
Total Syntheses of Pladienolide-Derived Spliceosome Modulators



1
College of Pharmacy, Chungnam National University, Daejeon 34134, Korea
2
Therapeutics and Biotechnology Division, Korea Research Institute of Chemical Technology, 141 Gajeong-ro, Yuseong-gu, Daejeon 34114, Korea
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(19), 5938; https://doi.org/10.3390/molecules26195938
Submission received: 1 September 2021
/
Revised: 27 September 2021
/
Accepted: 27 September 2021
/
Published: 30 September 2021
(This article belongs to the Special Issue Feature Review Papers in Organic Synthesis)
Abstract
:Pladienolides, an emerging class of naturally occurring spliceosome modulators, exhibit interesting structural features, such as highly substituted 12-membered macrocycles and epoxide-containing diene side chains. The potential of pladienolides as anti-cancer agents is confirmed by H3B-8800, a synthetic analog of this natural product class, which is currently under Phase I clinical trials. Since its isolation in 2004 and the first total synthesis in 2007, a dozen total syntheses and synthetic approaches toward the pladienolide class have been reported to date. This review focuses on the eight completed total syntheses of naturally occurring pladienolides or their synthetic analogs, in addition to a synthetic approach to the main framework of the natural product.
1. Introduction
Ribonucleic acid (RNA) post-transcriptional modification is a vital biological process in most eukaryotic cells. It allows the production of mature RNA that can perform normal and diverse functions in the cell [1]. One representative process in this modification is RNA splicing. Premature RNA consists of non-coding intron regions and coded exon regions. During RNA splicing, introns are removed, leaving exons that are re-ligated and can function as mature RNA. Because of its significant role in protein production, the mRNA splicing process in cells is tightly regulated. Indeed, splicing defects such as exon-skipping can induce changes in the levels of specific splicing isoforms, causing a variety of diseases, including cancer [2].
The splicing reaction is regulated by a spliceosome, which is a dynamic multimegadalton ribonucleoprotein (RNP) complex composed of five small nuclear RNA ribonucleoproteins (snRNPs: U1, U2, U4, U5, and U6) and numerous proteins [3]. Depending on the function of the spliceosome, a variety of mature mRNAs can be produced from the same pre-mRNA and can be translated to diverse proteins, such as antibodies, in a process called alternative splicing. Alternative splicing is an important mechanism for generating proteomic diversity from a relatively limited number of protein-coding genes [4]. Given the importance of RNA splicing and the fundamental role of the spliceosome in post-transcription, the spliceosome has gained attention as a target for fighting cancer. For example, the splicing factor 3b (SF3b) complex, a representative spliceosomal component, is the most frequently mutated splicing factor in cancers [5] such as myelodysplastic syndromes [6], acute myeloid leukemia (AML) [7], chronic lymphocytic leukemia (CLL) [8], and various solid tumors [9,10,11].
Most splicing modulators reported to date are naturally occurring molecules, including the FR class [12,13,14,15], herboxidiene class [16,17], and pladienolide class (Figure 1). Among them, pladienolides are structurally unique in that they possess a highly substituted macrocyclic core structure that has captured the attention of the synthetic chemistry community. Pladienolides are naturally occurring macrolides that were first isolated by Eisai Co. in 2004 from Mer-11107, an engineered strain of Streptomyces platensis [18,19,20]. This polyketide natural product was later reported to interfere with spliceosome function by targeting the SF3b subunit in a dose-dependent manner [21]. Due to this interesting biological function, several medicinal chemistry efforts have been made using simplified analogs [22,23] or synthetic molecules hybridized with another splicing modulator [24]. In addition, the biosynthetic production of novel pladienolide analogs has been recently reported by means of native expression of a pathway-specific activator [25,26].
Pladienolide B, one of the related macrolides, has been proven to be the most active congener, with nanomolar IC50 values against various human cancer cell lines [20,27,28,29]. The cocrystal structure of pladienolide B and the human SF3b core discovered in 2018 confirmed their inhibitory and modulatory effects via splicing [30]. In addition, pladienolide B has been recently reported to prevent SARS-CoV-2 replication at non-toxic concentrations in human cells by targeting the splicing process [31].
There are several natural pladienolide derivatives, including pladienolide A–G (1–7) [18,19,20], 6-deoxypladienolide D (8) [32], and FD-895 (9) [33,34], as well as synthetic derivatives E7107 (10) and H3B-8800 (11) (Figure 2). E7107 (10) is an analog of pladienolide D, developed by Eisai Co., and the first SF3B1 modulator that entered phase I clinical trials on patients with different types of solid tumors (NCT00459823 and NCT00499499) [35]. However, the trials were discontinued because of unexpected toxicity at higher doses, resulting in vision loss [36,37,38]. H3B-8800 (11), another analog developed by H3 Biomedicine, a subsidiary of Eisai Co., is an orally bioavailable drug currently under phase 1 clinical trials to treat patients with myelodysplastic syndromes, AML, and chronic myelomonocytic leukemia (NCT02841540) [39].
Owing to a growing interest in RNA splicing for drug discovery, a number of studies and patents have been reported, including review articles that (partially) cover the biological features and structure–activity relationship of pladienolides [40,41,42,43,44]. In this review, we provide a comprehensive overview of the total synthesis of pladienolides or their core structures, with a detailed analysis of the synthetic routes. We present a summary of the total syntheses of pladienolides from the time the first synthesis was reported to the present date (2007–2021). This review is divided into two main sections, based on the macrocyclization strategy.
2. Synthesis of Pladienolides
2.1. Synthetic Strategies Regarding Pladienolides
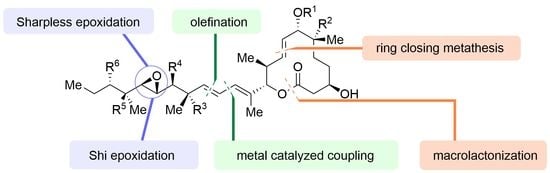
Pladienolides are composed of two fragments: a 12-membered macrolactone ring with five stereocenters and a side chain bearing up to six contiguous stereocenters, with a trans epoxide moiety at the center (Figure 3). Most of the reported pladienolide syntheses are based on the conjugation between these two fragments via metal-catalyzed coupling or olefination. The major macrolactone moiety has been established by ring-closing metathesis (RCM) or macrolactonization as the key last step. In addition, the absolute stereochemistry of trans epoxides has been controlled by external chiral sources, such as AD-mix (Sharpless asymmetric epoxidation) [45] or fructose-derived organocatalysts (Shi epoxidation) [46]. Pladienolides have methyl or hydroxy groups as substituents, the stereocenter of which is often controlled by asymmetric aldol reactions.
2.2. Macrocyclization via Ring-Closing Metathesis
Since the discovery of highly bench-stable and functional-group-tolerant metathesis catalysts, cross-metathesis has been considered a powerful tool for natural product synthesis [47]. In particular, RCM has become the most popular approach for synthetically challenging medium and macrocyclic natural products [48]. Unsurprisingly, a number of synthetic strategies for 12-membered core scaffold of pladienolides rely on RCM, including those published by Kotake [49], Ghosh [50], Burkart [34], Chandrasekhar [51], Keaney [52], Krische [53], and their coworkers.
2.2.1. Synthesis of Pladienolide B by Kotake and Coworkers (2007)
In 2007, the first total synthesis of pladienolide class natural products was reported by Kotake and coworkers [49]. Their approach for the construction of stereogenic centers is based on reagent-controlled stereoselective reactions to confirm the absolute configurations of the pladienolides. In their retrosynthetic plan (Scheme 1), the side chain unit and 12-membered macrolactone 12 of pladienolide B (2) could be disconnected by Julia–Kocienski olefination. Macrolactone 12 could be derived from 13 through RCM. Compound 13 was assembled by Yamaguchi esterification from fragments 14 and 15, and both were prepared in an asymmetric manner by anti-aldol and Reformatsky reactions, respectively.
The total synthesis commenced with the preparation of building blocks 14 and 15, as shown in Scheme 2. The absolute stereochemistry of 14 was established by Paterson anti-aldol condensation using ketone 17 [54] as an external chiral source, where aldehyde 16 was converted to compound 18 with excellent diastereoselectivity (98% de) after TBS protection. The resulting aldol product was then transformed to fragment 14 in several steps, including the removal of benzoyloxy ketone and Wittig olefination of the resulting aldehyde. Another fragment, 15, was also prepared in 10 steps from aldehyde 19 [55]. The Sm(II)-mediated asymmetric Reformatsky reaction of 19 with chiral auxiliary 20 afforded β-hydroxyamide 21, with acceptable diastereoselectivity (82% de) [56]. The diastereomers were later separated with column chromatography on silica gel. Removal of the chiral auxiliary, methylation, and TBS protection allowed the formation of product 22, which subsequently underwent asymmetric Sharpless dihydroxylation to afford compound 23, after benzylidene acetal formation and PMB ether deprotection. The Dess–Martin oxidation of 23, followed by the Wittig reaction and ester hydrolysis, finally provided key fragment 15.
Side-chain fragment 31, with five stereogenic centers at C16 and C18–21, was synthesized from the known syn-aldol product 24 [57]. Aldehyde 25 prepared from 24 through the Weinreb amide-mediated reduction was subjected to the Julia–Kocienski olefination with sulfone 26 to afford trans-olefin 27. Benzyl deprotection, Mitsunobu reaction with 28, and the resulting sulfide oxidation smoothly afforded sulfone 29, which underwent asymmetric Shi epoxidation with chiral ketone 30 to stereoselectively yield side chain fragment 31.
To complete the total synthesis, 14 was coupled with 15 under esterification conditions using Yamaguchi reagent 32, leading to 13 in a 93% yield (Scheme 3). RCM, followed by a sequence of PMB deprotection/oxidation, afforded the macrocyclic aldehyde 12. The Julia–Kocienski olefination between 12 and side-chain unit 31 afforded 33 upon silyl deprotection and reprotection of the resulting diol, with a dichloroacetyl group. Reprotection with the electron-deficient dichloroacetyl group was necessitated by the nucleophilic attack of the C21 hydroxy group on the proximal epoxide during benzylidene deprotection under acidic conditions. Finally, the removal of both benzylidene and dichloroacetyl groups provided pladienolide A (1), from which C7–OH was then selectively acetylated to yield pladienolide B (2), completing the first total syntheses of pladienolide-class natural products.
With the absolute structure of the macrocyclic core in hand, synthetic efforts were directed toward another natural anticancer analog, pladienolide D (4), whose structure differs from that of pladienolide B by one hydroxy group, located at C16. The absolute stereochemistry at C16 was confirmed to be R by the chemical degradation/derivatization of 4 and extensive NMR analysis. The conjugation of two fragments by the Julia–Kocienski olefination was expected to be ineffective in this case because of the presence of a quaternary C16. Thus, cross-metathesis (CM) was suggested to combine the fragments [58].
The synthesis of side-chain 38 commenced with the preparation of Julia–Kocienski reagent 35 from known alcohol 34 [59]. The olefination of 35 with 25 smoothly afforded trans-olefin 36, which was successfully transformed to epoxide 37 by Sharpless asymmetric epoxidation, with moderate diastereoselectivity (90% de) (Scheme 4). The second epoxidation of 37 by Shi’s ketone 30, followed by regioselective reductive epoxide cleavage, afforded the desired allylic alcohol 38. Another compound required for olefin cross-metathesis, 39, was obtained from 12 by sequential Tebbe olefination, global deprotection, and selective acylation. Finally, the cross-metathesis of 38 and 39 completed the first total synthesis of pladienolide D, with a 64% yield. All stereogenic centers in this synthesis were delivered and controlled with the aid of external chiral sources, such as chiral auxiliaries or chiral catalysis, to guarantee the absolute structure of complex natural products, despite the long linear steps required for this strategy.
2.2.2. Synthesis of Pladienolide B by Ghosh and Anderson (2012)
Ghosh and Anderson demonstrated a convergent synthetic pathway toward pladienolides to accelerate structural modification and structure–activity relationship studies (Scheme 5) [50]. Pladienolide B (2) was divided into small fragments with one or two stereogenic centers. Parallel to Kotake’s synthesis, the side chain unit could be connected to the core macrocycle by the Julia–Kocienski olefination, which could be accessed by an RCM of 40. The formation of 40 could be achieved by the Yamaguchi esterification between building blocks 41 and 42, which could be conveniently divided into small fragments, including 43.
First, building block 41 was prepared from prenol 44, which was protected with a trityl group and oxidized to 45 (Scheme 6). Asymmetric Brown crotylation [60] furnished 41 with a moderate enantiomeric excess (82% ee). In parallel, the preparation of 42 began with Sharpless asymmetric epoxidation of commercially available divinyl carbinol 46, which yielded 43 after PMB protection. β-Keto ester 47 was added to 43 to afford elongated chain 48, which was subjected to a series of reactions, including asymmetric reduction with l-tartaric acid [61], and a substrate-controlled diastereoselective Grignard reaction to provide key building block 42 with two newly generated stereogenic centers at C3 and C6.
To synthesize side-chain unit 52, Ghosh and Anderson employed cross-metathesis between two small fragments, namely, homoallylic alcohol 49 and mesylate 50, whereas the Kotake group used the Julia–Kocienski olefination (Scheme 2). The trans selectivity in cross-metathesis was moderate, with an E/Z ratio of 5:1 (Scheme 6). Subsequent TES protection, the 1-phenyl-1H-tetrazole-5-thiol 28 substitution of mesylate, followed by sulfur oxidation, produced sulfone 51. Asymmetric Shi epoxidation was also applied in this synthesis for the construction of epoxides at C18 and C19. TES protection completed the preparation of the Julia–Kocienski reagent 52.
With all of these building blocks, an approach similar to Kotake’s synthesis was adopted to complete the synthesis of pladienolide B (Scheme 7). Fragments 41 and 42 were connected through the Yamaguchi esterification to afford 53, which was macrocyclized by RCM to afford core 54. After acetylation of C7–OH, deprotection of the trityl group and IBX oxidation furnished aldehyde 55. Finally, the olefination of aldehyde 55 with sulfone 52 completed the total synthesis of pladienolide B. Ghosh and Anderson reported a convergent, scalable, and diversifiable synthesis involving 17 steps (LLS) and 1.4% overall yield.
2.2.3. Synthesis of FD-895 by Burkart and Coworkers (2012)
Concurrent with Ghosh and Anderson’s synthesis (Scheme 5, Scheme 6 and Scheme 7), Burkart and coworkers completed the total synthesis of FD-895 (9), which shares the identical macrocyclic core of pladienolide B but has a more substituted side chain [34]. Before the isolation of pladienolides in 2004, FD-895 was isolated in 1994 from Streptomyces hygroscopicus A-9561 [33]. Through intensive and combinatorial NMR studies, the full structure was confirmed, except for the stereochemistry at C16 and C17 positions. To confirm the absolute configuration of FD-895, a retrosynthesis was designed by the Burkart group, providing four diastereomers at C16 and C17. The stereoisomeric side chains could be prepared in a stereodivergent manner and then assembled by Stille coupling with a macrocyclic core (Scheme 8). The core unit would be accessed by an RCM of 56, which could be disconnected by esterification into two building blocks 57 and 58. Asymmetric aldol addition of 59 was proposed, to provide 58 with a C3 stereogenic center.
As shown in Scheme 9, the synthesis of macrocyclic core 64 began with the asymmetric Brown allylation of aldehyde 60 for generation of the stereogenic center C7 in 61 [62]. The induction of stereogenicity at C6 of FD-895 (9) was guided by a C7 stereocenter using the chelate-controlled Grignard addition method [63]. The ketone resulting from the DMP oxidation of 61 was converted to carbinol 62 by chelate-controlled methylation and diol protection, with concomitant deprotection of TBS. To elongate the carbon frameworks, Sammakia aldol addition was employed, along with chiral auxiliary 63 [64], to afford esterification substrate 58 after TBS protection and hydrolytic cleavage. The known alcohol 57 was coupled with acid 58 to produce ester 56, which was transformed into the 12-membered ring 64 via sequential reactions including RCM and acetylation. Although the synthetic strategy for the core scaffold was analogous to previous syntheses, Burkart and coworkers succeeded in shortening the purification steps.
In the next stage, they prepared four plausible diastereomeric side chains in a divergent manner (Scheme 10). This divergent strategy was based on Marshall’s propargylation of allenylstannane from the common intermediate aldehyde 70 [65]. The syn-aldol product 66, which contains two contiguous stereogenic centers (C20 and C21 of FD-895) was prepared from 65 using a method developed by Crimmins [66]. Conversion to the Weinreb amide, followed by methylation, and DIBAL-H reduction provided aldehyde 67, which was immediately transformed to allyl alcohol 69 via the HWE olefination with 68 and reduction to minimize epimerization at C20. The C18–C19 epoxide was installed by Sharpless asymmetric epoxidation, and the subsequent oxidation with IBX produced the common intermediate 70 for the diastereomeric side chains. Marshall’s propargylation using diverse allenylstannanes allowed the divergent synthesis of 72a–72d, in which 72a with a 16R,17R configuration was diastereoselectively prepared from 71.
In the final stage, hydrostannylation of the side chain isomers 72a–72d provided Stille reagents 73a–73d, which were utilized for cross-coupling reactions with macrocycle 64 to afford four isomers of FD-895 (9). As expected, the spectral data of one isomer (16R,17R) matched those of FD-895. Thus, Burkart and coworkers accomplished the first total synthesis of FD-895 and confirmed its absolute configuration at C16 and C17. Recently, a scalable synthetic method for 17S-FD-895 was also reported, which demonstrated anticancer activity 25 times stronger than that of FD-895 [67].
2.2.4. Synthesis of Pladienolide B by Kumar and Chandrasekhar (2013)
When biologically meaningful natural products are discovered and applied to drug discovery, synthetic and medicinal chemists sometimes attempt to truncate the structure to simplify the complexity of the compounds involved. Thus, Kumar and Chandrasekhar reported the enantioselective synthesis of pladienolide B, as well as its side-chain-truncated analogs bearing simple aromatic groups, rather than the complex and linear chain of the natural product [51]. In their retrosynthetic analysis (Scheme 11), pladienolide B (2) could be disconnected by Pd-catalyzed Stille coupling into the side chain and the macrocyclic part, the latter of which would be available via RCM of 74, similar to previous synthetic studies. Acid 75, which can be readily connected with 57 to produce 74, resulted from the oxidative adjustment of 76.
As depicted in Scheme 12, the construction of the macrocyclic core 82 began with the Sharpless asymmetric epoxidation of geraniol (77) to afford trans-epoxide 78. Benzyl protection and acid-mediated epoxide opening of 78 afforded 79 after diol protection [68]. Ozonolysis of trisubstituted olefin 79, followed by HWE olefination and DIBAL-H reduction, provided allylic alcohol 76, which underwent a second Sharpless asymmetric epoxidation to yield epoxy alcohol 80. Reductive regioselective epoxide opening, directed by the adjacent alcohol, provided 81 after protection steps [69]. Oxidation of the resulting alcohol 81 produced the corresponding aldehyde, which allowed a series of reactions including the Wittig olefination to afford building block 75. The connection of 75 with the known alcohol 57 was achieved under Yamaguchi conditions to afford 74. While the RCM of 74 with Grubbs first- and second-generation catalysts was ineffective, the use of the Hoveyda–Grubbs second-generation catalyst instead afforded a moderate yield (52%) in the absence of the acetal group. Subsequent acetylation was carried out at a low temperature (−10 °C) to selectively introduce the acetyl group on C7–OH, to afford the cyclic core 82 in good yield (90%).
In the next stage, side-chain synthesis commenced with the distinctive hydride transfer of the known epoxy alcohol 83 (Scheme 13) [70]. Intramolecular hydride transfer in the presence of TBSOTf and the amine base regioselectively opened the epoxide to afford aldehyde 84. Propionyl ester 85, a substrate of the Ireland–Claisen rearrangement, was generated via Grignard addition, oxidation, CBS asymmetric reduction, and propionylation. Rearrangement under basic conditions was carried out to afford carboxylic acid 86, delivering stereogenicity at C16 [71]. Further decorations, including aldehyde formation and the Corey–Fuchs reaction, provided alkyne 88 via 87. Finally, the hydrostannylation of 88 and coupling of the resulting product with core unit 82 completed the total synthesis of pladienolide B (4) [72].
The versatility of the esterification–RCM sequence discussed in this section was demonstrated by Kumar and Chandrasekhar’s synthesis of the side-chain-truncated analogs 91 (Scheme 14). Coupling of advanced intermediate 75 with Evans anti-aldol products 89 afforded dienes 90, which were converted to simplified pladienolide analogs 91. They demonstrated that the truncated analog 91 demonstrates biological activities comparable to those of the natural product in the A549 cell line, indicating that the macrocyclic core plays a pivotal role in anticancer activity.
2.2.5. Synthesis of 6-Deoxyoladienolide D by Keaney and Coworkers (2014)
Keaney and coworkers at Eisai Co. reported the total synthesis of 6-deoxypladienolide D (8) in 2014, scarcely accessed by the semisynthetic pathway, despite its potent splicing inhibitory activity [52]. Rather than using precise and delicate synthetic strategies, they focused on scalable and industrial-friendly synthetic routes. From a retrosynthetic perspective, 6-deoxypladienolde D (8) is divided into the macrocyclic core unit and the diene chain, which can be combined by Suzuki coupling (Scheme 15). The 12-membered ring could be constructed by the RCM of ester 92, which would be derived from 57 and 93 using a synthetic strategy akin to Burkart’s synthesis [34].
Preparation of building block 93 began with the commercially available citronellal (95) (Scheme 16). The Peterson olefination of the aldehyde group and oxidative cleavage of the olefin moiety produced aldehyde 94. Pinnick oxidation, followed by Claisen condensation, yielded β-keto ester 96, which underwent asymmetric reduction to afford diastereomeric mixture 97 with a ratio of 4:1. To cost-effectively isolate the pure diastereomer, a chiral resolution of 97 was achieved with (R)-(+)-α-methyl-benzylamine. The stereochemistry at C3 of 98 was confirmed by X-ray crystallography. Building block 93 was then obtained via a three-step sequence: acidification, silylation, and hydrolysis. One of the key building blocks in pladienolide synthesis, 57, was generated on a bulk scale by the Brown asymmetric crotylation of aldehyde 99 [73]. Additionally, the synthesis of the side-chain counterpart 101 utilized cross-metathesis between Kotake’s intermediate 38 [49] and vinyl boronate 100.
The final stage of the macrolide synthesis proceeded according to general synthetic procedures, including esterification of the two building blocks and RCM (Scheme 17). Allylic oxidation of the corresponding macrocycle 102, the key reaction in this synthesis, was preliminarily simulated via computational modeling. Influenced by the C6-methyl group, stereoselective oxidation at the C7 position was predicted and realized in moderate yield and complete diastereoselectivity. Sequential acetylation of 103, Suzuki coupling with 101, and TBS deprotection completed the total synthesis of 6-deoxypladienolide D (8). Keaney’s synthesis requires the use of inexpensive commercially available starting materials and late-stage functionalization to provide a sufficient quantity of scarce 6-deoxypladienolide B, to confirm the biological activity against mutant SF3b1.
2.2.6. Synthesis of Pladeinolide B by Yoo and Krische (2021)
Given the continuous demand for efficient and practical synthesis of complex pladienolide-type natural products, which have more than 11 stereogenic centers, Yoo and Krische designed a remarkably concise convergent synthesis of pladienolide B (2) [53]. By utilizing the state-of-the-art synthetic methodology developed by themselves, key fragments for the convergent synthesis were expected to be prepared [74,75]. As anticipated, the total synthesis of pladienolide B has been finished in only 10 longest-linear steps. Most stereogenic centers on the building blocks 103, 57, and 105 were established by their unique synthetic methods (Scheme 18).
Yoo and Krische’s synthesis is based on the parallel preparation of small fragments 57, 105, 106, and 107 (Scheme 19). Preparation of 105 commenced with the Sharpless asymmetric epoxidation of allylic alcohol 108 [76]. Regioselective epoxidation of more substituted alkenes in 108 afforded the highly enantiomerically rich epoxide 109 (dr = 20:1, 95% ee), which was regioselectively opened by the dienolate 47 to afford 110 after one-pot acetylation. Noyori asymmetric hydrogenation of 110 generated a stereogenic center at C3 with a moderate selectivity of 4:1. Protecting group adjustment successfully provided building block 105. The remaining building blocks were prepared via hydrogenative asymmetric crotylation. Allylic alcohol 57 was provided in a one-step reaction: Ir-catalyzed alcohol-mediated anti-crotylation of 111 and 112. Building blocks of the side-chain fragments 106 and 107 were also accessed by Ru-catalyzed syn-crotylation and Ir-catalyzed asymmetric allylation of 113 and 114, respectively.
After substitution of the hydroxy functional group in 107 with a methyl group under the Normant condition [77], cross-metathesis between the resulting alkynes 115 and 106 was conducted to provide the side chain carbon skeleton 116 (Scheme 20) [78]. Compound 116 was transformed to Suzuki reagent 103 via silyl deprotection, Shi epoxidation, and hydroboration [79]. In the final stage, Suzuki coupling of 103 and the core unit 104, prepared from 57 and 105 by Yamaguchi esterification and RCM, completed the total synthesis of pladienolide B (2). Yoo and Krische demonstrated the shortest stereodivergent synthetic routes for the pladienolide series by maximizing the usefulness of their distinctive chemistry.
2.3. Synthesis via Macrolactonization
Another strategy to construct the 12-membered macrocyclic pladienolide core is macrolactonization. Because macrocyclic lactone moieties are abundant in natural substances (e.g., 8-membered octalactins and 60-membered quinolidomicins) and natural macrocyclic lactones have exhibited a wide range of interesting properties such as medicinal/insecticide activity, fragrance production, and phytotoxicity, synthetic approaches have been extensively studied, particularly in the field of natural product synthesis [80]. Three syntheses of pladienolide B or its core structure that use macrolactonization have been reported by Skaanderup and Jensen [81], Maier and coworkers [82,83], and Rhoades et al. [84].
2.3.1. Synthesis of the Macrocyclic Core of ent-Pladienolide B by Skaanderup and Jensen (2008)
At the outset of the study by Skaanderup and Jensen (2008), the absolute and relative stereochemistry of pladienolides had not been completely revealed. This allowed them to synthesize the ent-pladienolide B core structure devoid of the side chain, because the structurally similar polyketide, 10-deoxymethynolide (117, Figure 4), was previously reported to have similar enantiomeric stereochemistry to that of pladienolide B.
As outlined in Scheme 21, the core structure of the ent-pladienolide B 118 could be accessed by macrolactonization to form a 12-membered ring, the linear substrate of which is conceivably disconnected by cross-metathesis into 119 and 120. The vicinal diol moiety 120 and its relative stereochemistry were built up by the Sharpless asymmetric dihydroxylation of 121. The C2–C3 bond formation is achievable via the asymmetric aldol reaction of aldehyde 122, with the aid of a chiral auxiliary 123.
The synthesis of core structure 118 by Skaanderup and Jensen began with commercially available acetate 124, which underwent selective epoxidation followed by oxidative cleavage to produce aldehyde 122 (Scheme 22). Chiral acetylthiazolidinethione 123 was then employed for asymmetric aldol addition with 122 to yield 121, albeit with a somewhat low diastereomeric ratio (dr = 4:1). After Sharpless asymmetric dihydroxylation to convert 121 to diol 125, the detachment of the chiral auxiliary, followed by Wittig methylenation, yielded a cross-metathesis substrate 120. Subsequent Hoveyda–Grubbs catalyst-mediated cross-coupling with homoallyl ether 119 afforded product 126 in moderate yield. Selective deprotection of the trityl ether in the presence of an acetal protecting group produced seco-acid 127 after methyl ester hydrolysis. Fine-tuning of the Yamaguchi condition eventually furnished 12-membered cycle 128, which was then modified to the core structure of ent-pladienolide B. This is the first example of access to a 12-membered ring via macrolactonization as the last key step, and core structure 118 was obtained in 15 steps with an overall yield of 10.0%.
2.3.2. Synthesis of Pladienolide B by Maier and Coworkers (2014)
In 2014, Maier and coworkers described a total synthesis of pladienolide B (2), incorporating the Horner–Wadsworth–Emmons olefination/macrolactonization sequence as key chemistry for the formation of 12-membered core 129 (Scheme 23) [82]. The precursors of 129, fragments 130 and 131, could be prepared by asymmetric aldol reactions, establishing their absolute and relative stereochemistry. The main skeleton of 131 was constructed by several transformations of commercially available (R)-linalool (132).
First, aldehyde fragment 130 with anti-stereochemistry was prepared by the Masamune–Abiko aldol reaction protocol using chiral ester 133 (Scheme 24) [85,86], after chiral auxiliary detachment and oxidation [83]. The synthesis of fragment 131 was carried out in parallel with (R)-linalool (132). Three steps, including the oxidative cleavage of the trisubstituted olefin, afforded 135, which was converted to 136 via the Nagao acetate aldol reaction with chiral methyl ketone 137 [87,88,89]. Following the establishment of the required stereochemistry, the chiral auxiliary was removed from compound 136, and the resulting compound was treated with 139 to introduce the methylphosphonate group, affording the HWE reaction substrate 131. Aldehyde 130 was then combined with methylphosphonate 131 using an HWE reaction to furnish elongated enone 140, which was reduced to produce 141 by a chelation-controlled reduction with a moderate diastereomeric ratio. Through several transformations, including Shiina-type macrolactonization using 2-methyl-6-nitrobenzoic anhydride (142) [90], the synthesis of the metal-catalyzed reaction substrate 143 was accomplished.
The required side chain, 144, was prepared, as shown in Scheme 25. The known TBS-protected aldol product 145 [57,91], produced using 146, was converted to allyl alcohol 147, which was then subjected to Sn2′ chlorination and the Finkelstein reaction, which yielded allyl iodide 148. The second employment of 146 for asymmetric allylation furnished 149, which underwent the redox sequence, Shi epoxidation, and Seyferth–Gilbert homologation to provide terminal alkyne 150. Alkyne 150 was subjected to palladium-catalyzed hydrostannylation to afford another counterpart 144, which, upon subsequent Stille coupling with 143, finally furnished pladienolide B (2). This total synthesis was completed in 18 steps (LLS) and 0.6% overall yield.
2.3.3. Total Synthesis of Pladienolide A, B, and H3B-8800 by Rhoades et al. (2021)
Rhoades et al. completed the total synthesis of pladienolides A and B as well as the synthetic analog H3B-8800 (13), which are currently under phase 1 clinical trials [84]. Their asymmetric synthesis featured a protecting group and chiral auxiliary-free approaches, resulting in 10-step (LLS)-total synthesis from commercially available building blocks. Retrosynthetically, the formation of pladienolide class products 1, 2, and 11 divergently results from vinyl iodide 151, via Suzuki coupling with diverse vinyl borane counterparts (Scheme 26). The diol moiety of 151 was introduced from 152 under the control of the site- and stereoselectivity. The 12-membered ring of 152 could be accessed by macrolactonization after the intermolecular Heck reaction between terminal olefin 153 and vinyl iodide 154 forming a C7–C8 bond with geometrical selectivity. The absolute and relative stereochemistry of homoallyl alcohol 153, identical to those of natural products, could be established via Krische’s crotylation protocol.
Building blocks 153 and 154, Heck coupling substrates, were prepared efficiently as shown in Scheme 27. Initially, the terminal olefin 153 was synthesized from enal 155, which can be obtained from commercially available propyne in two steps [92] and 3-buten-2-yl acetate 112 by Krische’s crotylation method [93] using a chiral Ir catalyst 156 in excellent diastereomeric and enantiomeric ratios. Dianion formation of β-keto ester 42, followed by the addition of allyl bromide 157, afforded vinyl iodide 154. Heck coupling between 153 and 154 in the presence of silver(I) salts afforded (E)-158 with (Z)-158 as a minor product (E:Z = 7:3). Heating 158 in toluene was effective in producing macrolactone 152, which was converted to alcohol 159 using stereoselective ketone reduction directed from outside the ring system. The subjection of 159 to the Sharpless asymmetric dihydroxylation with the use of AD-mix-β yielded triol 160 with good diastereoselectivity (dr = 10:1) and excellent regioselectivity toward the C6–C7 olefin despite the presence of other olefins. NIS-mediated silicon-iodine exchange [94] provided Suzuki reaction substrate 161 after selective acetylation of the 7-OH of 151.
Side-chain unit 103, a Suzuki coupling counterpart, was prepared by a cross-metathesis–alkyne hydroboration strategy, which was also adopted in Yoo and Krische’s synthesis (Scheme 28). Further, 103 was elongated by cross-metathesis between 49 and 162 using the Grubbs 2nd generation catalyst. Shi epoxidation of the corresponding internal olefin and primary alcohol oxidation afforded 163, the aldehyde of which was converted to a terminal alkyne with Bestmann–Ohira reagent (164). Hydroboration with N-heterocyclic carbene ligand 165 was effective in delivering the vinyl borane group of 103 [95]. The natural products pladienolide A (1) and B (2) were obtained by the Suzuki coupling of 103 with 151 and 161, respectively. The use of 164 during the workup resulted in the efficient removal of palladium metals [96]. For the synthesis of H3B-8800 (11), the core unit 151 was reacted with 165 in the presence of dibutyltin oxide to produce the carbamate, which upon Suzuki reaction with 103, was finally transformed to 11. It is the shortest synthesis of H3B-8800 (10 LLS from commercially available material, with 10.1% yield; compared with Eisai Co.: 12 LLS, 4.2% overall yield) [97].
3. Conclusions
This review focused on the total syntheses of pladienolide-class natural products that exhibit spliceosome-modulating activity by binding to the SF3b unit. These polyketides have become an attractive target for the synthetic community because of the synthetic challenges afforded by its 12-membered macrocyclic framework, as well as up to 10 stereogenic centers. Total syntheses for compounds of this class to date can be divided into two groups, according to the key method used to access the macrocyclic unit—RCM or macrolactonization. The yields of both cyclization steps are moderate to excellent (51–93%) as indicated in Table 1. The overall yields of LLS are also shown in Table 1, which reveals that Rhoades et al. completed the total synthesis of pladienolides with the best overall yield (ca. 12%) by virtue of the shortest steps (10 or 11 steps).
The synthetic feature of each synthesis is summarized in Table 2. The syntheses utilizing RCM reaction commonly share intermolecular esterification–RCM sequence for the construction of the macrocyclic core. On the other hand, macrolactonization-mediated syntheses utilize each different sequence for preparing macrocyclic cores. Most stereocenters on pladienolides were established in a reagent-controlled manner. The stereogenic methyl and hydroxy groups on both the macrocyclic core and the side chain fragment are mostly controlled by an asymmetric aldol reaction using a chiral auxiliary. The epoxide in the chain was installed using external chiral sources. Conjugation between the two fragments was conducted by metal-catalyzed coupling or olefination. These synthetic efforts finally elaborated the bioactive synthetic analogs, E7107 and H3B-8800, the latter of which is currently under phase 1 clinical trials to treat patients with various hematologic malignancies. Given the increasing interest in RNA splicing for drug discovery, as well as the promising potential as anti-cancer agents, the rapid development of synthetic and medicinal chemistry leveraging pladienolide-class natural products and their analogs is anticipated in the near future.
Author Contributions
Conceptualization, J.S. and H.J.; writing—original draft preparation, J.S., E.J., H.J.K. and H.J.; writing—review and editing, J.S. and H.J.; supervision, J.S. and H.J.; funding acquisition, J.S. and H.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Chungnam National University (2020–2021) and the grant of the Korea Research Institute of Chemical Technology (BSF21-502 and KK2131-30).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Baginski, B.; Wirecki, T.K.; De Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. Modomics: A database of RNA modification pathways. 2017 Update. Nucleic Acids Res. 2017, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Di, C.; Syafrizayanti, Z.Q.; Chen, Y.; Wang, Y.; Zhang, X.; Liu, Y.; Sun, C.; Zhang, H.; Hoheisel, J.D. Function, clinical application, and strategies of Pre-mRNA splicing in cancer. Cell Death Differ. 2018, 26, 1181–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Gong, Q.; Wang, Y.; Li, M.; Wang, L.; Ding, H.; Li, P. The biological function and clinical significance of SF3B1 mutations in cancer. Biomark. Res. 2020, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Eid, O.M.; Kader, R.M.A.A.; Fathalla, L.A.; Abdelrahman, A.H.; Rabea, A.; Mahrous, R.; Eid, M.M. Evaluation of MLPA as a comprehensive molecular cytogenetic tool to detect cytogenetic markers of chronic lymphocytic leukemia in Egyptian patients. J. Genet. Eng. Biotechnol. 2021, 19, 98–104. [Google Scholar] [CrossRef]
- Jiménez-Vacas, J.M.; Herrero-Aguayo, V.; Gómez-Gómez, E.; León-González, A.J.; Sáez-Martínez, P.; Alors-Pérez, E.; Fuentes-Fayos, A.C.; Martínez-López, A.; Sánchez-Sánchez, R.; González-Serrano, T.; et al. Spliceosome component SF3B1 as novel prognostic biomarker and therapeutic target for prostate cancer. Transl. Res. 2019, 212, 89–103. [Google Scholar] [CrossRef]
- Maguire, S.; Leonidou, A.; Wai, P.; Marchiò, C.; Ng, C.K.Y.; Sapino, A.; Salomon, A.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R.C. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J. Pathol. 2014, 235, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Huo, Y.; Yang, M.; Shen, Y.; Liu, D.; Fu, X.; Tao, L.; He, R.; Zhang, J.; Hua, R.; et al. SF3B1 mutation in pancreatic cancer contributes to aerobic glycolysis and tumor growth through a PP2A–c-Myc axis. Mol. Oncol. 2021, 1–15. [Google Scholar] [CrossRef]
- Donaldson, W.A. Syntheses of spliceostatins and thailanstatins: A review. Beilstein J. Org. Chem. 2020, 16, 1991–2006. [Google Scholar] [CrossRef]
- Lagisetti, C.; Pourpak, A.; Jiang, Q.; Cui, X.; Goronga, T.; Morris, S.W.; Webb, T.R. Antitumor compounds based on a natural product consensus pharmacophore. J. Med. Chem. 2008, 51, 6220–6224. [Google Scholar] [CrossRef] [Green Version]
- Lagisetti, C.; Pourpak, A.; Goronga, T.; Jiang, Q.; Cui, X.; Hyle, J.; Lahti, J.M.; Morris, S.W.; Webb, T.R. Synthetic mRNA splicing modulator compounds with In Vivo antitumor activity. J. Med. Chem. 2009, 52, 6979–6990. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Lagisetti, C.; Edwards, C.C.; Webb, T.R.; Potter, P.M. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem. Biol. 2011, 6, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Thirupathi, B.; Zilla, M.K. Syntheses and biological importance of herboxidiene/GEX1A. Chem. Select. 2019, 4, 11944–11958. [Google Scholar] [CrossRef]
- Pokhrel, A.R.; Dhakal, D.; Jha, A.; Sohng, J.K. Herboxidiene biosynthesis, production, and structural modifications: Prospect for hybrids with related polyketide. Appl. Microbiol. Biotechnol. 2015, 99, 8351–8362. [Google Scholar] [CrossRef]
- Sakai, T.; Sameshima, T.; Matsufuji, M.; Kawamura, N.; Dobashi, K.; Mizui, Y. Pladienolides, new substances from culture of streptomyces platensis Mer-11107 I. Taxonomy, fermentation, isolation and screening. In Vitro and In Vivo antitumor activities. J. Antibiot. 2004, 57, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Sakai, T.; Asai, N.; Okuda, A.; Kawamura, N.; Mizui, Y. Pladienolides, new substances from culture of Streptomyces platensis mer-11107. II. Physico-chemical properties and structure elucidation. J. Antibiot. 2004, 57, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Mizui, Y.; Sakai, T.; Iwata, M.; Uenaka, T.; Okamoto, K.; Shimizu, H.; Yamori, T.; Yoshimatsu, K.; Asada, M. Pladienolides, new substances from culture of streptomyces platensis mer-11107 III. In Vitro and In Vivo antitumor activities. J. Antibiot. 2004, 57, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Gundluru, M.K.; Pourpak, A.; Cui, X.; Morris, S.W.; Webb, T.R. Design, synthesis and initial biological evaluation of a novel pladienolide analog scaffold. Med. Chem. Comm. 2011, 2, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Mandel, A.L.; Jones, B.D.; La Clair, J.J.; Burkart, M.D. A synthetic entry to pladienolide B and FD-895. Bioorganic Med. Chem. Lett. 2007, 17, 5159–5164. [Google Scholar] [CrossRef] [Green Version]
- Lagisetti, C.; Yermolina, M.V.; Sharma, L.K.; Palacios, G.; Prigaro, B.J.; Webb, T.R. Pre-mRNA splicing-modulatory pharmacophores: The total synthesis of herboxidiene, a pladienolide-herboxidiene hybrid analog and related derivatives. ACS Chem. Biol. 2014, 9, 643–648. [Google Scholar] [CrossRef]
- Booth, T.J.; Kalaitzis, J.A.; Vuong, D.; Crombie, A.; Lacey, E.; Piggott, A.M.; Wilkinson, B. Production of novel pladienolide analogues through native expression of a pathway-specific activator. Chem. Sci. 2020, 11, 8249–8255. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Arisawa, A.; Takeda, S.; Tsuchida, T.; Aritoku, Y.; Yoshida, M.; Ikeda, H. Organization of the biosynthetic gene cluster for the polyketide antitumor macrolide, pladienolide, in Streptomyces platensis mer-11107. Biosci. Biotechnol. Biochem. 2008, 72, 2946–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, N.; Kotake, Y.; Niijima, J.; Fukuda, Y.; Uehara, T.; Sakai, T. Stereochemistry of pladienolide B. J. Antibiot. 2007, 60, 364–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, A.; Kotake, Y.; Takahashi, K.; Kadowaki, T.; Matsumoto, Y.; Minoshima, Y.; Sugi, N.H.; Sagane, K.; Hamaguchi, M.; Iwata, M.; et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 2011, 278, 4870–4880. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Muguruma, N.; Nakagawa, T.; Okamoto, K.; Kimura, T.; Kitamura, S.; Yano, H.; Sannomiya, K.; Goji, T.; Miyamoto, H.; et al. High antitumor activity of pladienolide B and its derivative in gastric cancer. Cancer Sci. 2014, 105, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Cretu, C.; Agrawal, A.A.; Cook, A.; Will, C.L.; Fekkes, P.; Smith, P.G.; Lührmann, R.; Larsen, N.; Buonamici, S.; Pena, V. Structural basis of splicing modulation by antitumor macrolide compounds. Mol. Cell 2018, 70, 265–273.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Kotake, Y.; Niijima, J.; Fukuda, Y.; Nagai, M.; Kanada, R.M.; Takeda, S.; Nakashima, T.; Yoshida, M.; Tsuchida, T.; Sameshima, T. Novel Physiologically Active Substances. U.S. Patent US/2007/7,256,178, 2007. [Google Scholar]
- Seki-Asano, M.; Okazaki, T.; Yamagishi, M.; Sakai, N.; Takayama, Y.; Hanada, K.; Morimoto, S.; Takatsuki, A.; Mizoue, K.; Morimoro, S. Isolation and characterization of a new 12-membered macrolide FD-895. J. Antibiot. 1994, 47, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Villa, R.; Mandel, A.L.; Jones, B.D.; La Clair, J.J.; Burkart, M.D. Structure of FD-895 revealed through total synthesis. Org. Lett. 2012, 14, 5396–5399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, M.; Ozawa, Y.; Uenaka, T.; Shimizu, H.; Niijima, J.; Kanada, R.M.; Fukuda, Y.; Nagai, M.; Kotake, Y.; Yoshida, M.; et al. E7107, a new 7-urethane derivative of pladienolide D, displays curative effect against several human tumor xenografts. Proc. Am. Assoc. Cancer Res. 2004, 45, 691. [Google Scholar]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef]
- Eskens, F.; Ramos, F.J.; Burger, H.; O’Brien, J.; Piera, A.; De Jonge, M.; Mizui, Y.; Wiemer, E.; Carreras, M.; Maria, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.M.; Zack, D.J. Alternative splicing and retinal degeneration. Clin. Genet. 2013, 84, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Results of a clinical trial of H3B-8800, a splicing modulator, in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) or chronic myelomonocytic leukemia (CMML). Blood 2019, 134, 673. [Google Scholar] [CrossRef]
- Webb, T.R.; Joyner, A.S.; Potter, P.M. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov. Today 2013, 18, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Salton, M.; Misteli, T. Small molecule modulators of pre-mRNA splicing in cancer therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Effenberger, K.A.; Urabe, V.K.; Jurica, M.S. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip. Rev. RNA 2017, 8, e1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León, B.; Kashyap, M.; Chan, W.C.; Krug, K.; Castro, J.E.; La Clair, J.J.; Burkart, M.D. A challenging pie to splice: Drugging the spliceosome. Angew. Chem. Int. Ed. 2017, 56, 12052–12063. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Meng, F. A comprehensive overview of structure-activity relationships of small-molecule splicing modulators targeting SF3B1 as anticancer agents. Chem. Med. Chem. 2020, 15, 2098–2120. [Google Scholar] [CrossRef] [PubMed]
- Katsuki, T.; Sharpless, K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Tu, Y.; Wang, Z.-X.; Shi, Y. An efficient asymmetric epoxidation method for trans-olefins mediated by a fructose-derived ketone. J. Am. Chem. Soc. 2010, 28, 9806–9807. [Google Scholar] [CrossRef]
- Cossy, J.; Arseniyadis, S.; Meyer, C. Metathesis in Natural Product Synthesis: Strategies, Substrates and Catalysts; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Gradillas, A.; Pérez-Castells, J. Macrocyclization by ring-closing metathesis in the total synthesis of natural products: Reaction conditions and limitations. Angew. Chem. Int. Ed. 2006, 45, 6086–6101. [Google Scholar] [CrossRef]
- Kanada, R.M.; Itoh, D.; Nagai, M.; Niijima, J.; Asai, N.; Mizui, Y.; Abe, S.; Kotake, Y. Total synthesis of the potent antitumor macrolides pladienolide B and D. Angew. Chem. Int. Ed. 2007, 46, 4350–4355. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Anderson, D.D. Enantioselective total synthesis of pladienolide B: A potent spliceosome inhibitor. Org. Lett. 2012, 14, 4730–4733. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.P.; Chandrasekhar, S. Enantioselective synthesis of pladienolide B and truncated analogues as new anticancer agents. Org. Lett. 2013, 15, 3610–3613. [Google Scholar] [CrossRef]
- Arai, K.; Buonamici, S.; Chan, B.; Corson, L.; Endo, A.; Gerard, B.; Hao, M.-H.; Karr, C.; Kira, K.; Lee, L.; et al. Total synthesis of 6-deoxypladienolide d and assessment of splicing inhibitory activity in a mutant sf3b1 cancer cell line. Org. Lett. 2014, 16, 5560–5563. [Google Scholar] [CrossRef]
- Yoo, M.; Krische, M.J. Total synthesis of the spliceosome modulator pladienolide B via. asymmetric alcohol-mediated syn- and anti-diastereoselective carbonyl crotylation. Angew. Chem. Int. Ed. 2021, 60, 13923–13928. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.; Wallace, D.J.; Cowden, C.J. Polyketide synthesis using the boron-mediated, anti-aldol reactions of lactate-derived ketones: Total synthesis of (-)-ACRL toxin IIIB. Synthesis 1998, 29, 639–652. [Google Scholar] [CrossRef]
- Grieco, P.; Masaki, Y.; Boxler, D. Sesterterpenes. I. Stereospecific total synthesis of moenocinol. J. Am. Chem. Soc. 1975, 97, 1597–1599. [Google Scholar] [CrossRef]
- Fukuzawa, S.-I.; Matsuzawa, H.; Yoshimitsu, S.-I. Asymmetric samarium-Reformatsky reaction of chiral α-bromoacetyl-2-oxazolidinones with aldehydes. J. Org. Chem. 2000, 65, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Gage, J.R.; Evans, D.A. Diastereoselective aldol condensation using a chiral oxazolidinone auxiliary: (2S*,3S*)-3-hydroxy-3-phenyl-2-methylpropanoic acid. Org. Synth. 2003, 68, 83. [Google Scholar] [CrossRef]
- Chatterjee, A.K.; Choi, T.-L.; Sanders, A.D.P.; Grubbs, R.H. A general model for selectivity in olefin cross metathesis. J. Am. Chem. Soc. 2003, 125, 11360–11370. [Google Scholar] [CrossRef] [Green Version]
- Nagano, H.; Nakanishi, E.; Takajo, S.; Sakuma, M.; Kudo, K. Synthesis of 6-(poly)prenyl-substituted polyprenols and their phosphates. Tetrahedron 1999, 55, 2591–2608. [Google Scholar] [CrossRef]
- Brown, H.C.; Bhat, K.S. Chiral synthesis via. organoboranes. Part 7. Diastereoselective and enantioselective synthesis of erythro-and threo-β-methylhomoallyl alcohols via enantiomeric (Z)- and (E)-crotylboranes. J. Am. Chem. Soc. 1987, 108, 5919–5923. [Google Scholar] [CrossRef]
- Yatagai, M.; Ohnuki, T. Asymmetric reduction of functionalized ketones with a sodium borohydride-(L)-tartaric acid system. J. Chem. Soc. Perkin Trans. 1990, 6, 1826–1828. [Google Scholar] [CrossRef]
- Ramachandran, P.V.; Liu, H.; Reddy, M.V.R.; Brown, H.C. Synthesis of homoallylic chiral tertiary alcohols via. chelation-controlled diastereoselective nucleophilic addition on α-alkoxyketones: Application for the synthesis of the C1−C11 subunit of 8-epi-fostriecin. Org. Lett. 2003, 5, 3755–3757. [Google Scholar] [CrossRef]
- Still, W.C.; McDonald, J.H. Chelation-controlled nucleophilic additions. 1. A highly effective system for asymmetric induction in the reaction of organometallics with α-alkoxyketones. Tetrahedron Lett. 1980, 21, 1031–1034. [Google Scholar] [CrossRef]
- Zhang, Y.; Phillips, A.A.J.; Sammakia, T. Highly selective asymmetric acetate aldol reactions of an N-Acetyl thiazolidinethione reagent. Org. Lett. 2004, 6, 23–25. [Google Scholar] [CrossRef]
- Marshall, J.A. Chiral allylic and allenic metal reagents for organic synthesis. J. Org. Chem. 2007, 72, 8153–8166. [Google Scholar] [CrossRef]
- Crimmins, M.T.; King, B.W.; Tabet, E.A. Asymmetric aldol additions with titanium enolates of acyloxazolidinethiones: Dependence of selectivity on amine base and Lewis acid stoichiometry. J. Am. Chem. Soc. 1997, 119, 7883–7884. [Google Scholar] [CrossRef]
- Chan, W.C.; La Clair, J.J.; León, B.; Trieger, K.A.; Slagt, M.Q.; Verhaar, M.T.; Bachera, D.U.; Rispens, M.T.; Hofman, R.M.; de Boer, V.L.; et al. Scalable synthesis of 17S-FD-895 expands the structural understanding of splice modulatory activity. Cell Rep. Phys. Sci. 2020, 1, 100277. [Google Scholar] [CrossRef]
- Gonzalez, I.C.; Forsyth, C.J. Total synthesis of thyrsiferyl 23-acetate, a specific inhibitor of protein phosphatase 2A and an anti-leukemic inducer of apoptosis. J. Am. Chem. Soc. 2000, 122, 9099–9108. [Google Scholar] [CrossRef]
- Viti, S.M. Regioselective reductions of 2,3-epoxy alcohols. Tetrahedron Lett. 1982, 23, 4541–4544. [Google Scholar] [CrossRef]
- Jung, M.E.; Lee, W.S.; Sun, D. Synthesis of four diastereomeric 3,5-dialkoxy-2,4-dimethylalkanals by a simple extension of the non-aldol aldol process to bis (propionates). Org. Lett. 1999, 1, 307–310. [Google Scholar] [CrossRef]
- Ireland, R.E.; Mueller, R.H.; Willard, A.K. The ester enolate Claisen rearrangement. Stereochemical control through stereoselective enolate formation. J. Am. Chem. Soc. 1976, 98, 2868–2877. [Google Scholar] [CrossRef]
- Kajikawa, T.; Aoki, K.; Singh, R.S.; Iwashita, T.; Kusumoto, T.; Frank, H.A.; Hashimoto, H.; Katsumura, S. Syntheses of allene-modified derivatives of peridinin toward elucidation of the effective role of the allene function in high energy transfer efficiencies in photosynthesis. Org. Biomol. Chem. 2009, 7, 3723–3733. [Google Scholar] [CrossRef] [Green Version]
- Brown, H.C.; Jadhav, P.K. Asymmetric carbon-carbon bond formation via β-allyldiisopinocampheylborane. Simple synthesis of secondary homoallylic alcohols with excellent enantiomeric purities. J. Am. Chem. Soc. 1983, 105, 2092–2093. [Google Scholar] [CrossRef]
- Kim, S.W.; Zhang, W.; Krische, M.J. Catalytic enantioselective carbonyl allylation and propargylation via. alcohol-mediated hydrogen transfer: Merging the chemistry of Grignard and Sabatier. Acc. Chem. Res. 2017, 50, 2371–2380. [Google Scholar] [CrossRef]
- Holmes, M.; Schwartz, L.A.; Krische, M.J. Intermolecular metal-catalyzed reductive coupling of dienes, allenes, and enynes with carbonyl compounds and imines. Chem. Rev. 2018, 118, 6026–6052. [Google Scholar] [CrossRef]
- Brito, G.A.; Jung, W.; Yoo, M.; Krische, M.J. Enantioselective iridium-catalyzed allylation of acetylenic ketones via 2-propanol-mediated reductive coupling of allyl acetate: C14-C23 of pladienolide D. Angew. Chem. Int. Ed. 2019, 58, 18803–18807. [Google Scholar] [CrossRef]
- Bertus, P.; Drouin, L.; Laroche, C.; Szymoniak, J. Pentadienyl transfer reagents based on zirconium: Preparation and reactions with carbonyl compounds. Tetrahedron 2004, 60, 1375–1383. [Google Scholar] [CrossRef]
- Overman, L.E.; Bell, K.L. Enantiospecific total synthesis of dendrobatid toxin 251D. A short chiral entry to the cardiac-active pumiliotoxin A alkaloids via. stereospecific iminium ion-vinylsilane cyclizations. J. Am. Chem. Soc. 1981, 103, 1851–1853. [Google Scholar] [CrossRef]
- Zhao, J.; Niu, Z.; Fu, H.; Li, Y. Ligand-free hydroboration of alkynes catalyzed by heterogeneous copper powder with high efficiency. Chem. Commun. 2013, 50, 2058–2060. [Google Scholar] [CrossRef]
- Parenty, A.; Moreau, X.; Campagne, J.-M. Macrolactonizations in the total synthesis of natural products. Chem. Rev. 2006, 106, 911–939. [Google Scholar] [CrossRef]
- Skaanderup, P.R.; Jensen, T. Synthesis of the macrocyclic core of (−)-pladienolide B. Org. Lett. 2008, 10, 2821–2824. [Google Scholar] [CrossRef]
- Müller, S.; Sasse, F.; Maier, M.E. Synthesis of pladienolide B and its 7-epimer with insights into the role of the allylic acetate. Eur. J. Org. Chem. 2014, 1025–1036. [Google Scholar] [CrossRef]
- Müller, S.; Mayer, T.; Sasse, F.; Maier, M.E. Synthesis of a pladienolide B analogue with the fully functionalized core structure. Org. Lett. 2011, 13, 3940–3943. [Google Scholar] [CrossRef]
- Rhoades, D.; Rheingold, A.L.; O’Malley, B.W.; Wang, J. Expedient total syntheses of pladienolide-derived spliceosome modulators. J. Am. Chem. Soc. 2021, 143, 4915–4920. [Google Scholar] [CrossRef]
- Abiko, A. Boron-mediated aldol reaction of carboxylic esters. Acc. Chem. Res. 2004, 37, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Menche, D.; Hassfeld, J.; Li, J.; Mayer, K.; Rudolph, S. Modular total synthesis of archazolid A and B. J. Org. Chem. 2009, 74, 7220–7229. [Google Scholar] [CrossRef]
- Nagao, Y.; Hagiwara, Y.; Kumagai, T.; Ochiai, M.; Inoue, T.; Hashimo, K.; Fujita, E. New C-4-chiral 1,3-thiazolidine-2-thiones: Excellent chiral auxiliaries for highly diastereo-controlled aldol-type reactions of acetic acid and α,β-unsaturated aldehydes. J. Org. Chem. 1986, 51, 2391–2393. [Google Scholar] [CrossRef]
- Nagao, Y.; Dai, W.M.; Ochiai, M.; Shiro, M. New general asymmetric synthesis of versatile γ-alkylated butenolides and its application to expeditious synthesis of the chiral Geissman-Waiss lactones useful for (+)-retronecine synthesis. J. Org. Chem. 1989, 54, 5211–5217. [Google Scholar] [CrossRef]
- Delaunay, D.; Toupet, L.; Corre, M.L. Reactivity of β-amino alcohols with carbon disulfide study on the synthesis of 2-oxazolidinethiones and 2-thiazolidinethiones. J. Org. Chem. 1995, 60, 6604–6607. [Google Scholar] [CrossRef]
- Shiina, I. Total synthesis of natural 8- and 9-membered lactones: Recent advancements in medium-sized ring formation. Chem. Rev. 2007, 107, 239–273. [Google Scholar] [CrossRef]
- Borisova, S.A.; Kim, H.J.; Pu, X.; Liu, H. Glycosylation of acyclic and cyclic aglycone substrates by macrolide glycosyltransferase DesVII/DesVIII: Analysis and implications. Chem. Bio. Chem. 2008, 9, 1554–1558. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.F.; Cirillo, P.F.; Schaus, J.V.; Panek, J.S. An efficient procedure for the preparation of chiral β-substituted (E)-crotylsilanes: Application of a rhodium(II) catalyzed silylformylation of terminal alkynes. Tetrahedron Lett. 1995, 36, 8723–8726. [Google Scholar] [CrossRef]
- Kim, I.S.; Han, S.B.; Krische, M.J. Anti-diastereo- and enantioselective carbonyl crotylation from the alcohol or aldehyde oxidation level employing a cyclometallated iridium catalyst: α-methyl allyl acetate as a surrogate to preformed crotylmetal reagents. J. Am. Chem. Soc. 2009, 131, 2514–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakarian, A.; Batch, A.; Holton, R.A. A convergent total synthesis of hemibrevetoxin B. J. Am. Chem. Soc. 2003, 125, 7822–7824. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Zhugralin, A.R.; Lee, Y.; Hoveyda, A.H. Highly selective methods for synthesis of internal (α-) vinylboronates through efficient NHC–Cu-catalyzed hydroboration of terminal alkynes. Utility in chemical synthesis and mechanistic basis for selectivity. J. Am. Chem. Soc. 2011, 133, 7859–7871. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, W.P.; Vo, A. Dithiocarbamates: Reagents for the removal of transition metals from organic reaction media. Org. Process. Res. Dev. 2014, 19, 1369–1373. [Google Scholar] [CrossRef]
- Keaney, G.F.; Wang, J.; Gerard, B.; Arai, K.; Liu, X.; Zheng, G.Z.; Kira, K.; Tivitmahaisoon, P.; Prajapati, S.; Gearhart, N.C.; et al. Pladienolide Pyridine Compounds and Methods of Use. U.S. Patent 2,015,032,952,8A1, 1 November 2016. [Google Scholar]
Figure 1.
Representative examples of naturally occurring spliceosome modulators.

Figure 2.
Chemical structure of pladienolides A–G (1–7), 6-deoxypladienolide D (8), FD-895 (9), and synthetic analogs (10 and 11). The colors show the part of each compound that is structurally different from the structure of pladienolide A (1).
Figure 2.
Chemical structure of pladienolides A–G (1–7), 6-deoxypladienolide D (8), FD-895 (9), and synthetic analogs (10 and 11). The colors show the part of each compound that is structurally different from the structure of pladienolide A (1).

Figure 3.
Brief summary of the disconnections of pladienolides as reported, 2007–2021.

Scheme 1.
Kotake’s retrosynthetic analysis of pladienolides A and B (2007).

Scheme 2.
Preparation of three fragments for the synthesis of pladienolide B (Kotake, 2007).

Scheme 3.
Completion of the total synthesis of pladienolides A and B (Kotake, 2007).

Scheme 4.
Preparation of the side chain fragment and completion of the total synthesis of pladienolide D (Kotake, 2007).
Scheme 4.
Preparation of the side chain fragment and completion of the total synthesis of pladienolide D (Kotake, 2007).

Scheme 5.
Ghosh and Anderson’s retrosynthetic analysis of pladienolide B (2012).

Scheme 6.
Preparation of the fragments for the synthesis of pladienolide B (Ghosh and Anderson, 2012).
Scheme 6.
Preparation of the fragments for the synthesis of pladienolide B (Ghosh and Anderson, 2012).

Scheme 7.
Final stage of the total synthesis of pladienolide B (2) (Ghosh and Anderson, 2012).

Scheme 8.
Burkart’s retrosynthetic analysis of FD-895 (2012).

Scheme 9.
Synthesis of macrocyclic core 64 (Burkart, 2012).

Scheme 10.
Final stage of the total synthesis of FD-895 (Burkart, 2012).

Scheme 11.
Kumar and Chandrasekhar’s retrosynthetic analysis of pladienolide B (2013).

Scheme 12.
Construction of the macrocyclic core unit 82 (Kumar and Chandrasekhar, 2013).

Scheme 13.
Final stage of the total synthesis of pladienolide B (Kumar and Chandrasekhar, 2013).

Scheme 14.
Synthesis of truncated pladienolide B analogs (Kumar and Chandrasekhar, 2013).

Scheme 15.
Keaney’s retrosynthetic analysis of 6-deoxypladienolide D (2014).

Scheme 16.
Total synthesis of 6-deoxypladienolide D (Keaney, 2014).

Scheme 17.
Final stage of the total synthesis of 6-deoxypladienolide D (Keaney, 2014).

Scheme 18.
Yoo and Krische’s retrosynthetic analysis of pladienolide B (2021).

Scheme 19.
Preparation of fragments required for the synthesis of pladienolide B (Yoo and Krische, 2021).
Scheme 19.
Preparation of fragments required for the synthesis of pladienolide B (Yoo and Krische, 2021).

Scheme 20.
The completion of the total synthesis of pladienolide B (Yoo and Krische, 2021).

Figure 4.
Structural comparison of pladienolide B (2) and 10-deoxymethynolide (117).

Scheme 21.
Skaanderup and Jensen’s retrosynthetic analysis (2008).

Scheme 22.
Synthesis of the core structure of ent-pladienolide B (Skaanderup and Jensen, 2008).

Scheme 23.
Maier’s retrosynthetic analysis of pladienolide B (2014).

Scheme 24.
Synthesis of the macrocyclic core unit 143 (Maier, 2014).

Scheme 25.
Preparation of the side-chain unit 144 and the final stage of the total synthesis of pladienolide B (Maier, 2014).
Scheme 25.
Preparation of the side-chain unit 144 and the final stage of the total synthesis of pladienolide B (Maier, 2014).

Scheme 26.
Rhoades et al.’s retrosynthetic analysis of pladienolide A, B, and H3B-8800 (2021).

Scheme 27.
Preparation of a 12-membered macrocyclic unit (Rhoades et al., 2021).

Scheme 28.
Final stage of the total synthesis of pladienolide A, B, and H3B-8800 (Rhoades et al., 2021).
Scheme 28.
Final stage of the total synthesis of pladienolide A, B, and H3B-8800 (Rhoades et al., 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of total syntheses of pladienolides.
| Cyclization Method | Year | Group | Synthetic Target | Cycliz-ation Yield | LLS | Overall Yield (LLS) | Scheme | Refs. |
|---|---|---|---|---|---|---|---|---|
| RCM | 2007 | Kotake | pladienolide A | 93% | 21 | 3.5% | 1–4 | [49] |
| pladienolide B | 22 | 2.9% | ||||||
| pladienolide D | 20 | 3.1% | ||||||
| 2012 | Ghosh | pladienolide B | 83% | 17 | 1.4% | 5–7 | [50] | |
| 2012 | Burkart | FD-895 | 48% | 15 | 0.8% | 8–10 | [34] | |
| 2013 | Chandrasekhar | pladienolide B | 52% | 21 | 7.0% | 11–14 | [51] | |
| 2014 | Keaney | 6-deoxypladienolide D | 77% | 18 | 0.6% | 15, 16 | [52] | |
| 2021 | Krische | pladienolide B | 51% | 10 | 0.8% | 18–20 | [53] | |
| Macrolactonization | 2008 | Skaanderup | macrocycle of pladienolide B | 63% | 15 | 10.0% | 21, 22 | [81] |
| 2014 | Maier | pladienolide B | 93% | 17 | 2.0% | 23–25 | [82,83] | |
| 2021 | Rhoades–O’Malley–Wang | pladienolide A | 70% | 9 | 11.8% | 26–28 | [84] | |
| pladienolide B | 10 | 11.9% | ||||||
| H3B-8800 | 10 | 10.1% |
Table 2.
Synthetic features of the total syntheses covered in this review.
| Cyclization Method | Group | Synthetic Features |
|---|---|---|
| RCM | Kotake (Section 2.2.1) |
|
| Ghosh (Section 2.2.2) |
| |
| Burkart (Section 2.2.3) |
| |
| Chandrasekhar (Section 2.2.4) |
| |
| Keaney (Section 2.2.5) |
| |
| Krische (Section 2.2.6) |
| |
| Macro-lactonization | Skaanderup (Section 2.3.1) |
|
| Maier (Section 2.3.2) |
| |
| Rhoades–O’Malley–Wang (Section 2.3.3) |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sim, J.; Jang, E.; Kim, H.J.; Jeon, H. Total Syntheses of Pladienolide-Derived Spliceosome Modulators. Molecules 2021, 26, 5938. https://doi.org/10.3390/molecules26195938
AMA Style
Sim J, Jang E, Kim HJ, Jeon H. Total Syntheses of Pladienolide-Derived Spliceosome Modulators. Molecules. 2021; 26(19):5938. https://doi.org/10.3390/molecules26195938
Chicago/Turabian StyleSim, Jaehoon, Eunbin Jang, Hyun Jin Kim, and Hongjun Jeon. 2021. "Total Syntheses of Pladienolide-Derived Spliceosome Modulators" Molecules 26, no. 19: 5938. https://doi.org/10.3390/molecules26195938