the most in vitro efficient, together with salicylidene hydrazone 2g. The introduction of unsubstituted benzylidene moiety into the molecule 1 resulted in a comparable activity (1 vs. 2a). The inhibition potency was improved when an electronegative substituent was present at the position 4: second CF3 group (–I effect; 2l), nitro group (–I and –M effects; 2m), or bromine (+M and predominant –I effect; 2k). The replacement of bromine by chlorine led to a mild drop in the activity (2k vs. 2b). On the other hand, electron-donating substituents were unfavorable (methyl 2i, methoxy 2j). We also evaluated chlorine and hydroxyl group positional isomers (2b–2d and 2e–2g, respectively); however, there was no clear structure–activity relationship. Interestingly, the combination of these two groups at their best positions led to the less potent compound within this study (2h).
IC50 values of 2 for BuChE were clearly higher. 2-Chlorobenzylidene analogue 2d showed the lowest value, followed by approximately 3-fold less active 4-CF3 (2l) and 4-NO2 (2k) substituted hydrazones. Contrarily, benzylidene (2a), salicylidene (2g), and 4-bromobenzylidene (2k) substituents were detrimental to inhibition. Remaining derivatives produced similar IC50 values. The positional isomers of 2d shared significantly lower inhibitory potency.
Among ketone-derived hydrazones 3, the inhibition of AChE was virtually uniform (59–70 µM); bigger ketones (camphor, cyclohexanone) were preferred over smaller ones (acetone, cyclopentanone). This structure–activity dependence was even more obvious in the case of BuChE inhibition (3a→3d, from 424 to 57 µM), favoring again cyclohexanone and especially camphor as starting scaffolds.
Then, we also evaluated additional derivatives 2n–2u. The positional isomers of the brominated derivative 2k provided an improved activity against BuChE for both ortho- and meta-isomers (up to 6.3×, 2k vs. 2o), although meta-isomer 2n was less effective against AChE. Analogous structure–activity relationships were also found for the novel nitro derivatives 2r and 2s. The hydrazones based on 2- and 3-CF3-benzaldehyde (2p and 2q) retained activity against AChE but were significantly more effective against BuChE, especially the ortho-isomer 2q with IC50 of 19.1 µM, i.e., the most active BuChE inhibitor in this study. In sum, especially 2-substituted benzaldehydes-based hydrazones were beneficial when compared to their original para-isomers in terms of improved activity against BuChE along with the same inhibition of AChE. Meta-isomers also led to a more balanced inhibition of both acetylcholine hydrolyzing enzymes.
Focusing on the removal of CF3 group (2t) and its replacement by CH3 group (2u), these modifications led to a decreased activity, although these two derivatives were also capable of inhibiting both AChE and BuChE. The benzohydrazide derivative 2t was a more potent inhibitor.
The modification of the parent hydrazide 1 led to 11 derivatives that were more active against AChE (2g, 2k–2m, 2o–2q, 2s, 2t, 3c, and 3d) and 5 others were fully comparable (2a, 2f, 2u, 3a, and 3b). For BuChE, 13 compounds (2d, 2l, 2m–2t, 3b–3d) exhibited lower IC50 than the hydrazide 1. Then, we compared the results with those obtained for galantamine, the highly active inhibitor of AChE approved for the treatment of Alzheimer’s disease-related dementia. However, none of the hydrazide–hydrazones exhibited comparable IC50 values.
Focusing on selectivity toward cholinesterases, the majority of the hydrazide–hydrazones 2–3 inhibited AChE more strongly (selectivity indexes, SI, ≥2). 2-Hydroxybenzylidene derivative 2g, the second most potent AChE inhibitor, was the most selective AChE inhibitor (SI = 18.6), followed by unsubstituted benzylidene 2a (8.7), propan-2-ylidene 3a (6.1), and 4-bromobenzylidene analogue 2k (SI of 5.5). On the other hand, only the presence of 2-chlorobenzylidene and 2-(trifluoromethyl)benzylidene substituents (2d and 2q) led to a higher inhibition of BuChE; however, not selectively (SI = 0.5 and 0.4, respectively). Two larger ketone-based hydrazones obtained from cyclohexanone (3c) and camphor (3d) produced a balanced inhibition of both acetylcholine hydrolyzing enzymes as well as the remaining hydrazones of isomeric 2-/3-bromo- and nitrobenzaldehydes (2n, 2o, 2r, and 2s) and 3-CF3-benzaldehyde (2p).
Moreover, all the original derivatives were without any signs of cytotoxicity for HepG2 cells at 100 µM (Table 1) [12].
Type of Inhibition
The reversible enzyme inhibitors can be classified into four groups: competitive, non-competitive, uncompetitive, and mixed type of inhibition. The type of inhibition could be distinguished using a Lineweaver–Burk plot [25] and corresponding comparison of two kinetic parameters: Michaelis constant (KM) and maximum velocity (Vm) of uninhibited and inhibited reaction. The Lineweaver–Burk plot of the most potent AChE inhibition with 2l is depicted in Figure 2. Based on the plot of KM and Vm, this hydrazide–hydrazone caused a mixed type of inhibition.
2.3. Prediction of BBB Permeability
In addition to their efficacy and low toxicity, compounds intended for treatment of dementias must be able to cross the blood-brain barrier (BBB) to reach their target site. To predict this crucial issue in silico, various empirical approaches have been proposed and validated. They are based on various physicochemical and structural parameters inferred from known CNS drugs.
Classical selection criteria include molecule size, lipophilicity, number of hydrogen bond donors and acceptors, aromatic rings, basic groups, heteroatoms, polar surface area, molar refractivity, molecular flexibility quantified by a number of rotatable single bonds, pKa, etc. [26,27,28,29]. In general, lipophilicity is regarded to be the most important property, and its increased value often results in an improved in vitro activity [27].
The data (Mw, consensus log P, number of hydrogen bond donors and acceptors, rotatable bonds, and topological polar surface area (tPSA)) of the derivatives 1–3 (Table 2) were compared with margins proposed as ideal for CNS drugs [29]. The vast majority of these parameters were within the suggested ranges; only several compounds (2j, 2l, 2m, and 2p–2s) had one more rotatable bond. Nevertheless, delivery across the BBB is not bounded sharply. Since it is a consequence of many factors, the cut-offs are not strict, and they are related to a higher probability of targeting the CNS.
In addition to this structural multiparameter appraisal, we also involved a newer BOILED-Egg (Brain Or IntestinaL EstimateD permeation) approach [26] based on lipophilicity and polarity of small molecules to predict penetration through the BBB via passive diffusion. Passive diffusion usually represents a key entrance route for centrally acting drugs, and it requires a certain degree of lipophilicity (log P) in combination with a mild polarity (tPSA). To estimate brain permeability using BOILED-Egg, the SwissADME [30] calculator was utilized.
The results are summarized in Table 2. With the exception of 4-CF3 derivative 2l and their isomers 2p and 2q (due to enhanced lipophilicity) and 4-NO2 compounds (2m, 2r, and 2s), the remaining hydrazide–hydrazones met the requirements for CNS active molecules. However, the BOILED-Egg approach-based prediction often failed in the prediction of BBB permeability of nitro group-containing drugs that are indicated far from the optimal physicochemical space, even in the case of typical CNS drugs and drugs exhibiting their pharmacologic effects in the brain (e.g., calcium channel blockers including nitrendipine, nilvadipine and nimodipine, non-steroidal anti-inflammatory agent nimesulide, tolcapone used as a levodopa booster, organophosphorus AChE inhibitors such as paraoxon and parathion, benzodiazepine hypnotics nitrazepam and flunitrazepam, antibiotic chloramphenicol, etc.). Thus, the prediction seems to be unreliable for nitro derivatives.
Furthermore, BOILED-Egg was useful for prediction of gastrointestinal absorption after oral administration based on the identical physicochemical descriptors. Hopefully, all the derivatives 1–3 meet in silico criteria for high gastrointestinal absorption; only for bis-CF3 analogues it was predicted to be lower.
3. Materials and Methods
3.1. Chemistry
General. All the reagents and solvents were purchased from Sigma-Aldrich (Darmstadt, Germany) and Penta Chemicals (Prague, Czech Republic), and they were used as received. The progress of the reactions and the purity of the products were monitored by thin layer chromatography using a mixture of toluene with ethyl acetate and tetrahydrofuran (4:1:0.3, v/v) as an eluent; plates were coated with 0.2 mm Merck 60 F254 silica gel (Merck Millipore, Darmstadt, Germany) and were visualized by UV irradiation (254 nm). Melting points were determined on a Büchi Melting Point B-540 apparatus (BÜCHI, Flawil, Switzerland) using open capillaries; the reported values are uncorrected.
Elemental analysis (C, H, N) was performed on a Vario MICRO Cube Element Analyzer (Elementar Analysensysteme, Hanau, Germany). Both calculated and found values are given as percentages. Infrared spectra (ATR) were recorded on a Nicolet 6700 FT-IR spectrometer (ThermoFisher Scientific, Waltham, MA, USA) in the range of 600 to 4000 cm−1. The NMR spectra were measured in DMSO-d6 at ambient temperature on a Varian V NMR S500 instrument (500 MHz for 1H and 125 MHz for 13C; Varian Comp., Palo Alto, CA, USA). The chemical shifts, δ, are given in ppm with respect to tetramethylsilane via signals of DMSO-d6 (2.49 for 1H and 39.7 for 13C spectra). The coupling constants (J) are reported in Hz.
The synthesis and characterization of the compounds 2a–2m and 3 were reported in depth by Krátký et al. [12].
The novel derivatives were prepared and purified according to an identical procedure. Briefly, an appropriate hydrazide (1 mmol) was dissolved in 6 mL of MeOH and then a substituted benzaldehyde was added in one portion (1.1 of equivalents) under vigorous stirring. The reaction mixture was stirred at rt for 5 min and then refluxed for 2 h. Resulting suspension was let to stay overnight at +4 °C and then was filtered off. The resulting crystals were washed with a small volume of cold MeOH and diethyl ether and then dried to obtain pure hydrazones 2n–2u.
N’-(3-Bromobenzylidene)-4-(trifluoromethyl)benzohydrazide 2n. White solid; yield 96%; mp 193–195 °C. IR (ATR): 3018, 1654, 1558, 1327, 1304, 1290, 1164, 1151, 1118, 1108, 1067, 1019, 962, 927, 853, 786, 770, 683 cm−1. 1H-NMR (500 MHz, DMSO-d6): δ 12.19 (1H, bs, NH), 8.47 (1H, s, CH=N), 8.12 (2H, d, J = 8.3 Hz, H2, H6), 7.96–7.90 (3H, m, H3, H5, H2’), 7.75 (1H, dt, J = 7.8, 1.3 Hz, H6´), 7.64 (1H, dt, J = 8.0, 1.5 Hz, H4´), 7.43 (1H, t, J = 7.8 Hz, H5´). 13C-NMR (126 MHz, DMSO): δ 162.58, 147.23, 137.49, 137.07, 133.25, 132.09 (q, J = 32.0 Hz), 131.50, 129.73, 129.09, 126.79, 125.96 (q, J = 3.8 Hz), 124.33 (q, J = 272.8 Hz), 122.66. Analytically calculated for C15H10BrF3N2O (371.15): C, 48.54; H, 2.72; N, 7.44. Found: C, 48.52; H, 2.77; N, 7.40.
N’-(2-Bromobenzylidene)-4-(trifluoromethyl)benzohydrazide 2o. White solid; yield 97%; mp 217–219.5 °C. 1H-NMR (500 MHz, DMSO-d6): δ 12.30 (1H, bs, NH), 8.84 (1H, s, CH=N), 8.15 (2H, d, J = 8.1 Hz, H2, H6), 8.02 (1H, dd, J = 7.9, 1.8 Hz, H6´), 7.92 (2H, d, J = 8.2 Hz, H3, H5), 7.70 (1H, dd, J = 8.0, 1.3 Hz, H3´), 7.50–7.45 (1H, m, H5´), 7.38 (1H, td, J = 7.6, 1.8 Hz, H4´). 13C-NMR (126 MHz, DMSO): δ 162.49, 147.27, 137.40, 133.67, 133.36, 132.41, 132.14 (q, J = 32.0 Hz), 129.11, 128.62, 127.79, 125.97 (q, J = 3.8 Hz), 124.33 (q, J = 272.3 Hz), 124.18. Analytically calculated for C15H10BrF3N2O (371.15): C, 48.54; H, 2.72; N, 7.44. Found: C, 48.50; H, 2.78; N, 7.48.
4-(Trifluoromethyl)-N’-[3-(trifluoromethyl)benzylidene]benzohydrazide 2p. White solid; yield 90%; mp 162.5–164.5 °C. IR (ATR): 3007, 1649, 1570, 1327, 1306, 1292, 1281, 1217, 1167, 1126, 1111, 1068, 1062, 1019, 945, 854, 803, 770, 698, 670 cm−1. 1H-NMR (500 MHz, DMSO-d6): δ 12.27 (1H, bs, NH), 8.55 (1H, s, CH=N), 8.13 (2H, d, J = 8.1 Hz, H2, H6), 8.09–8.03 (2H, m, H2´, H6´), 7.92 (2H, d, J = 8.1 Hz, H3, H5), 7.80 (1H, d, J = 7.9 Hz, H4´), 7.71 (1H, t, J = 7.8 Hz, H5´). 13C-NMR (126 MHz, DMSO): δ 162.43, 147.00, 137.20, 135.54, 131.87 (q, J = 31.9 Hz), 131.38, 130.29, 129.89 (q, J = 31.9 Hz), 128.86, 126.72 (q, J = 3.8 Hz), 125.73 (q, J = 3.9 Hz), 124.22 (q, J = 272.3 Hz), 124.07 (q, J = 272.8 Hz), 123.35 (q, J = 3.9 Hz). Analytically calculated for C16H10F6N2O (360.25): C, 53.34; H, 2.80; N, 7.78. Found: C, 53.32; H, 2.83; N, 7.90.
4-(Trifluoromethyl)-N’-[2-(trifluoromethyl)benzylidene]benzohydrazide 2q. White solid; yield 95%; mp 182.5–184 °C. IR (ATR): 3034, 1651, 1559, 1489, 1316, 1290, 1278, 1170, 1160, 1137, 1112, 1104, 1062,1032, 1020, 964, 860, 766, 671 cm−1. 1H-NMR (500 MHz, DMSO-d6): δ 12.36 (1H, bs, NH), 8.85 (1H, q, J = 2.2 Hz, CH=N), 8.25 (1H, d, J = 7.9 Hz, H6´), 8.14 (2H, d, J = 8.1 Hz, H2, H6), 7.93 (2H, d, J = 8.2 Hz, H3, H5), 7.83–7.76 (2H, m, H3´, H5´), 7.65 (1H, t, J = 7.7 Hz, H4´). 13C-NMR (126 MHz, DMSO): δ 162.62, 144.00, 137.29, 133.37, 132.46, 132.21 (q, J = 31.9 Hz), 130.77, 129.13, 127.40, 127.37 (q, J = 32.0 Hz), 126.42 (q, J = 3.9 Hz), 125.98 (q, J = 3.7 Hz), 124.62 (q, J = 272.7 Hz), 124.32 (q, J = 272.3 Hz). Analytically calculated for C16H10F6N2O (360.25): C, 53.34; H, 2.80; N, 7.78. Found: C, 53.39; H, 2.77; N, 7.72.
N’-(3-Nitrobenzylidene)-4-(trifluoromethyl)benzohydrazide 2r. Yellowish solid; yield 99%; mp 205–207 °C. IR (ATR): 3205, 3055, 1665, 1551, 1527, 1343, 1327, 1282, 1179, 1149, 1132, 1113, 1066, 1019, 949, 936, 898, 862, 814, 771, 738, 690, 669 cm−1. 1H-NMR (500 MHz, DMSO-d6): δ 12.31 (1H, bs, NH), 8.58–8.55 (2H, m, CH=N, H2´), 8.28–8.25 (1H, m, H4´), 8.17 (1H, dd, J = 7.8, 1.4 Hz, H6´), 8.13 (2H, d, J = 8.0 Hz, H2, H6), 7.92 (2H, d, J = 8.1 Hz, H3, H5), 7.76 (1H, t, J = 8.0 Hz, H5´). 13C-NMR (126 MHz, DMSO): δ 162.70, 148.68, 146.59, 137.36, 136.46, 133.95, 132.15 (q, J = 31.7 Hz), 130.95, 129.13, 125.98 (q, J = 3.9 Hz), 124.90, 124.32 (q, J = 272.4 Hz), 121.50. Analytically calculated for C15H10F3N3O3 (337.25): C, 52.42; H, 2.99; N, 12.46. Found: C, 52.43; H, 2.95; N, 12.53.
N’-(2-Nitrobenzylidene)-4-(trifluoromethyl)benzohydrazide 2s. Yellowish solid; yield 98%; mp 215.5–216.5 °C. IR (ATR): 3201, 3089, 1651, 1554, 1522, 1365, 1343, 1323, 1292, 1175, 1152, 1131, 1113, 1065, 1018, 955, 922, 855, 788, 744, 702, 687 cm−1. 1H-NMR (500 MHz, DMSO-d6): δ 12.35 (1H, bs, NH), 8.86 (1H, s, CH=N), 8.13–8.04 (4H, m, H3, H5, H3´, H6´), 7.89 (2H, d, J = 8.1 Hz, H2, H6), 7.79 (1H, d, J = 7.6 Hz, H5´), 7.66 (1H, t, J = 7.8 Hz, H4´). 13C-NMR (126 MHz, DMSO): δ 162.73, 148.84, 144.44, 137.33, 134.32, 132.29 (q, J = 32.2 Hz), 131.41, 129.29, 129.13, 128.51, 126.06 (q, J = 3.8 Hz), 125.30, 124.43 (q, J = 272.3 Hz). Analytically calculated for C15H10F3N3O3 (337.25): C, 52.42; H, 2.99; N, 12.46. Found: C, 52.37; H, 3.06; N, 12.55.
N’-[4-(Trifluoromethyl)benzylidene]benzohydrazide 2t. White solid; yield 99%; mp 188–189 °C. IR (ATR): 3290, 1664, 1548, 1377, 1325, 1305, 1281, 1171, 1156, 1147, 1107, 1067, 1018, 953, 856, 839, 718, 692, 670 cm−1. 1H-NMR (500 MHz, DMSO-d6): δ 12.06 (1H, bs, NH), 8.54 (1H, s, CH=N), 7.98–7.91 (4H, m, H2, H6, H2´, H6´), 7.82 (2H, d, J = 8.0 Hz, H3´, H5´), 7.61 (1H, d, J = 7.3 Hz, H4), 7.54 (2H, t, J = 7.5 Hz, H3, H5). 13C-NMR (126 MHz, DMSO): δ 163.81, 146.44, 138.79, 133.66, 132.41, 130.15 (q, J = 31.6 Hz), 128.99, 128.16, 128.12, 126.20 (q, J = 3.9 Hz), 124.57 (q, J = 272.2 Hz). Analytically calculated for C15H11F3N2O (292.26): C, 61.65; H, 3.79; N, 9.59. Found: C, 61.60; H, 3.85; N, 9.63.
4-Methyl-N’-[4-(trifluoromethyl)benzylidene]benzohydrazide 2u. White solid; yield 94%; mp 212–213.5 °C. IR (ATR): 3277, 1661, 1545, 1386, 1369, 1323, 1280, 1159, 1146, 1120, 1105, 1066, 1017, 950, 838, 750, 672 cm−1. 1H-NMR (500 MHz, DMSO-d6): δ 11.93 (1H, bs, NH), 8.49 (1H, s, CH=N), 7.90 (2H, d, J = 8.0 Hz, H2, H6), 7.83–7.76 (4H, m, H2´, H3´, H5´, H6´), 7.30 (2H, d, J = 7.9 Hz, H3, H5), 2.34 (3H, s, CH3). 13C-NMR (126 MHz, DMSO): δ 163.69, 146.32, 142.57, 138.95, 130.85, 130.16 (q, J = 31.5 Hz), 129.58, 128.26, 128.15, 126.26 (q, J = 3.8 Hz), 124.66 (q, J = 272.2 Hz), 21.57. Analytically calculated for C16H13F3N2O (306.29): C, 62.74; H, 4.28; N, 9.15. Found: C, 62.69; H, 4.24; N, 9.12.
3.2. Evaluation of Acetyl- and Butyrylcholinesterase Inhibition
The activities of the investigated compounds 2–3 were measured via Ellman’s spectrophotometric method that was modified according to Zdražilová et al. [31] to determine the IC50 values. Ellman’s method has been widely applied for the screening of the efficiency of potential AChE and BuChE inhibitors. The activity of cholinesterases was assessed indirectly by quantifying the amount of 5-mercapto-2-nitrobenzoic acid ion formed in the reaction of the thiol reagent 5,5′-disulfanediylbis(2-nitrobenzoic acid) with thiocholine, a product of the hydrolysis of acetyl- (ATCh) or butyrylthiocholine (BTCh) catalyzed by AChE and BuChE, respectively.
AChE used in this assay was obtained from electric eel (Electrophorus electricus L.) and BuChE from equine serum. The benzazepine alkaloid drug galantamine was used as a reference enzyme inhibitor. All the enzymes, the thiol reagent, ATCh, BTCh, and galantamine were purchased from Sigma-Aldrich (Prague, Czech Republic).
The enzyme activity in final reaction mixture (2 mL) was 0.2 U/mL, the concentration of ATCh or BTCh was 40 µM, and the concentration of 5,5′-disulfanediylbis(2-nitrobenzoic acid) was 0.1 mM for all reactions. The investigated hydrazide–hydrazones were dissolved in DMSO and then diluted by demineralized water (conductivity 3 μS, equipment supplier BKG Water Treatment, Hradec Králové, Czech Republic) to the concentration of 1 mM. For all of them as well as galantamine, five different concentrations of inhibitor in final reaction mixture were used. All measurements were carried out in triplicates, and the average values of reaction rate (v0-uninhibited reaction, vi-inhibited reaction) were used for construction of the dependence v0/vi vs. concentration of inhibitor. From the obtained equation of the regression curve, IC50 values were calculated.
For the determination of the type of inhibition of the most active AChE inhibitor 2l, a Lineweaver–Burk plot [25] was used, and the measuring procedure was analogous to the determination of IC50. The enzyme activity in the final reaction mixture (2 mL) was 0.2 U/mL, a concentration of ATCh was 20–80 µM, and a concentration of 5,5′-disulfanediylbis(2-nitrobenzoic acid) was 0.1 mM. For each of the substrate concentrations, four different concentrations of the inhibitor were used (10, 20, 30, and 40 µM). The dependence absorbance vs. time was observed and the reaction rate was calculated. All measurements were carried out in duplicate and the average values of reaction rate were used for the construction of the Lineweaver–Burk plot. Furthermore, the dependencies vs. concentration of ATCh in absence and presence of inhibitor 2l were analyzed by nonlinear regression using GraphPad Prism 9.0.1 for Windows (GraphPad Software, San Diego, CA, USA, www.graphpad.com). From obtained equations of regression curves in the Lineweaver–Burk plot, the values of Michaelis constant (KM) and maximum velocity (Vm) were calculated. Using nonlinear regression (chosen panel: enzyme kinetics—inhibition/mixed model inhibition) the values of KI (54.63 µM) and alpha (1.28) were determined. Based on all obtained results, the type of inhibition was identified.
3.3. Determination of Parameters for CNS Delivery
The parameters for in silico prediction of CNS delivery (log P as a value of consensus Log Po/w, number of rotatable bonds, and topological polar surface area, tPSA) as well as BOILED-Egg [26] method estimation of brain permeability were calculated using a free web tool SwissADME [30]; http://www.swissadme.ch/index.php.
4. Conclusions
We screened twenty-five hydrazide–hydrazones for their in vitro potency to inhibit AChE and BuChE. These compounds were derivatives of 4-(trifluoromethyl)benzohydrazide and various aromatic aldehydes and aliphatic ketones of choice. In addition, we evaluated the influence of various substitution patterns of benzaldehyde. IC50 values found were in the micro molar range from 46.8 µM for AChE and 19.1 µM for BuChE. Most of the derivatives were better inhibitors of AChE. 2-/3-/4-(Trifluoromethyl)-, 2-/4-nitro-, 2-/4-bromo-, and 2-hydroxybenzaldehydes together with camphor and cyclohexanone represented the most successful carbonyl compounds used for the modification of the parent hydrazide. Focusing on the type of inhibition, the most potent AChE inhibitor 2l derived from 4-(trifluoromethyl)benzaldehyde exhibited a mixed inhibition of the enzyme. After in silico evaluation, the hydrazide–hydrazones shared generally convenient properties for CNS delivery and gastrointestinal absorption.
Supplementary Materials
The following are available online. Table S1: Kinetic parameters for 2l inhibiting eeAChE calculated from L–B plot; Figure S1: The dependence v0/vi vs. concentration of 2a inhibiting eeAChE; Figure S2: The dependence v0/vi vs. concentration of 2g inhibiting eeAChE; Figure S3: The dependence v0/vi vs. concentration of 2k inhibiting eeAChE; Figure S4: The dependence v0/vi vs. concentration of 3c inhibiting eeAChE; Figure S5: The dependence v0/vi vs. concentration of 2d inhibiting eqBuChE; Figure S6: The dependence v0/vi vs. concentration of 3d inhibiting eqBuChE.
Author Contributions
Conceptualization, M.K.; methodology, M.K. and Š.Š.; investigation, M.K., K.S., and Q.A.V.; writing—original draft preparation, M.K., Š.Š., and J.V.; writing—review and editing, K.S. and Š.Š.; supervision, M.K., Š.Š., and J.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Czech Science Foundation, grant number 20-19638Y,SVV 260 547, and the project EFSA-CDN (No. CZ.02.1.01/0.0/0.0/16_019/0000841) co-funded by ERDF.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available on request due to restrictions. The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy.
Acknowledgments
The authors wish to acknowledge financial support from the University of Pardubice, Faculty of Chemical Technology.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 1, 2a–2u, and 3a–3d are available from the authors.
References
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anticancer agents. J. Fluorine Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Wang, J.; Sanchez-Rosello, M.; Acena, L.J.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Ojima, I. Strategic incorporation of fluorine into taxoid anticancer agents for medicinal chemistry and chemical biology studies. J. Fluor. Chem. 2017, 198, 10–23. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 611, 45822–45880. [Google Scholar] [CrossRef]
- Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications. Available online: https://www.worldscientific.com/doi/pdf/10.1142/9781848166363_fmatter (accessed on 2 January 2021).
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Acena, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent advances in the trifluoromethylation methodology and new CF3-containing drugs. J. Fluor. Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Yale, H.L. The Trifluoromethyl Group in Medicinal Chemistry. J. Med. Pharm. Chem. 1959, 1, 121–133. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef]
- Rollas, S.; Küçükgüzel, S.G. Biological activities of hydrazone derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, G.; Marella, A.; Shaquiquzzaman, M.; Akhtar, M.; Ali, M.R.; Alam, M.M. A review exploring biological activities of hydrazones. J. Pharm. Bioallied Sci. 2014, 6, 69–80. [Google Scholar] [PubMed]
- Krátký, M.; Bősze, S.; Baranyai, Z.; Stolaříková, J.; Vinšová, J. Synthesis and biological evolution of hydrazones derived from 4-(trifluoromethyl)benzohydrazide. Bioorg. Med. Chem. Lett. 2017, 27, 5185–5189. [Google Scholar] [CrossRef] [PubMed]
- He, L.-Y.; Qiu, X.-Y.; Cheng, J.-Y.; Liu, S.-J.; Wu, S.-M. Synthesis, characterization and crystal structures of vanadium(V) complexes derived from halido-substituted tridentate hydrazone compounds with antimicrobial activity. Polyhedron 2018, 156, 105–110. [Google Scholar] [CrossRef]
- Yang, Y.-S.; Su, M.-M.; Zhang, X.-P.; Liu, Q.-X.; He, Z.-X.; Xu, C.; Zhu, H.-L. Developing potential Helicobacter pylori urease inhibitors from novel oxoindoline derivatives: Synthesis, biological evaluation and in silico study. Bioorg. Med. Chem. Lett. 2018, 28, 3182–3186. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-S.; Su, M.-M.; Xu, J.-F.; Liu, Q.-X.; Bai, L.-F.; Hu, X.-W.; Zhu, H.-L. Discovery of novel oxoindolin derivatives as atypical dual inhibitors for DNA Gyrase and FabH. Bioorg. Chem. 2019, 93, 103309. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.-D.; Li, D.; Sun, Y.; Han, X.-Y.; Yi, F.; Sha, Y.; Ren, Y.-L.; Ding, M.-W.; Feng, L.-L.; Wan, J. Design and syntheses of novel N′-((4-oxo-4H-chromen-3-yl)methylene)benzohydrazide as inhibitors of cyanobacterial fructose-1,6-/sedoheptulose-1,7-bisphosphatase. Bioorg. Med. Chem. 2013, 21, 2826–2831. [Google Scholar] [CrossRef]
- Karaman, N.; Sıcak, Y.; Taşkın-Tok, T.; Öztürk, M.; Karaküçük-İyidoğan, A.; Dikmen, M.; Koçyiğit-Kaymakçıoğlu, B.; Oruç-Emre, E.E. New piperidine-hydrazone derivatives: Synthesis, biological evaluations and molecular docking studies as AChE and BChE inhibitors. Eur. J. Med. Chem. 2016, 124, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Aktar, B.S.K.; Sıcak, Y.; Tok, T.T.; Oruç-Emre, E.E.; Yağlıoğlu, A.S.; Iyidoğan, A.K.; Öztürk, M.; Demirtaş, I. Designing heterocyclic chalcones, benzoyl/sulfonyl hydrazones: An insight into their biological activities and molecular docking study. J. Mol. Struct. 2020, 1211, 128059. [Google Scholar] [CrossRef]
- Javaid, S.; Saad, S.M.; Zafar, H.; Malik, R.; Khan, K.M.; Choudhary, M.I.; Rahman, A.U. Thymidine phosphorylase and prostrate cancer cell proliferation inhibitory activities of synthetic 4-hydroxybenzohydrazides: In vitro, kinetic, and in silico studies. PLoS ONE 2020, 15, e0227549. [Google Scholar] [CrossRef] [Green Version]
- Taha, M.; Aldhamin, E.A.J.; Almandil, N.B.; Anouar, E.; Uddin, N.; Alomari, M.; Rahim, F.; Adalat, B.; Ibrahim, M.; Nawaz, F.; et al. Synthesis of indole based acetohydrazide analogs: Their in vitro and in silico thymidine phosphorylase studies. Bioorg. Chem. 2020, 98, 103745. [Google Scholar] [CrossRef]
- Leigh, M.; Raines, D.J.; Castillo, C.E.; Duhme-Klair, A.K. Inhibition of xanthine oxidase by thiosemicarbazones, hydrazones and dithiocarbazates derived from hydroxy-substituted benzaldehydes. ChemMedChem 2011, 6, 1107–1118. [Google Scholar] [CrossRef]
- Maniak, H.; Talma, M.; Matyja, K.; Trusek, A.; Giurg, M. Synthesis and structure-activity relationship studies of hydrazide-hydrazones as inhibitors of laccase from Trametes versicolor. Molecules 2020, 25, 1255. [Google Scholar] [CrossRef] [Green Version]
- Krátký, M.; Baranyai, Z.; Štěpánková, Š.; Svrčková, K.; Švarcová, M.; Stolaříková, J.; Horváth, L.; Bősze, S.; Vinšová, J. N-Alkyl-2-[4-(trifluoromethyl)benzoyl]hydrazine-1-carboxamides and their analogues: Synthesis and multitarget biological activity. Molecules 2020, 25, 2268. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Štěpánková, Š.; Brablíková, M.; Svrčková, K.; Švarcová, M.; Vinšová, J. Novel iodinated hydrazide-hydrazones and their analogues as acetyl- and butyrylcholinesterase Inhibitors. Curr. Top. Med. Chem. 2020, 20, 2106–2117. [Google Scholar] [CrossRef] [PubMed]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016, 7, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Ghose, A.K.; Ott, G.R.; Hudkins, R.L. Technically extended multiparameter optimization (TEMPO): An advanced robust scoring scheme to calculate central nervous system druggability and monitor lead optimization. ACS Chem. Neurosci. 2017, 8, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Mikitsh, J.L.; Chacko, A.M. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspect. Medicin. Chem. 2014, 6, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Zdrazilova, P.; Stepankova, S.; Komers, K.; Ventura, K.; Cegan, A. Half-inhibition concentrations of new cholinesterase inhibitors. Z. Naturforsch. C J. Biosci. 2004, 59, 293–296. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Examples of trifluoromethyl motif containing CNS-acting drugs.

Scheme 1.
Synthesis of the 4-(trifluoromethyl)benzohydrazide 1.

Scheme 2.
Synthesis of the hydrazide–hydrazones 2 and 3 [X = CF3 (2a-2s), H (2t), CH3 (2u)].

Figure 2.
Lineweaver–Burk plot for 2l inhibiting eeAChE (ATCh = acetylthiocholine; [I] = concentration of inhibitor).
Figure 2.
Lineweaver–Burk plot for 2l inhibiting eeAChE (ATCh = acetylthiocholine; [I] = concentration of inhibitor).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Inhibition of AChE and BuChE, selectivity and toxicity data of 1–3.
 | |||||
|---|---|---|---|---|---|
| Code | R | IC50 AChE (µM) | IC50 BuChE (µM) | Selectivity BuChE/AChE | IC50 (HepG2) [12] |
| 2a | H | 75.90 ± 1.6 | 651.3 ± 31.5 | 8.7 | >100 |
| 2b | 4-Cl | 88.5 ± 1.4 | 253.8 ± 3.3 | 2.9 | >100 |
| 2c | 3-Cl | 77.1 ± 0.03 | 260.9 ± 40.7 | 3.4 | >100 |
| 2d | 2-Cl | 128.5 ± 10.0 | 63.6 ± 0.9 | 0.5 | >100 |
| 2e | 4-OH | 101.9 ± 3.8 | 229.2 ± 14.5 | 2.3 | >100 |
| 2f | 3-OH | 70.4 ± 2.8 | 249.7 ± 7.5 | 3.5 | >100 |
| 2g | 2-OH | 47.4 ± 1.8 | 881.1 ± 8.0 | 18.6 | >100 |
| 2h | 2-OH-5-Cl | 137.7 ± 3.0 | 287.2 ± 12.6 | 2.1 | >100 |
| 2i | 4-CH3 | 91.4 ± 6.2 | 253.1 ± 9.6 | 2.8 | >100 |
| 2j | 4-CH3O | 108.5 ± 2.2 | 279.1 ± 2.9 | 2.6 | >100 |
| 2k | 4-Br | 62.1 ± 0.02 | 342.3 ± 11.3 | 5.5 | >100 |
| 2l | 4-CF3 | 46.8 ± 2.1 | 175.0 ± 8.3 | 3.7 | >100 |
| 2m | 4-NO2 | 60.0 ± 0.4 | 190.6 ± 1.1 | 3.2 | >100 |
| 2n | 3-Br | 96.9 ± 1.3 | 166.3 ± 0.8 | 1.7 | ND |
| 2o | 2-Br | 62.6 ± 3.2 | 54.2 ± 0.1 | 0.9 | ND |
| 2p | 3-CF3 | 49.9 ± 1.4 | 63.6 ± 2.8 | 1.3 | ND |
| 2q | 2-CF3 | 50.0 ± 1.6 | 19.1 ± 0.5 | 0.4 | ND |
| 2r | 3-NO2 | 81.3 ± 2.6 | 132.9 ± 10.2 | 1.6 | ND |
| 2s | 2-NO2 | 59.7 ± 5.8 | 93.1 ± 4.9 | 1.6 | ND |
 | |||||
| 2t | H | 62.9 ± 0.8 | 189.8 ± 1.7 | 3.0 | ND |
| 2u | CH3 | 75.3 ± 1.5 | 244.9 ± 7.0 | 3.3 | ND |
 | |||||
| 3a | propan-2-ylidene | 69.7 ± 5.2 | 424.3 ± 1.4 | 6.1 | >100 |
| 3b | cyclopentylidene | 69.2 ± 1.0 | 199.8 ± 13.2 | 2.9 | >100 |
| 3c | cyclohexylidene | 61.2 ± 1.3 | 74.7 ± 2.7 | 1.2 | >100 |
| 3d | 1,7,7-trimethylbicyclo-[2.2.1]heptan-2-ylidene | 59.3 ± 1.8 | 57.2 ± 3.4 | 1.0 | >100 |
| 1 [23] | - | 69.4 ± 1.4 | 204.0 ± 2.7 | 2.9 | >100 |
| Galantamine | 1.54 ± 0.02 | 2.77 ± 0.15 | 1.8 | ND | |
IC50 values are expressed as the mean ± SD (n = three independent experiments). ND: not determined. The lowest IC50 values for each enzyme are given in bold as well as the most selective inhibitors for both enzymes.
Table 2.
Blood-brain barrier (BBB) permeability parameters.
| Code | Mw | log P | H-Bond Donors | H-Bond Acceptors | tPSA (Å2) | Rotatable Bonds | BOILED-Egg |
|---|---|---|---|---|---|---|---|
| 2a | 292.26 | 3.66 | 1 | 3 | 41.46 | 5 | ✔ |
| 2b | 326.70 | 4.19 | 1 | 3 | 41.46 | 5 | ✔ |
| 2c | 326.70 | 4.20 | 1 | 3 | 41.46 | 5 | ✔ |
| 2d | 326.70 | 4.37 | 1 | 3 | 41.46 | 5 | ✔ |
| 2e | 308.26 | 3.26 | 2 | 4 | 61.69 | 5 | ✔ |
| 2f | 308.26 | 3.26 | 2 | 4 | 61.69 | 5 | ✔ |
| 2g | 308.26 | 3.43 | 2 | 4 | 61.69 | 5 | ✔ |
| 2h | 342.70 | 4.01 | 2 | 4 | 61.69 | 5 | ✔ |
| 2i | 306.28 | 4.00 | 1 | 3 | 41.46 | 5 | ✔ |
| 2j | 322.28 | 3.67 | 1 | 4 | 50.69 | 6 | ✔ |
| 2k | 371.15 | 4.28 | 1 | 3 | 41.46 | 5 | ✔ |
| 2l | 360.25 | 4.72 | 1 | 3 | 41.46 | 6 | - |
| 2m | 337.25 | 3.06 | 1 | 5 | 87.28 | 6 | - |
| 2n | 371.15 | 4.29 | 1 | 3 | 41.46 | 5 | ✔ |
| 2o | 371.15 | 4.45 | 1 | 3 | 41.46 | 5 | ✔ |
| 2p | 360.25 | 4.71 | 1 | 3 | 41.46 | 6 | - |
| 2q | 360.25 | 4.77 | 1 | 3 | 41.46 | 6 | - |
| 2r | 337.25 | 3.05 | 1 | 5 | 87.28 | 6 | - |
| 2s | 337.25 | 3.23 | 1 | 5 | 87.28 | 6 | - |
| 2t | 292.26 | 3.65 | 1 | 3 | 41.46 | 5 | ✔ |
| 2u | 306.28 | 3.91 | 1 | 3 | 41.46 | 5 | ✔ |
| 3a | 244.21 | 2.93 | 1 | 3 | 41.46 | 4 | ✔ |
| 3b | 270.25 | 3.35 | 1 | 3 | 41.46 | 4 | ✔ |
| 3c | 284.28 | 3.69 | 1 | 3 | 41.46 | 4 | ✔ |
| 3d | 338.37 | 4.60 | 1 | 3 | 41.46 | 4 | ✔ |
| 1 | 204.15 | 1.70 | 2 | 3 | 55.12 | 3 | ✔ |
| Breakpoint [29] | ≤500 | ≤6.0 | <5 | <10 | ≤90 | ≤5 | - |
Mw: molecular weight, tPSA: topological polar surface area.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Krátký, M.; Svrčková, K.; Vu, Q.A.; Štěpánková, Š.; Vinšová, J. Hydrazones of 4-(Trifluoromethyl)benzohydrazide as New Inhibitors of Acetyl- and Butyrylcholinesterase. Molecules 2021, 26, 989. https://doi.org/10.3390/molecules26040989
AMA Style
Krátký M, Svrčková K, Vu QA, Štěpánková Š, Vinšová J. Hydrazones of 4-(Trifluoromethyl)benzohydrazide as New Inhibitors of Acetyl- and Butyrylcholinesterase. Molecules. 2021; 26(4):989. https://doi.org/10.3390/molecules26040989
Chicago/Turabian StyleKrátký, Martin, Katarína Svrčková, Quynh Anh Vu, Šárka Štěpánková, and Jarmila Vinšová. 2021. "Hydrazones of 4-(Trifluoromethyl)benzohydrazide as New Inhibitors of Acetyl- and Butyrylcholinesterase" Molecules 26, no. 4: 989. https://doi.org/10.3390/molecules26040989