Development of a Unified Reversed-Phase HPLC Method for Efficient Determination of EP and USP Process-Related Impurities in Celecoxib Using Analytical Quality by Design Principles



Abstract
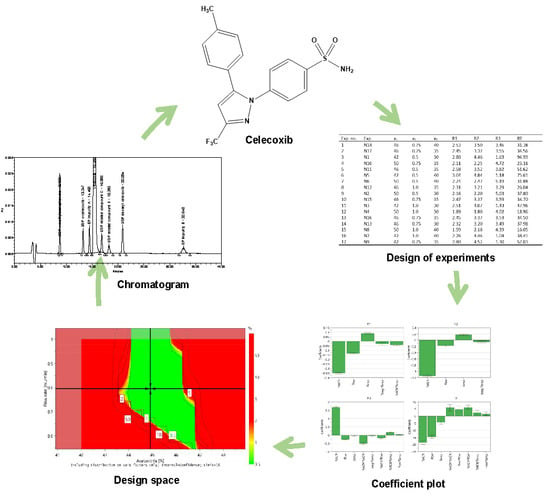
:
1. Introduction
2. Results and Discussion
2.1. Initial Testing of Existing Key HPLC Methods
2.2. Method Scouting
2.3. Analytical Target Profile, Critical Method Parameters, and Attributes
2.4. Method Optimization: Design of Experiments
- Chromatographic column: Chiralpak IA-3, 250 × 4.6 mm
- Column temperature: 40 °C
- Mobile phase: acetonitrile/water = 45/55% (v/v)
- Flow rate: 0.8 mL/min
- Detection: spectrophotometer at 250 nm
- Injection volume: 10 μL
2.5. Method Verification
2.5.1. Precision
2.5.2. LOD and LOQ
2.5.3. Linearity
2.5.4. Accuracy
2.5.5. Response Factors
3. Materials and Methods
3.1. Reagents and Standards
3.2. Diluent, Mobile Phase, Column, and Chromatographic Conditions
3.3. HPLC System and Software
3.4. Preparation of Solutions
3.4.1. Preparation of Standard and Sensitivity Solutions
3.4.2. Preparation of Sample Solution
3.4.3. Preparation of Impurity Solutions
3.4.4. Preparation of Spiked Solution for Method Optimization
3.5. Method Verification
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clemett, D.; Goa, K.L. Celecoxib. Drugs 2000, 59, 957–980. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E.; Keating, G.M. Celecoxib. Drugs 2007, 67, 2433–2474. [Google Scholar] [CrossRef] [PubMed]
- McCormack, P.L. Celecoxib. Drugs 2011, 71, 2457–2489. [Google Scholar] [CrossRef]
- Antoniou, K.; Malamas, M.; Drosos, A.A. Clinical pharmacology of celecoxib, a COX-2 selective inhibitor. Expert Opin. Pharmacother. 2007, 8, 1719–1732. [Google Scholar] [CrossRef]
- Papageorgiou, N.; Zacharia, E.; Briasoulis, A.; Charakida, M.; Tousoulis, D. Celecoxib for the treatment of atherosclerosis. Expert Opin. Invest. Drugs 2016, 25, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Krasselt, M.; Baerwald, C. Celecoxib for the treatment of musculoskeletal arthritis. Expert Opin. Pharmacother. 2019, 20, 1689–1702. [Google Scholar] [CrossRef]
- Attimarad, M.; Venugopala, K.N.; SreeHarsha, N.; Aldhubiab, B.E.; Nair, A.B. Validation of rapid RP-HPLC method for concurrent quantification of amlodipine and celecoxib in pure and formulation using an experimental design. Microchem. J. 2020, 152, 104365. [Google Scholar] [CrossRef]
- Bapatu, H.R.; Maram, R.K.; Murthy, R.S. Stability-indicating HPLC method for quantification of celecoxib and diacerein along with its impurities in capsule dosage form. J. Chrom. Sci. 2015, 53, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Srinivasulu, D.; Sastry, B.S.; Rajendra Prasad, Y.; Om Prakash, G. Separation and determination of process-related impurities of celecoxib in bulk drugs using reversed phase liquid chromatography. Farmacia 2012, 60, 436–447. [Google Scholar]
- Baboota, S.; Faiyaz, S.; Ahuja, A.; Ali, J.; Shafiq, S.; Ahmad, S. Development and optimization of a stability-indicating HPLC method for analyses of celecoxib (CXB) in bulk drug and microemulsion formulations. Acta Chromatogr. 2007, 18, 116–129. [Google Scholar]
- Rao, R.N.; Meena, S.; Nagaraju, D.; Rao, A.R.; Ravikanth, S. Liquid-chromatographic separation and determination of process-related impurities, including a regio-specific isomer of celecoxib on reversed-phase C18 column dynamically coated with hexamethyldisilazane. Anal. Sci. 2006, 22, 1257–1260. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, A.S.; Shingare, M.S. A new stability-indicating RP-HPLC method to determine assay and known impurity of celecoxib API. Drug. Dev. Ind. Pharm. 2005, 31, 779–783. [Google Scholar] [CrossRef]
- Satyanarayana, U.; Rao, D.S.; Kumar, Y.R.; Babu, J.M.; Kumar, P.R.; Reddy, J.T. Isolation, synthesis and characterization of impurities in celecoxib, a COX-2 inhibitor. J. Pharm. Biomed. Anal. 2004, 35, 951–957. [Google Scholar] [CrossRef]
- Dhabu, P.M.; Akamanchi, K.G. A stability-indicating HPLC method to determine celecoxib in capsule formulations. Drug. Dev. Ind. Pharm. 2002, 28, 815–821. [Google Scholar] [CrossRef]
- Saha, R.N.; Sajeev, C.; Jadhav, P.R.; Patil, S.P.; Srinivasan, N. Determination of celecoxib in pharmaceutical formulations using UV spectrophotometry and liquid chromatography. J. Pharm. Biomed. Anal. 2002, 28, 741–751. [Google Scholar] [CrossRef]
- Rao, D.S.; Srinivasu, M.K.; Narayana, C.L.; Reddy, G.O. LC separation of ortho and meta isomers of celecoxib in bulk and formulations using a chiral column. J. Pharm. Biomed. Anal. 2001, 25, 21–30. [Google Scholar] [CrossRef]
- Srinivasu, M.K.; Narayana, C.L.; Rao, D.S.; Reddy, G.O. A validated LC method for the quantitative determination of celecoxib in pharmaceutical dosage forms and purity evaluation in bulk drugs. J. Pharm. Biomed. Anal. 2000, 22, 949–956. [Google Scholar] [CrossRef]
- Sankar, D.G.; Priya, K.D.; Krishna, M.V.; Latha, P.V.M. Selective RP-HPLC determination of celecoxib in capsules. Asian J. Chem. 2006, 18, 803–806. [Google Scholar]
- Jadhav, P.S.; Jamkar, P.M.; Avachat, A.M. Stability indicating method development and validation for simultaneous estimation of atorvastatin calcium and celecoxib in bulk and niosomal formulation by RP-HPLC. Braz. J. Pharm. Sci. 2015, 51, 653–661. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.; Kim, D.; Lee, S.H.; Jung, B.H. Simultaneous quantitative determination of celecoxib and its two metabolites using liquid chromatography-tandem mass spectrometry in alternating polarity switching mode. J. Pharm. Biomed. Anal. 2015, 107, 32–39. [Google Scholar] [CrossRef]
- Ma, Y.; Gao, S.; Hu, M. Quantitation of celecoxib and four of its metabolites in rat blood by UPLC-MS/MS clarifies their blood distribution patterns and provides more accurate pharmacokinetics profiles. J. Chrom. B 2015, 1001, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Dongari, N.; Sauter, E.R.; Tande, B.M.; Kubátová, A. Determination of celecoxib in human plasma using liquid chromatography with high resolution time of flight-mass spectrometry. J. Chrom. B 2014, 955–956, 86–92. [Google Scholar] [CrossRef] [PubMed]
- EP Monograph on Celecoxib, EP 9.8. 2017. Available online: http://online6.edqm.eu/ep908/ (accessed on 24 January 2020).
- USP Monograph for Celecoxib, USP42-NF37 1S. 2019. Available online: https://online.uspnf.com/uspnf/document/1_GUID-259720A0-ACDC-41A1-99C8-4B685226D65B_3_en-US?source=Search%20Results&highlight=celecoxib (accessed on 24 January 2020).
- USP-PF, In-Process Revision, Celecoxib Capsules USP, 43 (3). 2017. Available online: http://www.usppf.com/pf/pub/index.html (accessed on 24 January 2020).
- Tome, T.; Žigart, N.; Časar, Z.; Obreza, A. Development and optimization of liquid chromatography analytical methods by using AQbD principles: Overview and recent advances. Org. Process. Res. Dev. 2019, 23, 1784–1802. [Google Scholar] [CrossRef] [Green Version]
- Wingert, N.R.; Ellwanger, J.B.; Bueno, L.M.; Gobetti, C.; Garcia, C.V.; Steppe, M.; Schapoval, E.E.S. Application of quality by design to optimize a stability-indicating LC method for the determination of ticagrelor and its impurities. Eur. J. Pharm. Sci. 2018, 118, 208–215. [Google Scholar] [CrossRef]
- Schmidtsdorff, S.; Schmidt, A.H.; Parr, M.K. Structure assisted impurity profiling for rapid method development in liquid chromatography. J. Chromatogr. A 2018, 1577, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Zacharis, C.K.; Vastardi, E. Application of analytical quality by design principles for the determination of alkyl p-toluenesulfonates impurities in aprepitant by HPLC. Validation using total-error concept. J. Pharm. Biomed. Anal. 2018, 150, 152–161. [Google Scholar] [CrossRef]
- Rácz, N.; Molnár, I.; Zöldhegyi, A.; Rieger, H.-J.; Kormány, R. Simultaneous optimization of mobile phase composition and pH using retention modeling and experimental design. J. Pharm. Biomed. Anal. 2018, 160, 336–343. [Google Scholar] [CrossRef]
- Zöldhegyi, A.; Rieger, H.-J.; Molnár, I.; Fekhretdinova, L. Automated UHPLC separation of 10 pharmaceutical compounds using software-modeling. J. Pharm. Biomed. Anal. 2018, 156, 379–388. [Google Scholar] [CrossRef]
- Edlabadkar, A.; Rajput, A. AQbD approach- RP-HPLC method for optimization, development and validation of garenoxacin mesylate in bulk and in tablets. Eurasian J. Anal. Chem. 2018, 13. [Google Scholar] [CrossRef]
- Kormány, R.; Molnár, I.; Fekete, J. Renewal of an old European Pharmacopoeia method for terazosin using modeling with mass spectrometric peak tracking. J. Pharm. Biomed. Anal. 2017, 135, 8–15. [Google Scholar] [CrossRef]
- Kormány, R.; Tamás, K.; Guillarme, D.; Fekete, S. A workflow for column interchangeability in liquid chromatography using modeling software and quality-by-design principles. J. Pharm. Biomed. Anal. 2017, 146, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Tumpa, A.; Stajić, A.; Jančić-Stojanović, B.; Medenica, M. Quality by design in the development of hydrophilic interaction liquid chromatography method with gradient elution for the analysis of olanzapine. J. Pharm. Biomed. Anal. 2017, 134, 18–26. [Google Scholar] [CrossRef]
- Dobričić, V.; Vukadinović, D.; Jančić-Stojanović, B.; Vladimirov, S.; Čudina, O. AQbD-oriented development of a new LC method for simultaneous determination of telmisartan and its impurities. Chromatographia 2017, 80, 1199–1209. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, C. Application of quality by design concept to develop a dual gradient elution stability-indicating method for cloxacillin forced degradation studies using combined mixture-process variable models. J. Chromatogr. A 2017, 1514, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.G.; Patel, A.T.; Shah, P.A.; Gandhi, T.R. Multivariate optimization for simultaneous determination of aspirin and simvastatin by reverse phase liquid chromatographic method using AQbD approach. Bull. Fac. Pharmacy Cairo Univ. 2017, 55, 293–301. [Google Scholar] [CrossRef]
- Bossunia, M.T.I.; Urmi, K.F.; Shaha, C.K. Quality-by-design approach to stability indicating RP-HPLC analytical method development for estimation of canagliflozin API and its validation. Pharm. Methods 2017, 8, 92–101. [Google Scholar] [CrossRef]
- Sunitha, G.; Anumolu, P.D.; Subrahmanyam, C.V.S.; Mounika, G.; Priyanka, B. Stability indicating liquid chromatographic assessment of dolutegravir by AQbD approach—Central composite design. Indian Drugs 2017, 54, 44–51. [Google Scholar]
- Ganorkar, S.B.; Dhumal, D.M.; Shirkhedkar, A.A. Development and validation of simple RP-HPLC-PDA analytical protocol for zileuton assisted with design of experiments for robustness determination. Arab. J. Chem. 2017, 10, 273–282. [Google Scholar] [CrossRef] [Green Version]
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Q2(R2)/Q14 EWG Analytical Procedure Development and Revision of Q2(R1) Analytical Validation (Business Plan, Concept Paper, Work Plan). 2018. Available online: https://www.ich.org/page/quality-guidelines (accessed on 24 January 2020).
- Sahu, P.K.; Ramisetti, N.R.; Cecchi, T.; Swain, S.; Patro, C.S.; Panda, J. An overview of experimental designs in HPLC method development and validation. J. Pharm. Biomed. Anal. 2018, 147, 590–611. [Google Scholar] [CrossRef]
- Lundstedt, T.; Seifert, E.; Abramo, L.; Thelin, B.; Nyström, Å.; Pettersen, J.; Bergman, R. Experimental design and optimization. Chemom. Intell. Lab. Syst. 1998, 42, 3–40. [Google Scholar] [CrossRef]
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Q2(R1) Validation of Analytical Procedures: Text and Methodology. 2005. Available online: https://www.ich.org/page/quality-guidelines (accessed on 11 February 2020).
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp. No. | Exp. | x1 | x2 | x3 | R1 | R2 | R3 | RT |
|---|---|---|---|---|---|---|---|---|
| 1 | N14 | 46 | 0.75 | 40 | 2.53 | 3.50 | 3.46 | 31.38 |
| 2 | N17 | 46 | 0.75 | 35 | 2.45 | 3.37 | 3.55 | 34.56 |
| 3 | N1 | 42 | 0.5 | 30 | 2.80 | 4.46 | 1.63 | 94.93 |
| 4 | N10 | 50 | 0.75 | 35 | 2.11 | 2.25 | 4.72 | 23.16 |
| 5 | N11 | 46 | 0.5 | 35 | 2.58 | 3.52 | 3.82 | 51.62 |
| 6 | N5 | 42 | 0.5 | 40 | 3.04 | 4.84 | 1.18 | 75.61 |
| 7 | N6 | 50 | 0.5 | 40 | 2.24 | 2.47 | 5.19 | 31.88 |
| 8 | N12 | 46 | 1.0 | 35 | 2.31 | 3.21 | 3.29 | 26.04 |
| 9 | N2 | 50 | 0.5 | 30 | 2.16 | 2.20 | 5.03 | 37.80 |
| 10 | N15 | 46 | 0.75 | 35 | 2.47 | 3.37 | 3.59 | 34.70 |
| 11 | N3 | 42 | 1.0 | 30 | 2.51 | 4.07 | 1.43 | 47.96 |
| 12 | N4 | 50 | 1.0 | 30 | 1.89 | 1.80 | 4.02 | 18.96 |
| 13 | N16 | 46 | 0.75 | 35 | 2.45 | 3.37 | 3.53 | 34.50 |
| 14 | N13 | 46 | 0.75 | 30 | 2.32 | 3.20 | 3.49 | 37.98 |
| 15 | N8 | 50 | 1.0 | 40 | 1.99 | 2.18 | 4.39 | 16.05 |
| 16 | N7 | 42 | 1.0 | 40 | 2.76 | 4.46 | 1.04 | 38.41 |
| 17 | N9 | 42 | 0.75 | 35 | 2.80 | 4.52 | 1.30 | 57.03 |
| Model (Response) | p-Value (Regression) | R2 | R2 Adjusted | Q2 | Reproducibility |
|---|---|---|---|---|---|
| R1 | 8.5259 × 10−16 | 0.999 | 0.998 | 0.997 | 0.9986 |
| R2 | 2.1878 × 10−18 | 0.999 | 0.999 | 0.998 | 1.0000 |
| R3 | 5.9027 × 10−13 | 1.000 | 0.999 | 0.997 | 0.9995 |
| RT | 5.8067 × 10−9 | 0.996 | 0.993 | 0.963 | 1.0000 |
| CMA (Response) | Predicted | Observed |
|---|---|---|
| R1 | 2.59317 | 2.58367 |
| R2 | 3.76787 | 3.73814 |
| R3 | 2.89245 | 2.91747 |
| RT | 32.3846 | 32.648 |
| Exp. No. | Exp. | x1 | x2 | x3 | R1 | R2 | R3 | RT |
|---|---|---|---|---|---|---|---|---|
| 1 | N6 | 46.5 | 0.75 | 40 | 2.47 | 3.37 | 3.69 | 29.67 |
| 2 | N5 | 43.5 | 0.75 | 40 | 2.76 | 4.21 | 2.13 | 41.41 |
| 3 | N10 | 45 | 0.8 | 38.5 | 2.57 | 3.7 | 2.97 | 33.77 |
| 4 | N1 | 43.5 | 0.75 | 37 | 2.71 | 4.1 | 2.23 | 44.19 |
| 5 | N4 | 46.5 | 0.85 | 37 | 2.38 | 3.25 | 3.66 | 27.84 |
| 6 | N7 | 43.5 | 0.85 | 40 | 2.69 | 4.12 | 2.06 | 36.63 |
| 7 | N11 | 45 | 0.8 | 38.5 | 2.56 | 3.69 | 2.95 | 33.64 |
| 8 | N3 | 43.5 | 0.85 | 37 | 2.65 | 4.03 | 2.17 | 39.11 |
| 9 | N8 | 46.5 | 0.85 | 40 | 2.41 | 3.3 | 3.59 | 26.21 |
| 10 | N2 | 46.5 | 0.75 | 37 | 2.44 | 3.31 | 3.76 | 31.47 |
| 11 | N9 | 45 | 0.8 | 38.5 | 2.58 | 3.71 | 2.97 | 33.82 |
| Impurity | Concentration Level (%) | Added Conc. (µg/mL) | Found Conc. (µg/mL) | Recovery (%) |
|---|---|---|---|---|
| EP impurity A | 0.05 | 0.2548 | 0.2632 | 103.31 |
| 0.40 | 2.0380 | 2.0309 | 99.65 | |
| 0.48 | 2.4456 | 2.4255 | 99.18 | |
| EP impurity B | 0.05 | 0.2498 | 0.2378 | 95.23 |
| 0.15 | 0.7493 | 0.7468 | 99.67 | |
| 0.18 | 0.8991 | 0.8850 | 98.43 | |
| USP related compound C | 0.05 | 0.2551 | 0.2073 | 81.26 |
| 0.15 | 0.7653 | 0.6920 | 90.42 | |
| 0.18 | 0.9184 | 0.8442 | 91.92 | |
| USP related compound D | 0.05 | 0.2637 | 0.2484 | 94.21 |
| 0.15 | 0.7911 | 0.7753 | 98.00 | |
| 0.18 | 0.9493 | 0.9546 | 100.56 | |
| USP o-celecoxib | 0.05 | 0.2690 | 0.2699 | 100.32 |
| 0.15 | 0.8071 | 0.8128 | 100.71 | |
| 0.18 | 0.9685 | 0.9794 | 101.12 | |
| USP desaryl celecoxib | 0.05 | 0.2455 | 0.2449 | 99.79 |
| 0.15 | 0.7364 | 0.7375 | 100.16 | |
| 0.18 | 0.8836 | 0.8785 | 99.42 | |
| USP 4-methylacetophenone | 0.05 | 0.2356 | 0.2371 | 100.65 |
| 0.15 | 0.7067 | 0.7066 | 99.98 | |
| 0.18 | 0.8481 | 0.8452 | 99.66 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tome, T.; Časar, Z.; Obreza, A. Development of a Unified Reversed-Phase HPLC Method for Efficient Determination of EP and USP Process-Related Impurities in Celecoxib Using Analytical Quality by Design Principles. Molecules 2020, 25, 809. https://doi.org/10.3390/molecules25040809
Tome T, Časar Z, Obreza A. Development of a Unified Reversed-Phase HPLC Method for Efficient Determination of EP and USP Process-Related Impurities in Celecoxib Using Analytical Quality by Design Principles. Molecules. 2020; 25(4):809. https://doi.org/10.3390/molecules25040809
Chicago/Turabian StyleTome, Tim, Zdenko Časar, and Aleš Obreza. 2020. "Development of a Unified Reversed-Phase HPLC Method for Efficient Determination of EP and USP Process-Related Impurities in Celecoxib Using Analytical Quality by Design Principles" Molecules 25, no. 4: 809. https://doi.org/10.3390/molecules25040809