
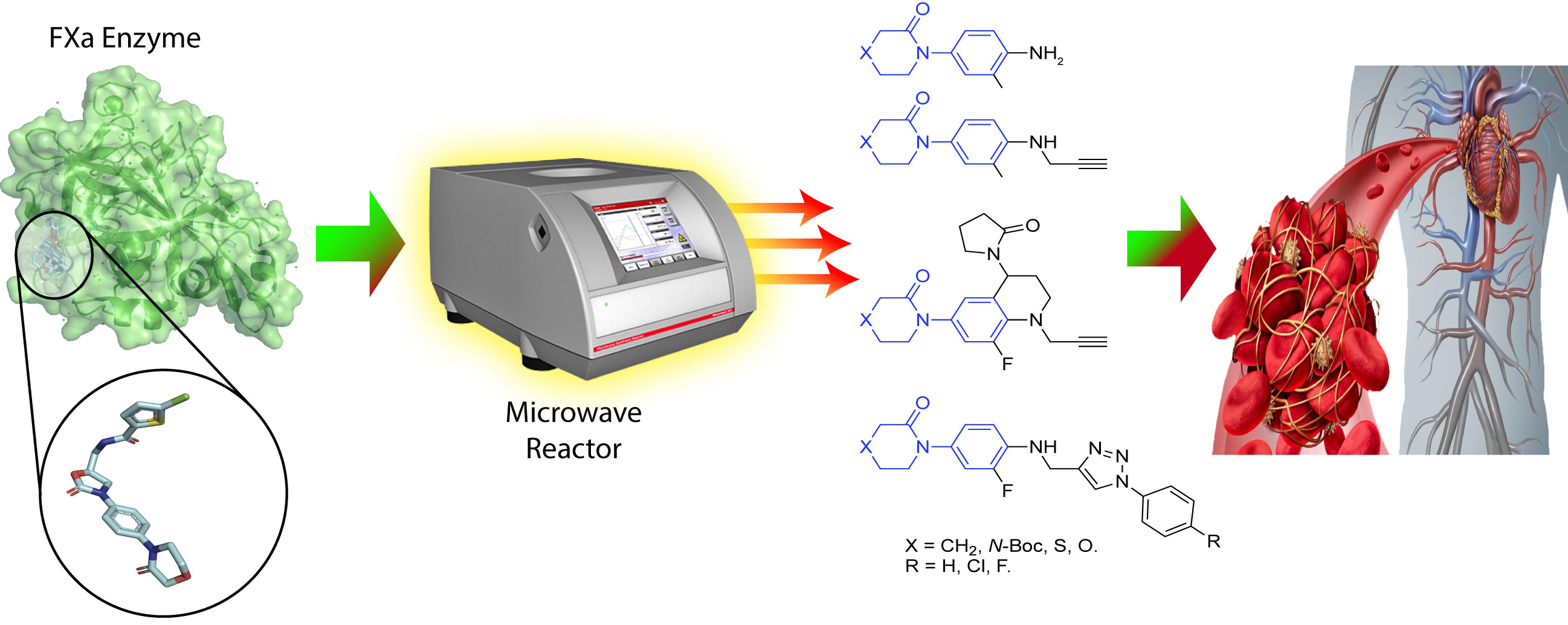
Innovative Three-Step Microwave-Promoted Synthesis of N-Propargyltetrahydroquinoline and 1,2,3-Triazole Derivatives as a Potential Factor Xa (FXa) Inhibitors: Drug Design, Synthesis, and Biological Evaluation

 ,
,  , , ,
, , ,
Abstract
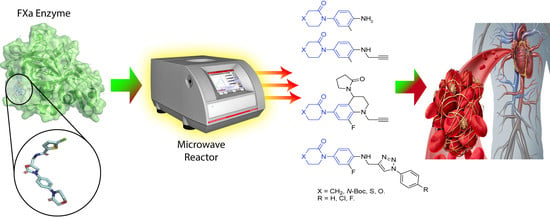
:
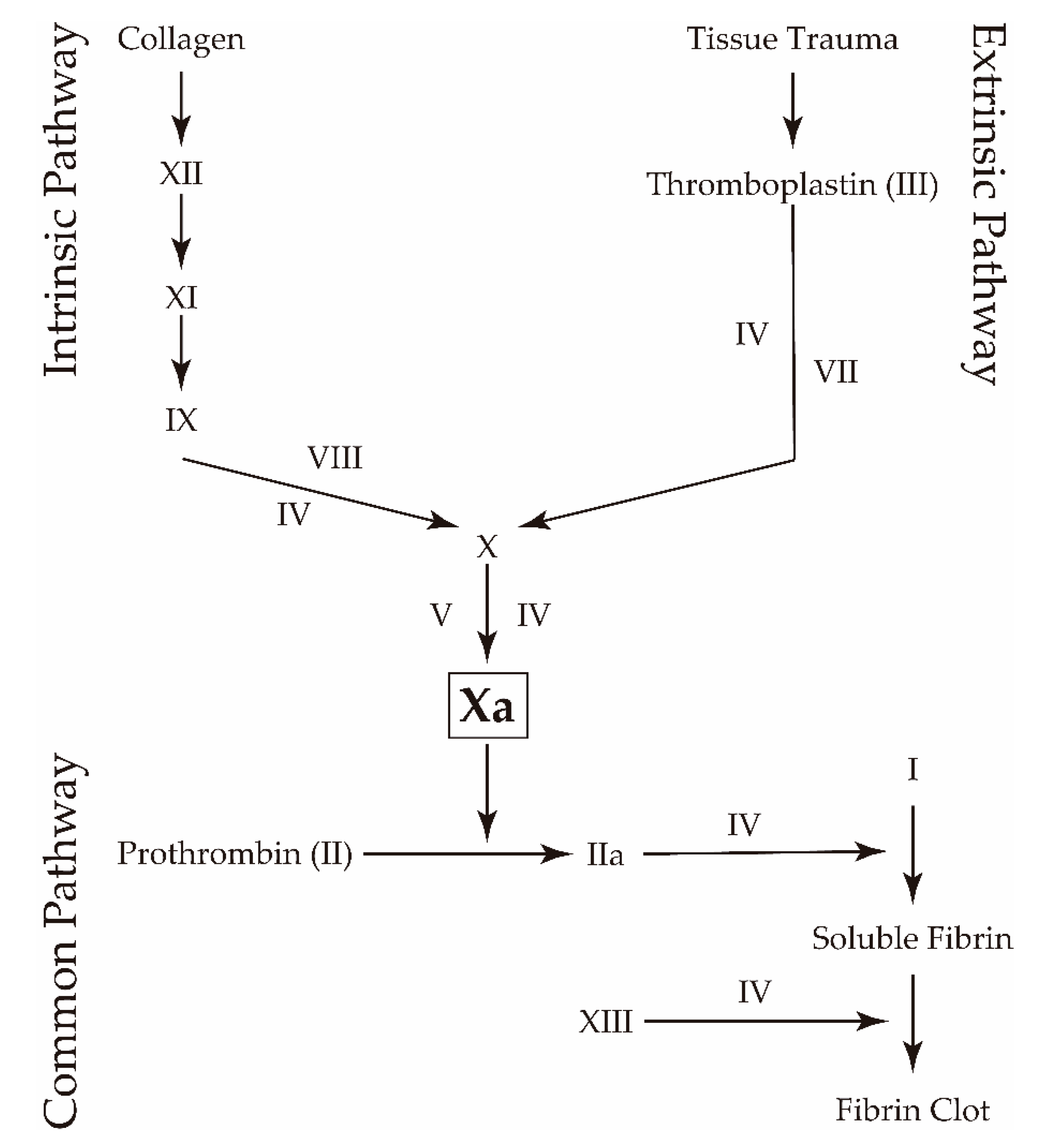
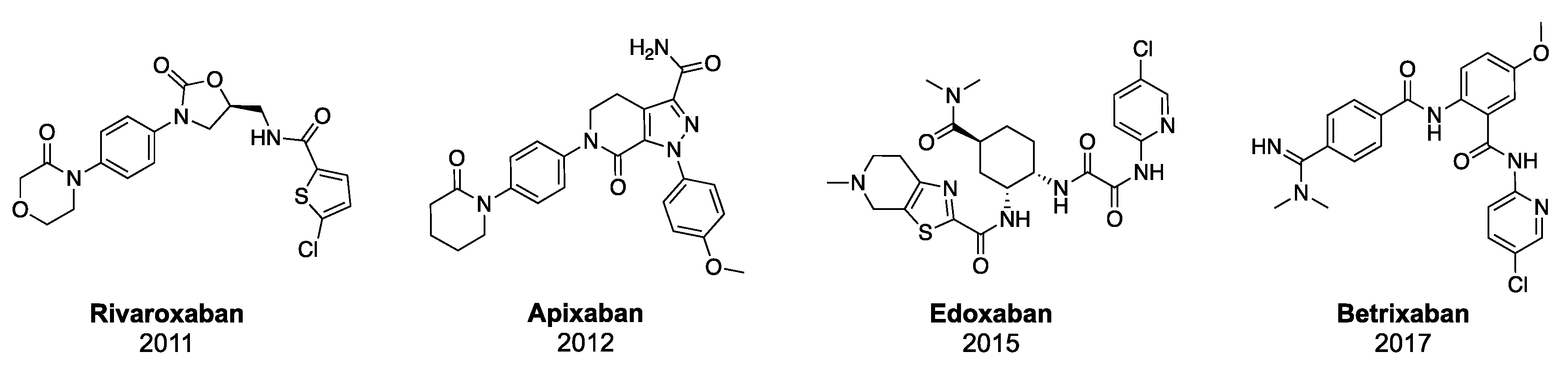
1. Introduction
2. Results and Discussion
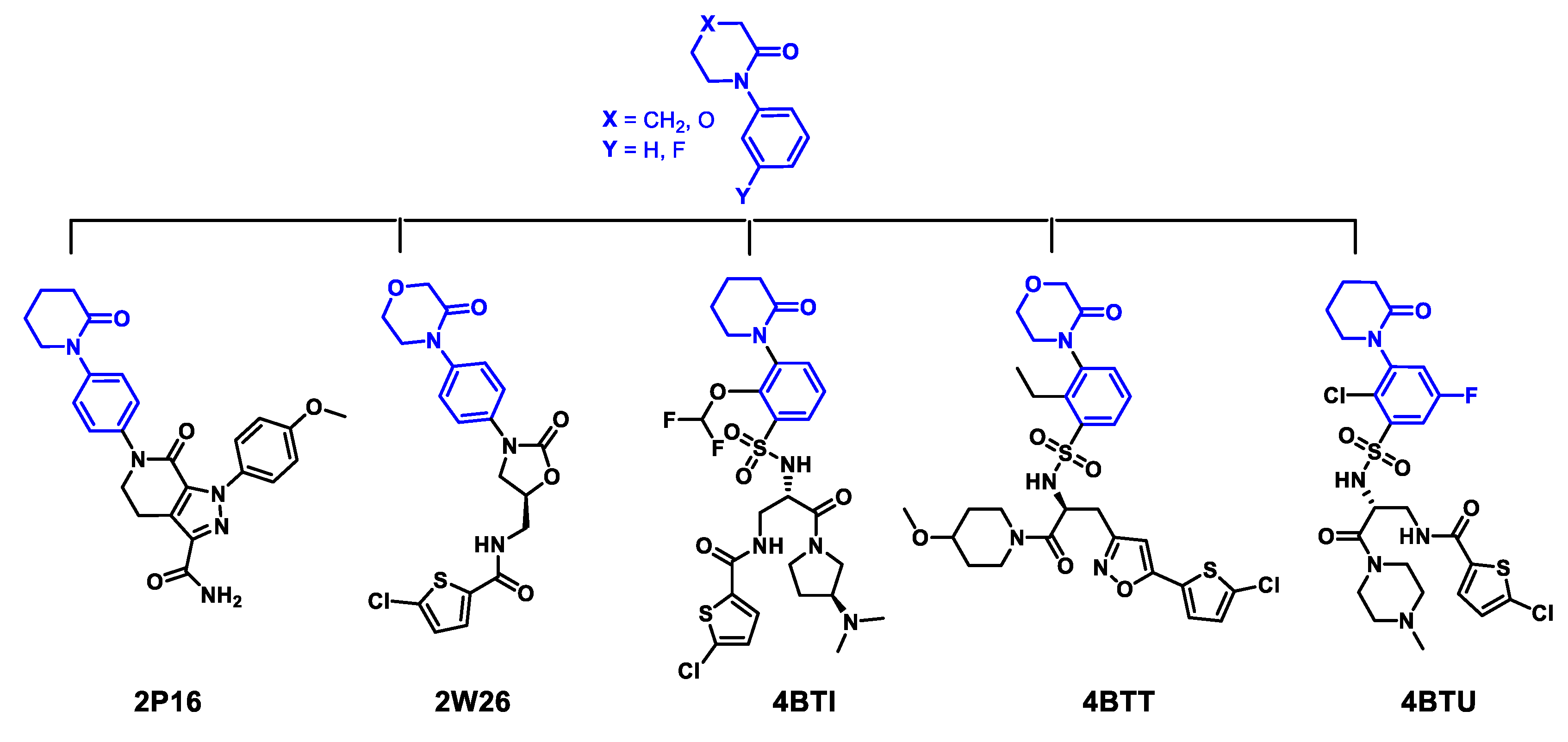
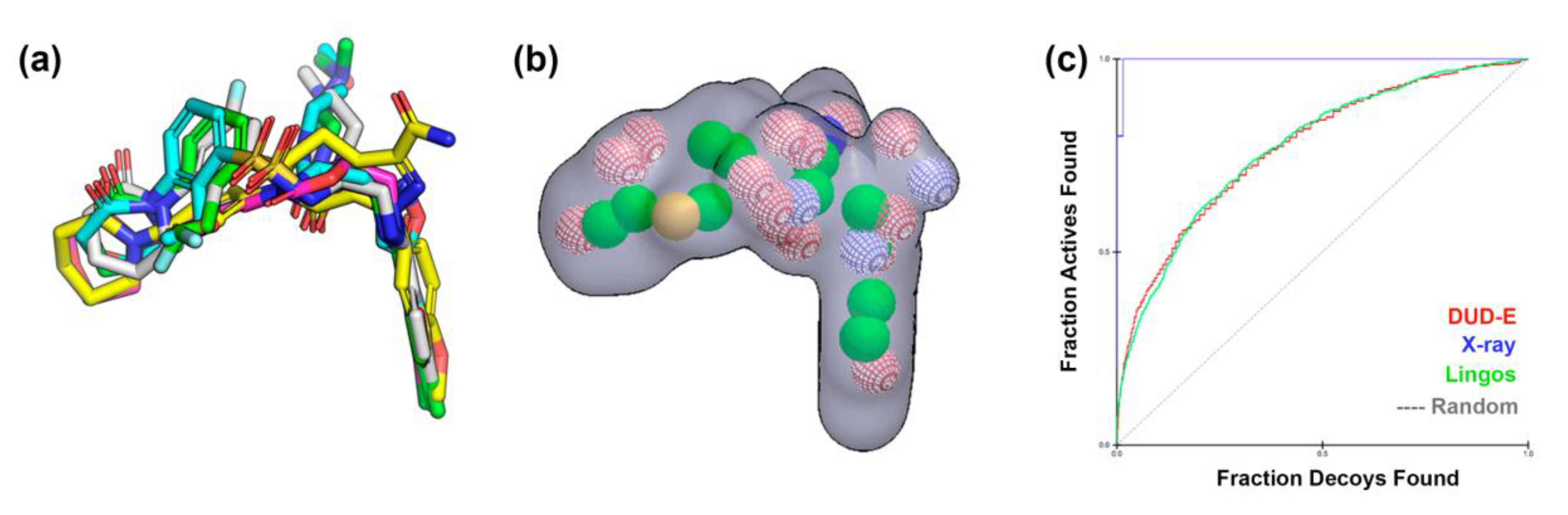
2.1. Protein-Ligand Complexes Selection and Ligand Clustering
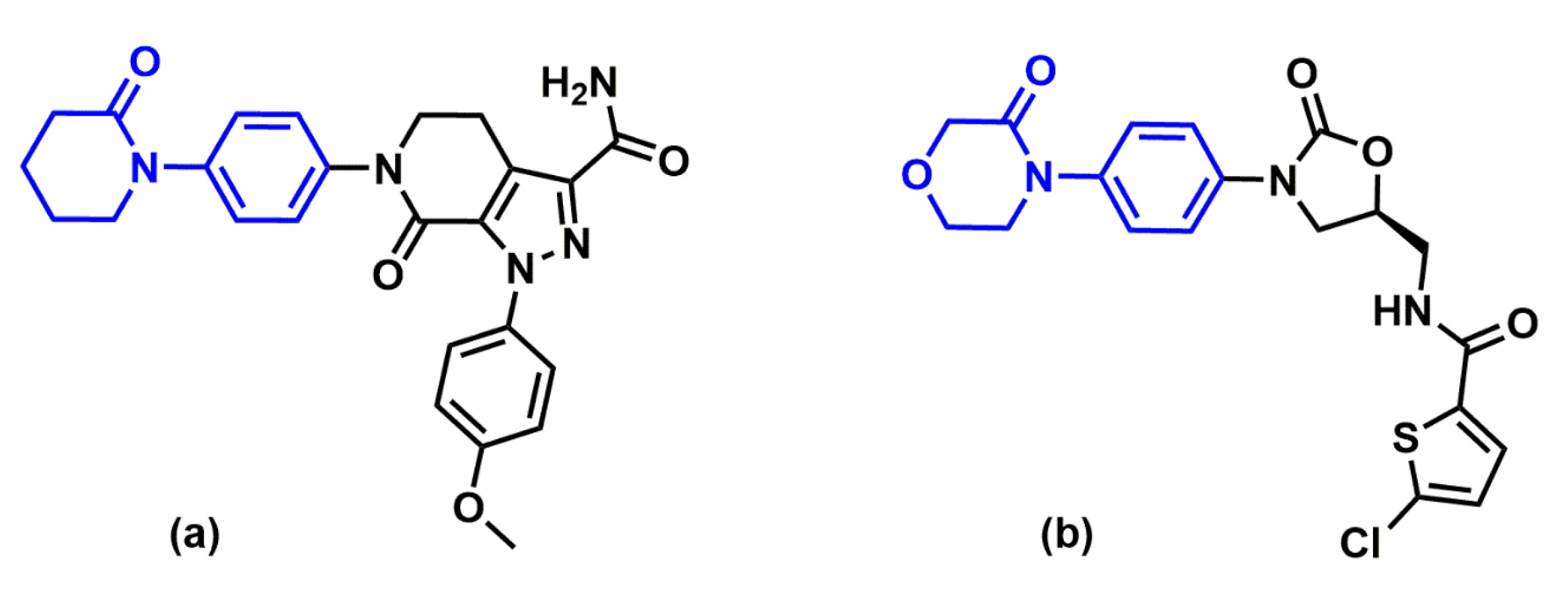
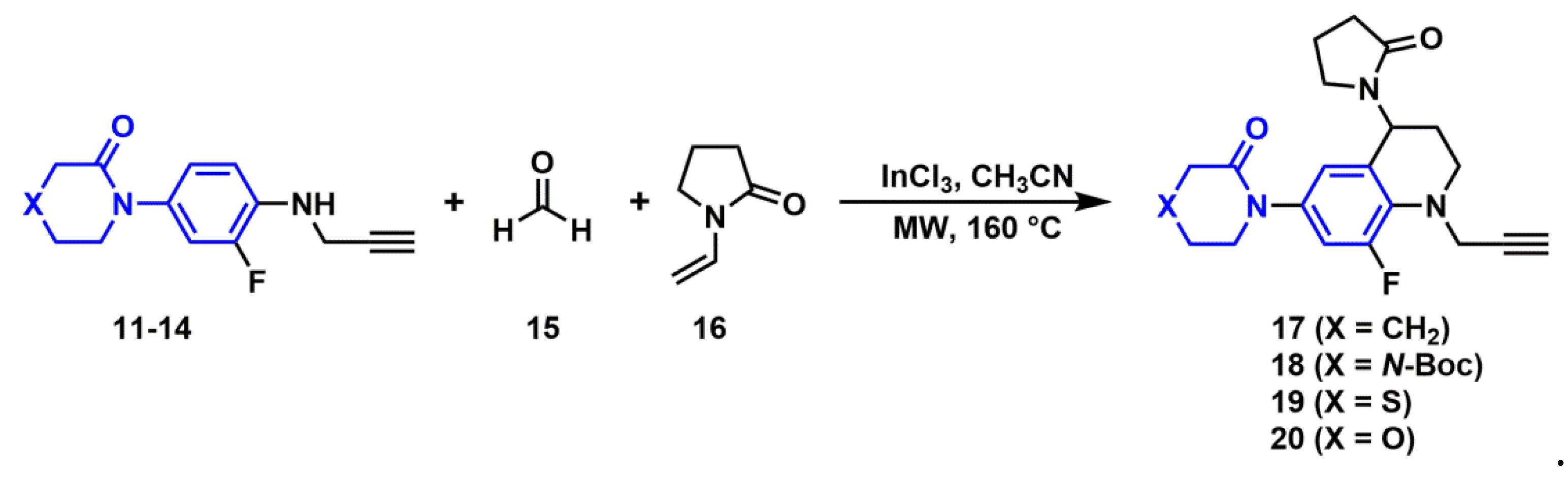
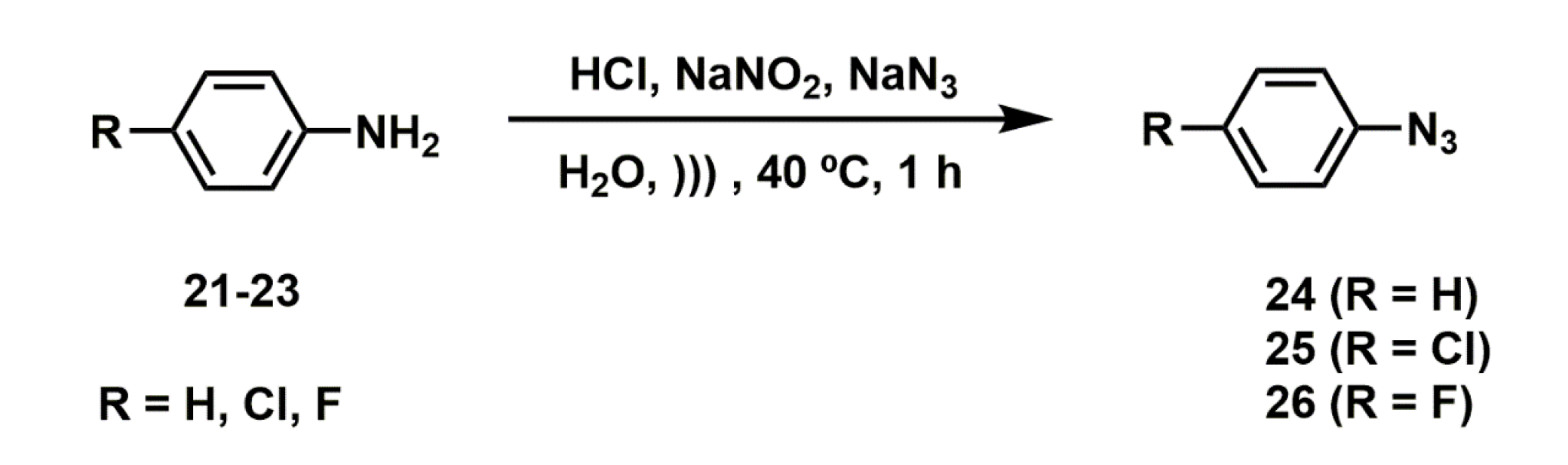
2.2. Synthesis of Novel Derivatives
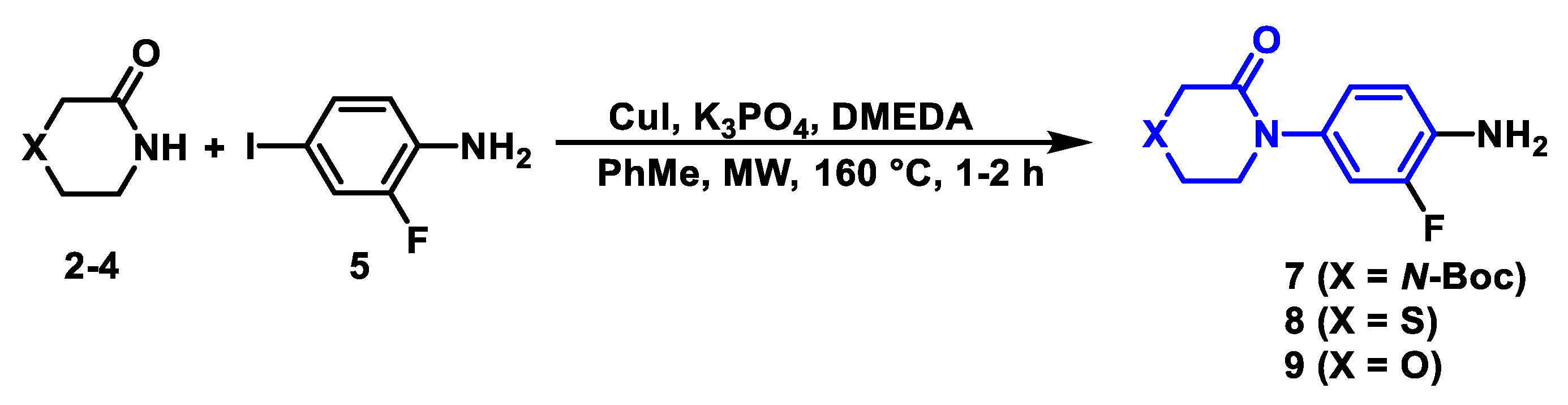
2.2.1. Synthetic Methodology Improvements for Aniline Precursors
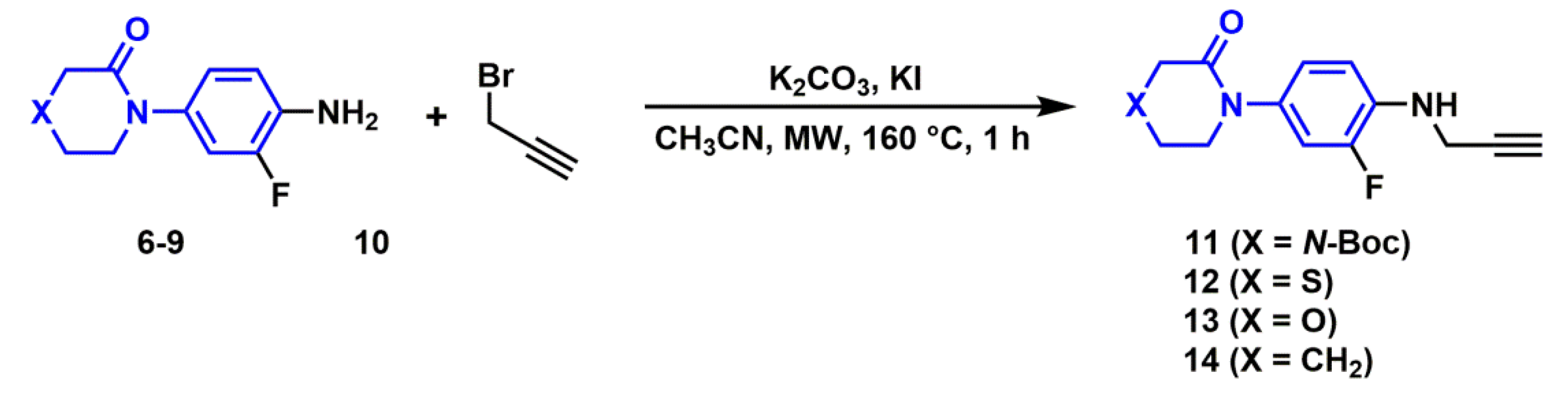
2.2.2. Synthesis of N-propargylanilines
2.2.3. Synthesis of N-Propargyl Tetrahydroquinolines
2.2.4. Synthesis of Aryl Azides
2.2.5. Synthesis of 1H-1,2,3-triazole derivatives
2.3. Theoretical Study
2.3.1. X-Ray and Theoretical Optimization Correlation
2.3.2. Energetic Behavior and Thermochemistry
2.4. Biological Analysis
2.4.1. FXa Inhibition in Vitro Assay
2.4.2. Anti FXa assay, Prothrombin Time (PT) and Activated Partial Thromboplastin Time (aPTT) ex Vivo Assay
2.4.3. Cell Viability Assays
2.5. Computational Analysis
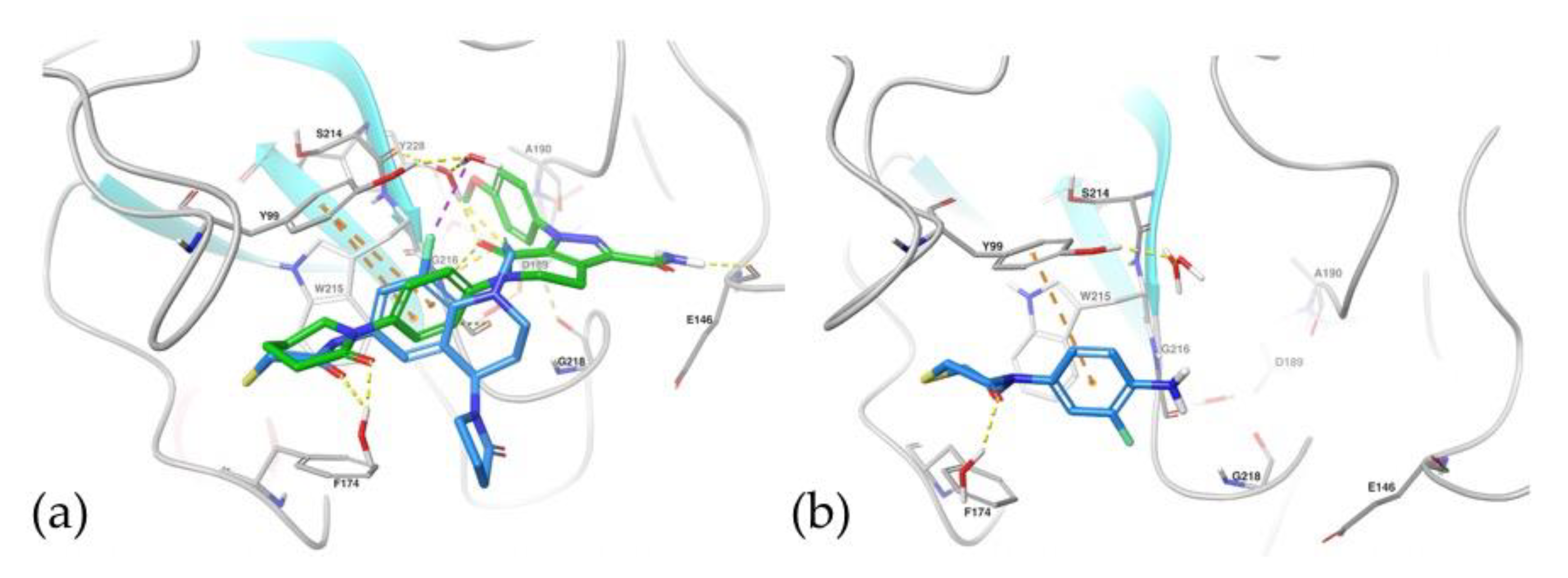
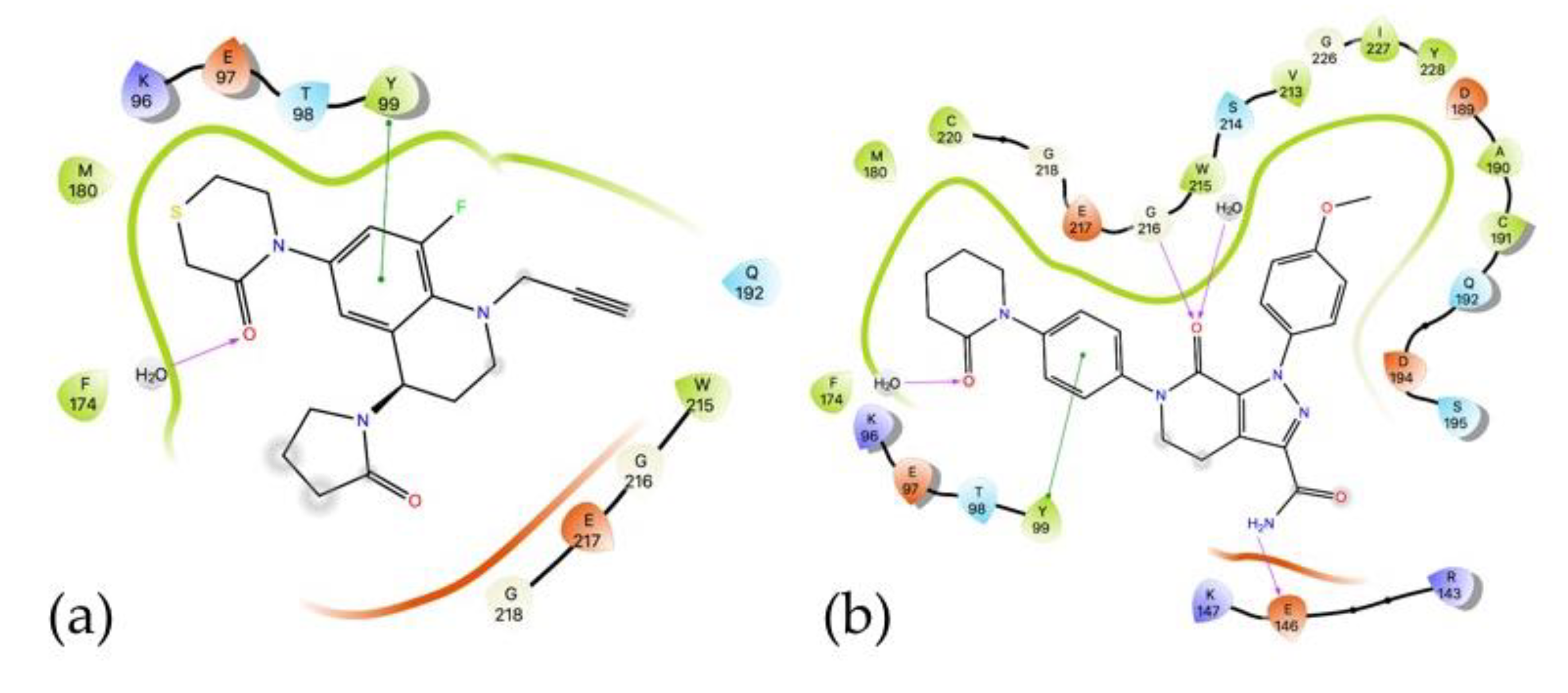
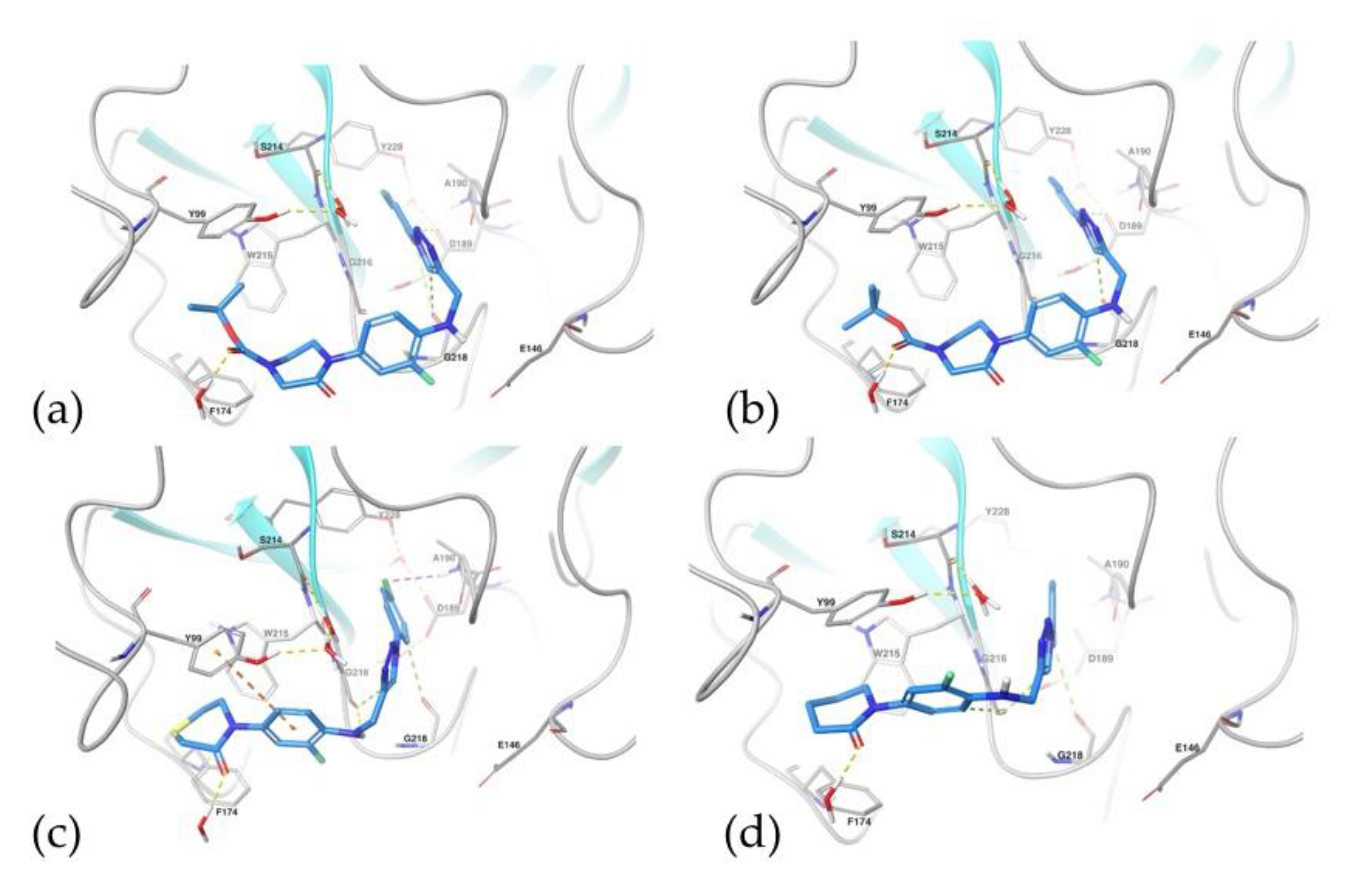
2.5.1. Docking Studies
2.5.2. Molecular Dynamics Simulations
2.5.3. In Silico Pharmacokinetics Prediction
3. Materials and Methods
3.1. FXa-Ligand Structure Retrieval and Standardization
3.2. Shape-Based Query Generation and Validation
3.3. Synthesis of Derivatives
3.3.1. Instrumentation and Chemicals
3.3.2. General Procedure for the Synthesis of Aniline Derivatives (6–9)
3.3.3. General Procedure for the Synthesis of N-propargyl Aniline Derivatives (11–14)
3.3.4. General Procedure for the Synthesis of N-propargyl tetrahydroquinoline Derivatives (17–20)
3.3.5. General Procedure for the Synthesis of Azides (24–26)
3.3.6. General Procedure for the synthesis of 1H-1,2,3-triazole Derivatives (27–38)
Synthesis of Copper Nanoparticles Supported on ZnO
3.4. Theoretical Study
3.5. Biological Analysis
3.5.1. FXa in Vitro Inhibition Assay
3.5.2. Coagulation Parameters Analyses: ex Vivo Assay
Anti-FXa Assay
Prothrombin Time (PT) Test
Activated Partial Thromboplastin Time (aPTT) Test
3.5.3. Cell Viability Assays
3.6. Computational Analysis
3.6.1. Docking Studies
3.6.2. Molecular Dynamics Simulations
3.6.3. In Silico Pharmacokinetics Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Instituto Nacional de Estadísticas. Principales Causas de Muerte en Chile por regiones 1997–2017. Available online: https://ine.cl/estadisticas/sociales/demografia-y-vitales/nacimientos-matrimonios-y-defunciones (accessed on 13 December 2019).
- Feigin, V.L.; Lawes, C.M.; Bennett, D.A.; Anderson, C.S. Stroke epidemiology: A review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003, 2, 43–53. [Google Scholar] [CrossRef]
- Ministerio de Salud del Gobiernno de Chile Estrategia Nacional de Salud 2011–2020. Available online: https://www.minsal.cl/portal/url/item/c4034eddbc96ca6de0400101640159b8.pdf (accessed on 13 December 2019).
- Ministerio de Salud del Gobiernno de Chile Normas de Seguridad del Paciente y Calidad de Atención Respecto de Prevención Enfermedad Tromboembólica. Available online: https://www.minsal.cl/portal/url/item/cede67f930f982cce040010164012d43.pdf (accessed on 13 December 2019).
- Bombin F., J.; Kotlik, A.A.; Díaz G., A.; Vera O., R.; Contreras T., J.; Vásquez Z., D. Anticoagulant Drugs. Rev. Chil. Cirugía 2005, 57, 311–319. [Google Scholar]
- Palomo G, I.; Pereira G, J.; Alarcón L, M.; Pinochet P, C.; Vélez SM, M.T.; Hidalgo P, P.; Skagerberg, K.; Poblete C, F. Factor V Leiden y mutación de la protrombina G20210A en pacientes con trombosis venosa y arterial. Rev. Med. Chil. 2005, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raskob, G.E.; Angchaisuksiri, P.; Blanco, A.N.; Buller, H.; Gallus, A.; Hunt, B.J.; Hylek, E.M.; Kakkar, A. ISTH Steering Committee for World Thrombosis Day Thrombosis: A major contributor to the global disease burden. J. Thromb. Haemost. 2014, 12, 1580–1590. [Google Scholar]
- Oldenburg, J.; Marinova, M.; Müller-Reible, C.; Watzka, M. The Vitamin K Cycle. In Vitamins & Hormones; Academic Press: London, UK, 2008; Volume 78, pp. 35–54. [Google Scholar]
- Gustafsson, D.; Bylund, R.; Antonsson, T.; Nilsson, I.; Nyström, J.-E.; Eriksson, U.; Bredberg, U.; Teger-Nilsson, A.-C. A new oral anticoagulant: The 50-year challenge. Nat. Rev. Drug Discov. 2004, 3, 649–659. [Google Scholar] [CrossRef]
- Wolf, P.A.; Abbott, R.D.; Kannel, W.B. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991, 22, 983–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flisfisch, H.; Aguiló, J.; Lillo Cuevas, D. Trombosis Venosa Profunda. Rev. Med. y Humanidades 2014, IV, 46–50. [Google Scholar]
- Higgs, E.A.; Higgs, G.A.; Moncada, S.; Vane, J.R. Prostacyclin (PGI2) Inhibits the Formation of Platelet Thrombin Arterioles and Venules of the Hamster Cheek Pouch. Br. J. Pharmacol. 1978, 63, 535–539. [Google Scholar] [CrossRef]
- Esmon, C.T. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J. Biol. Chem. 1989, 264, 4743–4746. [Google Scholar] [CrossRef]
- Dahlbäck, B.; Villoutreix, B.O. The anticoagulant protein C pathway. FEBS Lett. 2005, 579, 3310–3316. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Pedersen, L.C. Anticoagulant heparan sulfate: Structural specificity and biosynthesis. Appl. Microbiol. Biotechnol. 2007, 74, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collen, D.; Lijnen, H.R. The Tissue-Type Plasminogen Activator Story. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1151–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, Z.M. Von Willebrand factor, platelets and endothelial cell interactions. J. Thromb. Haemost. 2003, 1, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; Gardiner, E.E.; Shen, Y.; Whisstock, J.C.; Berndt, M.C. Glycoprotein Ib–IX–V. Int. J. Biochem. Cell Biol. 2003, 35, 1170–1174. [Google Scholar] [CrossRef]
- Smith, W.L.; Marnett, L.J.; DeWitt, D.L. Prostaglandin and thromboxane biosynthesis. Pharmacol. Ther. 1991, 49, 153–179. [Google Scholar] [CrossRef]
- Heijnen, H.; van der Sluijs, P. Platelet secretory behaviour: As diverse as the granules … or not? J. Thromb. Haemost. 2015, 13, 2141–2151. [Google Scholar] [CrossRef]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the Extrinsic Pathway of Blood Coagulation in Hemostasis and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, M. Remodeling the Blood Coagulation Cascade. J. Thromb. Thrombolysis 2003, 16, 17–20. [Google Scholar] [CrossRef]
- Ansell, J. Factor Xa or thrombin: Is factor Xa a better target? J. Thromb. Haemost. 2007, 5, 60–64. [Google Scholar] [CrossRef]
- Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Misselwitz, F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat. Rev. Drug Discov. 2011, 10, 61–75. [Google Scholar] [CrossRef]
- Kanuri, S.H.; Kreutz, R.P. Pharmacogenomics of Novel Direct Oral Anticoagulants: Newly Identified Genes and Genetic Variants. J. Pers. Med. 2019, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, R.K.; Bender, D.A.; Botham, K.M.; Kennelly, P.J.; Rodwell, V.W.; Weil, P.A. Harper Bioquímica Aplicada, 29th ed.; McGraw-Hill: New York, NY, USA, 2013. [Google Scholar]
- Chen, M.-C.; Wong, H.-S.; Lin, K.-J.; Chen, H.-L.; Wey, S.-P.; Sonaje, K.; Lin, Y.-H.; Chu, C.-Y.; Sung, H.-W. The characteristics, biodistribution and bioavailability of a chitosan-based nanoparticulate system for the oral delivery of heparin. Biomaterials 2009, 30, 6629–6637. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, R.M.; Fareed, J. The effect of molecular weight on the bioavailability of heparin. Thromb. Res. 1987, 48, 591–596. [Google Scholar] [CrossRef]
- Ankrom, W.; Wood, H.B.; Xu, J.; Geissler, W.; Bateman, T.; Chatterjee, M.S.; Feng, K.-I.; Metzger, J.M.; Strapps, W.R.; Tadin-Strapps, M.; et al. Preclinical and translational evaluation of coagulation factor IXa as a novel therapeutic target. Pharmacol. Res. Perspect. 2016, 4, e00207. [Google Scholar] [CrossRef]
- Lip, G.Y.H.; Pan, X.; Kamble, S.; Kawabata, H.; Mardekian, J.; Masseria, C.; Bruno, A.; Phatak, H. Major bleeding risk among non-valvular atrial fibrillation patients initiated on apixaban, dabigatran, rivaroxaban or warfarin: A “real-world” observational study in the United States. Int. J. Clin. Pract. 2016, 70, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Vene, N.; Mavri, A.; Gubenšek, M.; Tratar, G.; Vižintin Cuderman, T.; Pohar Perme, M.; Blinc, A. Risk of Thromboembolic Events in Patients with Non-Valvular Atrial Fibrillation After Dabigatran or Rivaroxaban Discontinuation–Data from the Ljubljana Registry. PLoS ONE 2016, 11, e0156943. [Google Scholar] [CrossRef]
- Acanfora, D.; Acanfora, C.; Scicchitano, P.; Longobardi, M.; Furgi, G.; Casucci, G.; Lanzillo, B.; Dentamaro, I.; Zito, A.; Incalzi, R.A.; et al. Safety and Feasibility of Treatment with Rivaroxaban for Non-Canonical Indications: A Case Series Analysis. Clin. Drug Investig. 2016, 36, 857–862. [Google Scholar] [CrossRef]
- Kohrt, J.T.; Bigge, C.F.; Bryant, J.W.; Casimiro-Garcia, A.; Chi, L.; Cody, W.L.; Dahring, T.; Dudley, D.A.; Filipski, K.J.; Haarer, S.; et al. The Discovery of (2R,4R)-N-(4-chlorophenyl)-N- (2-fluoro-4-(2-oxopyridin-1(2H)-yl)phenyl)-4-methoxypyrrolidine-1,2-dicarboxamide (PD 0348292), an Orally Efficacious Factor Xa Inhibitor. Chem. Biol. Drug Des. 2007, 70, 100–112. [Google Scholar] [CrossRef]
- Roehrig, S.; Straub, A.; Pohlmann, J.; Lampe, T.; Pernerstorfer, J.; Schlemmer, K.-H.; Reinemer, P.; Perzborn, E. Discovery of the Novel Antithrombotic Agent 5-Chloro- N -({(5 S )-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene- 2-carboxamide (BAY 59-7939): An Oral, Direct Factor Xa Inhibitor. J. Med. Chem. 2005, 48, 5900–5908. [Google Scholar] [CrossRef]
- Heo, Y.-A. Andexanet Alfa: First Global Approval. Drugs 2018, 78, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Sanchis-Gomar, F.; Favaloro, E.J. Andexanet: Effectively Reversing Anticoagulation. Trends Pharmacol. Sci. 2016, 37, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Rawal, A.; Ardeshna, D.; Minhas, S.; Cave, B.; Ibeguogu, U.; Khouzam, R. Current status of oral anticoagulant reversal strategies: A review. Ann. Transl. Med. 2019, 7, 411. [Google Scholar] [CrossRef] [PubMed]
- Dobesh, P.P.; Bhatt, S.H.; Trujillo, T.C.; Glaubius, K. Antidotes for reversal of direct oral anticoagulants. Pharmacol. Ther. 2019, 204, 107405. [Google Scholar] [CrossRef] [PubMed]
- Khadse, A.N.; Sharma, M.K.; Murumkar, P.R.; Rajput, S.J.; Yadav, M.R. Advances in the Development of Novel Factor Xa Inhibitors: A Patent Review. Mini-Reviews Med. Chem. 2018, 18, 1332–1353. [Google Scholar] [CrossRef]
- Zbinden, K.G.; Anselm, L.; Banner, D.W.; Benz, J.; Blasco, F.; Décoret, G.; Himber, J.; Kuhn, B.; Panday, N.; Ricklin, F. Design of novel aminopyrrolidine factor Xa inhibitors from a screening hit. Eur. J. Med. Chem. 2009, 44, 2787–2795. [Google Scholar] [CrossRef]
- Jacquemond-Collet, I.; Benoit-Vical, F.; Valentin, A.; Stanislas, E.; Mallié, M.; Fourasté, I. Antiplasmodial and Cytotoxic Activity of Galipinine and other Tetrahydroquinolines from Galipea officinalis. Planta Med. 2002, 68, 68–69. [Google Scholar] [CrossRef]
- Lam, K.-H.; Lee, K.K.-H.; Gambari, R.; Wong, R.S.-M.; Cheng, G.Y.-M.; Tong, S.-W.; Chan, K.-W.; Lau, F.-Y.; Lai, P.B.-S.; Wong, W.-Y.; et al. Preparation of Galipea officinalis Hancock type tetrahydroquinoline alkaloid analogues as anti-tumour agents. Phytomedicine 2013, 20, 166–171. [Google Scholar] [CrossRef]
- Jacquemond-Collet, I.; Hannedouche, S.; Fourasté, I.; Moulis, C. Novel quinoline alkaloid from trunk bark of Galipea officinalis. Fitoterapia 2000, 71, 605–606. [Google Scholar] [CrossRef]
- Sugiyama, R.; Nishimura, S.; Ozaki, T.; Asamizu, S.; Onaka, H.; Kakeya, H. 5-Alkyl-1,2,3,4-tetrahydroquinolines, New Membrane-Interacting Lipophilic Metabolites Produced by Combined Culture of Streptomyces nigrescens and Tsukamurella pulmonis. Org. Lett. 2015, 17, 1918–1921. [Google Scholar] [CrossRef]
- Gulder, T.A.M.; Moore, B.S. Salinosporamide Natural Products: Potent 20 S Proteasome Inhibitors as Promising Cancer Chemotherapeutics. Angew. Chemie Int. Ed. 2010, 49, 9346–9367. [Google Scholar] [CrossRef]
- Neff, S.A.; Lee, S.U.; Asami, Y.; Ahn, J.S.; Oh, H.; Baltrusaitis, J.; Gloer, J.B.; Wicklow, D.T. Aflaquinolones A–G: Secondary Metabolites from Marine and Fungicolous Isolates of Aspergillus spp. J. Nat. Prod. 2012, 75, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-S.; He, H.-P.; Yang, J.-H.; Di, Y.-T.; Hao, X.-J. New Monoterpenoid Coumarins from Clausena anisum-olens. Molecules 2008, 13, 931–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwan, J.; Kleoff, M.; Hartmayer, B.; Heretsch, P.; Christmann, M. Synthesis of Quinolinone Alkaloids via Aryne Insertions into Unsymmetric Imides in Flow. Org. Lett. 2018, 20, 7661–7664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedoya, L.M.; Abad, M.J.; Calonge, E.; Saavedra, L.A.; Gutierrez C., M.; Kouznetsov, V.V.; Alcami, J.; Bermejo, P. Quinoline-based compounds as modulators of HIV transcription through NF-κB and Sp1 inhibition. Antiviral Res. 2010, 87, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Su, D.-S.; Lim, J.J.; Tinney, E.; Wan, B.-L.; Young, M.B.; Anderson, K.D.; Rudd, D.; Munshi, V.; Bahnck, C.; Felock, P.J.; et al. Substituted tetrahydroquinolines as potent allosteric inhibitors of reverse transcriptase and its key mutants. Bioorg. Med. Chem. Lett. 2009, 19, 5119–5123. [Google Scholar] [CrossRef]
- Chander, S.; Wang, P.; Ashok, P.; Yang, L.-M.; Zheng, Y.-T.; Murugesan, S. Rational design, synthesis, anti-HIV-1 RT and antimicrobial activity of novel 3-(6-methoxy-3,4-dihydroquinolin-1(2H)-yl)-1-(piperazin-1-yl)propan-1-one derivatives. Bioorg. Chem. 2016, 67, 75–83. [Google Scholar] [CrossRef]
- Quan, M.L.; Wong, P.C.; Wang, C.; Woerner, F.; Smallheer, J.M.; Barbera, F.A.; Bozarth, J.M.; Brown, R.L.; Harpel, M.R.; Luettgen, J.M.; et al. Tetrahydroquinoline Derivatives as Potent and Selective Factor XIa Inhibitors. J. Med. Chem. 2014, 57, 955–969. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Mehta, A.Y.; Desai, U.R. Potent direct inhibitors of factor Xa based on the tetrahydroisoquinoline scaffold. Eur. J. Med. Chem. 2012, 54, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Khalid, W.; Badshah, A.; Khan, A.; Nadeem, H.; Ahmed, S. Synthesis, characterization, molecular docking evaluation, antiplatelet and anticoagulant actions of 1,2,4 triazole hydrazone and sulphonamide novel derivatives. Chem. Cent. J. 2018, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- de Andrade Moura, L.; de Almeida, A.C.; da Silva, A.V.; de Souza, V.R.; Ferreira, V.F.; Menezes, M.V.; Kaiser, C.R.; Ferreira, S.B.; Fuly, A.L. Synthesis, Anticlotting and Antiplatelet Effects of 1,2,3-Triazoles Derivatives. Med. Chem. (Los. Angeles). 2016, 12, 733–741. [Google Scholar]
- Yuan, J.; Liu, K.; Li, L.; Yuan, Y.; Liu, X.; Li, Y. A Novel Synthesis of the Oxazolidinone Antithrombotic Agent Rivaroxaban. Molecules 2014, 19, 14999–15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Ji, Y. Alternate Synthesis of Apixaban (BMS-562247), an Inhibitor of Blood Coagulation Factor Xa. Synth. Commun. 2013, 43, 72–79. [Google Scholar] [CrossRef]
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- de la Hoz, A.; Loupy, A. Microwaves in Organic Synthesis, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2012; Volumes 1–2. [Google Scholar]
- van der Eycken, E.; Kappe, C.O. Topics in Heterocyclic Chemistry 01: Microwave-Asisted Synthesis of Heterocycles, 1st ed.; Springer: Berlin, Germany, 2006. [Google Scholar]
- Larhed, M.; Olofsson, K. Topics in Current Chemistry 266: Microwave Methods in Organic Synthesis, 1st ed.; Springer: Berlin, Germany, 2006. [Google Scholar]
- Ameta, S.; Punjabi, P.; Ameta, R.; Ameta, C. Microwave-Assited Organic Synthesis: A Green Chemical Approach, 1st ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Bogdal, D. Microwave-assisted Organic Synthesis: One Hundred Reaction Procedures, 1st ed.; Tetrahedron Organic Chemistry Series; Elsevier Ltd.: Oxford, UK, 2005. [Google Scholar]
- García-Muñoz, M.J.; Zacconi, F.; Foubelo, F.; Yus, M. Indium-Promoted Diastereo- and Regioselective Propargylation of Chiral Sulfinylimines. Eur. J. Org. Chem. 2013, 2013, 1287–1295. [Google Scholar] [CrossRef]
- Zacconi, F.; Arias, H.R. Biocatalysts. J. Thermodyn. Catal. 2013, 04, e121. [Google Scholar] [CrossRef] [Green Version]
- Zacconi, F.; Arias, H.R. Innovation and Catalysis. J. Thermodyn. Catal. 2013, 4, e116. [Google Scholar] [CrossRef]
- Núñez-Navarro, N.; Segovia, G.; Burgos, R.; Lagos, C.; Fuentes-Ibacache, N.; Faúndez, M.; Zacconi, F. Microwave Assisted Synthesis of Novel Six-Membered 4-C, 4-O and 4-S Lactams Derivatives: Characterization and in vitro Biological Evaluation of Cytotoxicity and Anticoagulant Activity. J. Braz. Chem. Soc. 2017, 28, 203–207. [Google Scholar] [CrossRef]
- Lagos, C.; Segovia, G.; Nuñez-Navarro, N.; Faúndez, M.; Zacconi, F. Novel FXa Inhibitor Identification through Integration of Ligand- and Structure-Based Approaches. Molecules 2017, 22, 1588. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Meneyrol, J.; Follmann, M.; Lassalle, G.; Wehner, V.; Barre, G.; Rousseaux, T.; Altenburger, J.-M.; Petit, F.; Bocskei, Z.; Schreuder, H.; et al. 5-Chlorothiophene-2-carboxylic Acid [( S )-2-[2-Methyl-3-(2-oxopyrrolidin-1-yl)benzenesulfonylamino]-3-(4-methylpiperazin-1-yl)-3-oxopropyl]amide (SAR107375), a Selective and Potent Orally Active Dual Thrombin and Factor Xa Inhibitor. J. Med. Chem. 2013, 56, 9441–9456. [Google Scholar] [CrossRef]
- Pinto, D.J.P.; Orwat, M.J.; Koch, S.; Rossi, K.A.; Alexander, R.S.; Smallwood, A.; Wong, P.C.; Rendina, A.R.; Luettgen, J.M.; Knabb, R.M.; et al. Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro- 1 H -pyrazolo[3,4- c ]pyridine-3-carboxamide (Apixaban, BMS-562247), a Highly Potent, Selective, Efficacious, and Orally Bioavailable Inhibitor of Blood Coagulati. J. Med. Chem. 2007, 50, 5339–5356. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, A.; McGaughey, G.B.; Sheridan, R.P.; Good, A.C.; Warren, G.; Mathieu, M.; Muchmore, S.W.; Brown, S.P.; Grant, J.A.; Haigh, J.A.; et al. Molecular Shape and Medicinal Chemistry: A Perspective. J. Med. Chem. 2010, 53, 3862–3886. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Peleg, M.; Normand, M.D.; Corradini, M.G. The Arrhenius equation revisited. Crit. Rev. Food Sci. Nutr. 2012, 52, 830–851. [Google Scholar] [CrossRef]
- Gurjar, K.K.; Sharma, R.K. Mechanistic Studies of Ullmann-Type C-N Coupling Reactions: Carbonate-Ligated Copper(III) Intermediates. ChemCatChem 2017, 9, 862–869. [Google Scholar] [CrossRef]
- Li, J.J. Ullmann coupling. In Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications Fifth Edition; Li, J.J., Ed.; Springer International Publishing: San Francisco, CA, USA, 2014; pp. 611–612. ISBN 978-3-319-03979-4. [Google Scholar]
- Soria-Castro, S.M.; Andrada, D.M.; Caminos, D.A.; Argüello, J.E.; Robert, M.; Peñéñory, A.B. Mechanistic Insight into the Cu-Catalyzed C–S Cross-Coupling of Thioacetate with Aryl Halides: A Joint Experimental–Computational Study. J. Org. Chem. 2017, 82, 11464–11473. [Google Scholar] [CrossRef]
- Durán, R.; Núñez-Navarro, N.; Zacconi, F.C.; Herrera, B. Theoretical study of C-arylations with aryl halides to determine the reaction mechanism, the effect of substituents and heteroatoms. Phys. Chem. Chem. Phys. 2019, 21, 10163–10170. [Google Scholar] [CrossRef]
- Santana-Romo, F.; Duarte, Y.; Castillo, F.; Maestro, M.A.; Zacconi, F.C. Microwave-mediated Synthesis of N-allyl/Propargyl Derivatives: Enzymatic Analysis as a Potential Factor Xa (FXa) Inhibitor, Theoretical and Computational Molecular Docking. Int. J. Chem. Eng. Appl. in press.
- Li, J.J. Ullmann reaction. In Name Reactions: A Collection of Detailed Reaction Mechanisms; Springer: Berlin/Heidelberg, Germay, 2006; pp. 599–600. ISBN 978-3-540-30030-4. [Google Scholar]
- Batool, T.; Rasool, N.; Gull, Y.; Noreen, M.; Nasim, F.-H.; Yaqoob, A.; Zubair, M.; Rana, U.A.; Khan, S.U.-D.; Zia-Ul-Haq, M.; et al. A Convenient Method for the Synthesis of (Prop-2-Ynyloxy)Benzene Derivatives via Reaction with Propargyl Bromide, Their Optimization, Scope and Biological Evaluation. PLoS ONE 2015, 9, e115457. [Google Scholar] [CrossRef]
- Gao, L.-H.; Zhang, J.-Y.; Song, S.-Z.; Cao, T.-T.; Ge, G.-P.; Li, Q.; Wei, W.-T. Base-promoted domino radical cyclization of 1,6-enynes. Org. Biomol. Chem. 2019, 17, 7674–7678. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Bi, W.; Li, X.; Chen, X.; Qu, L.; Zhao, Y. A Practical Method to Synthesize 1,2,3-Triazole-Amino-Bisphosphonate Derivatives. Phosphorus. Sulfur. Silicon Relat. Elem. 2015, 190, 1735–1742. [Google Scholar] [CrossRef]
- Malmir, M.; Heravi, M.M.; Sadjadi, S.; Hosseinnejad, T. Ultrasonic and bio-assisted synthesis of Ag@HNTs-T as a novel heterogeneous catalyst for the green synthesis of propargylamines: A combination of experimental and computational study. Appl. Organomet. Chem. 2018, 32, e4291. [Google Scholar] [CrossRef]
- Selvaraj, V.; Rajendran, V. Propargylation of indene-1,3-dione under a new phase-transfer catalyst combined with ultrasonication–A kinetic study. Ultrason. Sonochem. 2014, 21, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Bohórquez, A.R.R.; Romero-Daza, J.; Acelas, M. Versatile and mild HCl-catalyzed cationic imino Diels-Alder reaction for the synthesis of new tetrahydroquinoline derivatives. Synth. Commun. 2016, 46, 338–347. [Google Scholar] [CrossRef]
- Sridharan, V.; Suryavanshi, P.A.; Menéndez, J.C. Advances in the Chemistry of Tetrahydroquinolines. Chem. Rev. 2011, 111, 7157–7259. [Google Scholar] [CrossRef]
- Rodríguez, Y.A.; Gutiérrez, M.; Ramírez, D.; Alzate-Morales, J.; Bernal, C.C.; Güiza, F.M.; Romero Bohórquez, A.R. Novel N -allyl/propargyl tetrahydroquinolines: Synthesis via Three-component Cationic Imino Diels-Alder Reaction, Binding Prediction, and Evaluation as Cholinesterase Inhibitors. Chem. Biol. Drug Des. 2016, 88, 498–510. [Google Scholar] [CrossRef]
- Muthukrishnan, I.; Sridharan, V.; Menéndez, J.C. Progress in the Chemistry of Tetrahydroquinolines. Chem. Rev. 2019, 119, 5057–5191. [Google Scholar] [CrossRef]
- Tejería, A.; Pérez-Pertejo, Y.; Reguera, R.M.; Carbajo-Andrés, R.; Balaña-Fouce, R.; Alonso, C.; Martin-Encinas, E.; Selas, A.; Rubiales, G.; Palacios, F. Antileishmanial activity of new hybrid tetrahydroquinoline and quinoline derivatives with phosphorus substituents. Eur. J. Med. Chem. 2019, 162, 18–31. [Google Scholar] [CrossRef]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chemie Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Alonso, F.; Moglie, Y.; Radivoy, G. Copper Nanoparticles in Click Chemistry. Acc. Chem. Res. 2015, 48, 2516–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, L.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, L.J. ORTEP -3 for Windows—A version of ORTEP -III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chemie Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Balawender, R.; Komorowski, L. Atomic Fukui function indices and local softness ab initio. J. Chem. Phys. 1998, 109, 5203–5211. [Google Scholar] [CrossRef]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef]
- Kessinger, C.W.; Kim, J.W.; Henke, P.K.; Thompson, B.; McCarthy, J.R.; Hara, T.; Sillesen, M.; Margey, R.J.P.; Libby, P.; Weissleder, R.; et al. Statins Improve the Resolution of Established Murine Venous Thrombosis: Reductions in Thrombus Burden and Vein Wall Scarring. PLoS ONE 2015, 10, e0116621. [Google Scholar] [CrossRef] [Green Version]
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the Utility of Alerts for Pan-Assay INterference CompoundS. J. Chem. Inf. Model. 2017, 57, 417–427. [Google Scholar] [CrossRef]
- Dahlin, J.L.; Nissink, J.W.M.; Strasser, J.M.; Francis, S.; Higgins, L.; Zhou, H.; Zhang, Z.; Walters, M.A. PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Lindhoff-Last, E.; Samama, M.M.; Ortel, T.L.; Weitz, J.I.; Spiro, T.E. Assays for Measuring Rivaroxaban: Their Suitability and Limitations. Ther. Drug Monit. 2010, 32, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Crain, E.J.; Pinto, D.J.; Watson, C.A. Dose-Dependent Antithrombotic Effects of Apixaban, an Oral Direct Factor Xa Inhibitor, in Prevention and Treatment of Thrombosis in Rabbit Models of Arteriovenous-Shunt and Venous Thrombosis at Doses That Preserve Hemostasis. Blood 2007, 110, 933. [Google Scholar] [CrossRef]
- McCarty, D.; Robinson, A. Factor Xa inhibitors: A novel therapeutic class for the treatment of nonvalvular atrial fibrillation. Ther. Adv. Cardiovasc. Dis. 2016, 10, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskandari, K.; Lesani, M. Does Fluorine Participate in Halogen Bonding? Chem. A Eur. J. 2015, 21, 4739–4746. [Google Scholar] [CrossRef]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. Fluorine-Centered Halogen Bonding: A Factor in Recognition Phenomena and Reactivity. Cryst. Growth Des. 2011, 11, 4238–4246. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Supramolecular Chemistry. Angew. Chemie Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Horowitz, S.; Trievel, R.C. Carbon-Oxygen Hydrogen Bonding in Biological Structure and Function. J. Biol. Chem. 2012, 287, 41576–41582. [Google Scholar] [CrossRef] [Green Version]
- Scheiner, S. Weak H-bonds. Comparisons of CH⋯O to NH⋯O in proteins and PH⋯N to direct P⋯N interactions. Phys. Chem. Chem. Phys. 2011, 13, 13860–13872. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Mulakala, C.; Viswanadhan, V.N. Could MM-GBSA be accurate enough for calculation of absolute protein/ligand binding free energies? J. Mol. Graph. Model. 2013, 46, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-1: Desmond Molecular Dynamics System, D.E. Shaw Research; Schrödinger: New York, NY, USA, 2019.
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2019-1: QikProp; Schrödinger, LLC: New York, NY, USA, 2019.
- Dagan-Wiener, A.; Nissim, I.; Ben Abu, N.; Borgonovo, G.; Bassoli, A.; Niv, M.Y. Bitter or not? BitterPredict, a tool for predicting taste from chemical structure. Sci. Rep. 2017, 7, 12074. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks Iii, C.L.; Mackerell Jr, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- vROCS, Version 3.3.1.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2019; Available online: www.eyesopen.com (accessed on 2 January 2020).
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Vijai Kumar Reddy, T.; Prabhavathi Devi, B.L.A.; Prasad, R.B.N.; Sujitha, P.; Ganesh Kumar, C. Total synthesis and biological evaluation of clavaminol-G and its analogs. Eur. J. Med. Chem. 2013, 67, 384–389. [Google Scholar] [CrossRef]
- Gutierrez, V.; Mascaró, E.; Alonso, F.; Moglie, Y.; Radivoy, G. Direct synthesis of β-ketophosphonates and vinyl phosphonates from alkenes or alkynes catalyzed by CuNPs/ZnO. RSC Adv. 2015, 5, 65739–65744. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02-SMP; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dwivedi, A.; Baboo, V.; Bajpai, A. Fukui Function Analysis and Optical, Electronic, and Vibrational Properties of Tetrahydrofuran and Its Derivatives: A Complete Quantum Chemical Study. J. Theor. Chem. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Bhunia, S.S.; Roy, K.K.; Saxena, A.K. Profiling the Structural Determinants for the Selectivity of Representative Factor-Xa and Thrombin Inhibitors Using Combined Ligand-Based and Structure-Based Approaches. J. Chem. Inf. Model. 2011, 51, 1966–1985. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-1: Small-Molecule Drug Discovery Suite; Protein Preparation Wizard; Epik, Impact, Prime; Schrödinger, LLC: New York, NY, USA, 2019; Available online: https://www.schrodinger.com/ (accessed on 2 January 2020).
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: a software program for pKaprediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-4. Schrödinger, LLC: New York, NY, USA. Available online: https://www.schrodinger.com/ (accessed on 2 January 2020).
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinforma. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neria, E.; Fischer, S.; Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Ishikawa, M.; Hashimoto, Y. Improvement in Aqueous Solubility in Small Molecule Drug Discovery Programs by Disruption of Molecular Planarity and Symmetry. J. Med. Chem. 2011, 54, 1539–1554. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [Green Version]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID (Reference) | Resolution (Å) | RMSD Cluster Center (Å) | 1 RMSD Binding Site (6Å) | 2 Tanimoto Distance |
|---|---|---|---|---|
| 4BTI [70] | 2.30 | ----- | ----- | 0.000 |
| 4BTU [70] | 2.37 | 0.440 | 0.222 | 0.426 |
| 4BTT [70] | 2.59 | 0.530 | 0.381 | 0.582 |
| 2W26 Rivaroxaban [34] | 2.08 | 0.276 | 0.207 | 0.618 |
| 2P16 Apixaban [71] | 2.30 | 0.290 | 0.381 | 0.689 |
| Entry | CuI (equiv.) | DMEDA (equiv.) | T (°C) | Time (h) | Heating Source | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 0.05 | 0.1 | 20 | 96 | r.t. | - |
| 2 | 0.05 | 0.1 | 100 | 72 | conventional | - |
| 3 | 0.1 | 0.2 | 100 | 72 | conventional | 1 |
| 4 | 0.05 | 0.5 | 100 | 72 | conventional | - |
| 5 | 0.05 | 0.1 | 120 | 48 | conventional | 1 |
| 6 | 0.1 | 0.2 | 120 | 48 | conventional | 4 |
| 7 | 0.05 | 0.5 | 120 | 48 | conventional | 8 |
| 8 | 0.05 | 0.1 | 60 | 6 | sonication | - |
| 9 | 0.1 | 0.2 | 60 | 6 | sonication | 5 |
| 10 | 0.05 | 0.5 | 60 | 6 | sonication | 20 |
| 11 | 0.05 | 0.1 | 120 | 2 | microwave | 6 |
| 12 | 0.1 | 0.2 | 120 | 2 | microwave | 8 |
| 13 | 0.15 | 0.3 | 120 | 2 | microwave | 11 |
| 14 | 0.25 | 0.5 | 120 | 2 | microwave | 17 |
| 15 | 0.5 | 0.5 | 120 | 2 | microwave | 19 |
| 16 | 1.00 | 1.0 | 120 | 2 | microwave | 26 |
| 17 | 2.00 | 2.0 | 120 | 2 | microwave | 46 |
| 18 | 2.00 | 2.0 | 160 | 1.5 | microwave | 50 |
| 193 | 2.00 | 2.0 | 160 | 1.5 | microwave | 62 |
| Entry | Heating Source | T (°C) | Time (h) | Yield (%) | |||
|---|---|---|---|---|---|---|---|
| 11 | 12 | 13 | 14 | ||||
| 1 | r.t. | 20 | 96 | 13 | 19 | 10 | 9 |
| 2 | conventional | 80 | 24 | 31 | 40 | 29 | 15 |
| 3 | conventional | 80 | 72 | 36 | 47 | 34 | 30 |
| 4 | conventional | 90 | 24 | 37 | 49 | 37 | 34 |
| 5 | sonication | 40 | 15 | 43 | 57 | 45 | 39 |
| 6 | sonication | 80 | 4 | 55 | 67 | 51 | 43 |
| 7 | microwave | 160 | 0.5 | 69 | 72 | 70 | 67 |
| Entry | Heating Source | T (°C) | Time (h) | Yield (%) | |||
|---|---|---|---|---|---|---|---|
| 17 | 18 | 19 | 20 | ||||
| 1 | r.t. | 20 | 96 | 32 | - | 34 | 36 |
| 2 | conventional | 80 | 24 | 47 | - | 45 | 52 |
| 3 | conventional | 80 | 72 | 53 | - | 54 | 59 |
| 4 | conventional | 90 | 24 | 59 | - | 56 | 62 |
| 5 | sonication | 40 | 8 | 69 | - | 64 | 67 |
| 6 | sonication | 80 | 2 | 72 | - | 72 | 73 |
| 7 | microwave | 160 | 0.5 | 85 | - | 89 | 83 |
| Bond Length | CALC | EXP | Crystal Structure 1 |
|---|---|---|---|
| F(1)-C(10) | 1.366 | 1.339 | 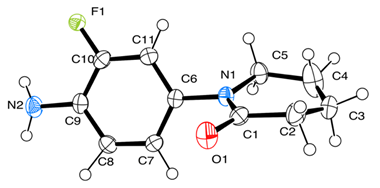 |
| O(1)-C(1) | 1.220 | 1.232 | |
| N(1)-C(1) | 1.382 | 1.354 | |
| N(1)-C(6) | 1.435 | 1.438 | |
| N(1)-C(5) | 1.477 | 1.480 | |
| N(2)-C(9) | 1.391 | 1.382 | |
| C(2)-C(3) | 1.529 | 1.540 | |
| C(3)-C(4) | 1.301 | 1.497 | |
| Bond Angles | CALC | EXP | |
| C(4)-C(3)-C(2) | 109.2 | 109.4 | |
| C(3)-C(2)-C(1) | 115.9 | 115.24 | |
| C(5)-N(1)-C(6) | 116.3 | 115.67 | |
| C(1)-N(1)-C(6) | 118.8 | 119.09 | |
| C(11)-C(10)-F(1) | 119.2 | 119.26 | |
| C(11)-C(6)-N(1)-C(1) | 112.7 | 116.25 | |
| C(4)-C(5)-C(1)-C(2) | 14.8 | 9.1 |
| Bond Length | CALC | EXP | Crystal Structure 1 |
|---|---|---|---|
| F(1)-C(7) | 1.365 | 1.364 |  |
| O(2)-C(2) | 1.219 | 1.231 | |
| N(1)-C(2) | 1.377 | 1.352 | |
| N(1)-C(5) | 1.433 | 1.436 | |
| N(1)-C(3) | 1.474 | 1.479 | |
| N(2)-C(8) | 1.390 | 1.383 | |
| C(1)-O(1) | 1.418 | 1.418 | |
| O(1)-C(4) | 1.417 | 1.430 | |
| Bond Angles | CALC | EXP | |
| C(4)-O(1)-C(1) | 110.6 | 109.4 | |
| O(1)-C(1)-C(2) | 116.7 | 115.6 | |
| C(3)-N(1)-C(5) | 117.6 | 117.2 | |
| C(2)-N(1)-C(5) | 120.8 | 120.7 | |
| C(6)-C(7)-F(1) | 119.0 | 118.3 | |
| C(6)-C(5)-N(1)-C(2) | 50.0 | 50.2 | |
| C(4)-C(3)-C(2)-C(1) | 18.7 | 16.0 | |
| C(10)-C(5)-N(1)-C(2) | 50.2 | 131.7 |
| Bond Length | CALC | EXP | Crystal Structure 1 |
|---|---|---|---|
| F(1)-C(4) | 1.365 | 1.363 | 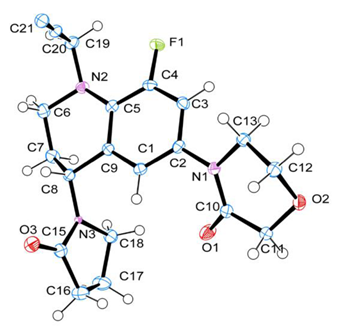 |
| O(1)-C(10) | 1.219 | 1.222 | |
| N(1)-C(2) | 1.432 | 1.436 | |
| N(1)-C(10) | 1.378 | 1.368 | |
| N(1)-C(13) | 1.474 | 1.475 | |
| N(2)-C(5) | 1.393 | 1.411 | |
| N(2)-C(6) | 1.465 | 1.474 | |
| N(3)-C(8) | 1.474 | 1.457 | |
| N(3)-C(18) | 1.464 | 1.471 | |
| N(3)-C(15) | 1.372 | 1.356 | |
| O(3)-C(15) | 1.219 | 1.231 | |
| O(2)-C(12) | 1.417 | 1.426 | |
| O(2)-C(11) | 1.418 | 1.421 | |
| C(8)-N(3) | 1.474 | 1.457 | |
| Bond Angles | CALC | EXP | |
| C(12)-O(2)-C(11) | 110.7 | 108.3 | |
| O(2)-C(11)-C(10) | 116.5 | 115.1 | |
| C(13)-N(1)-C(2) | 117.6 | 117.4 | |
| C(10)-N(1)-C(2) | 120.5 | 120.1 | |
| C(3)-C(4)-F(1) | 116.5 | 115.8 | |
| C(5)-N(2)-C(19) | 122.1 | 117.8 | |
| C(3)-C(2)-N(1)-C(10) | 126.8 | 137.7 | |
| C(12)-C(13)-C(10)-C(11) | 18.2 | 13.1 | |
| C(5)-N(2)-C(19)-C(20) | 128.5 | 55.3 | |
| C(9)-C(8)-N(3)-C(15) | 153.8 | 111.2 | |
| C(21)-N(2)-C(8)-N(3) | 108.7 | 158.0 |
| Compound | HOMO | LUMO | GAP | HARD. (η) | C. POT. (μ) | ELEC. (ω) |
|---|---|---|---|---|---|---|
| 11 | −5.3917 | −0.3859 | 5.0058 | 2.5029 | −2.8888 | 1.6671 |
| 12 | −5.5550 | −0.5519 | 5.0031 | 2.5016 | −3.0534 | 1.8635 |
| 13 | −5.8366 | −0.5010 | 5.3357 | 2.6678 | −3.1688 | 1.8819 |
| 14 | −5.5593 | −0.5954 | 4.9639 | 2.4820 | −3.0774 | 1.9078 |
| 27 | −5.4418 | −1.5467 | 3.8951 | 1.9475 | −3.4942 | 3.1347 |
| 30 | −5.5892 | −1.5913 | 3.9979 | 1.9989 | −3.5903 | 3.2242 |
| 33 | −5.8339 | −1.6205 | 4.2134 | 2.1067 | −3.7272 | 3.2970 |
| 36 | −5.5982 | −1.6003 | 3.9979 | 1.9990 | −3.5993 | 3.2404 |
| 28 | −5.5354 | −1.8583 | 3.6771 | 1.8386 | −3.6968 | 3.7167 |
| 31 | −5.6674 | −1.8931 | 3.7742 | 1.8871 | −3.7802 | 3.7862 |
| 34 | −5.9278 | −1.9249 | 4.0028 | 2.0014 | −3.9264 | 3.8514 |
| 37 | −5.6973 | −1.9111 | 3.7862 | 1.8931 | −3.8042 | 3.8222 |
| 29 | −5.5147 | −1.7138 | 3.8009 | 1.9005 | −3.6142 | 3.4367 |
| 32 | −5.6524 | −1.7557 | 3.8967 | 1.9484 | −3.7040 | 3.5209 |
| 35 | −5.9082 | −1.7843 | 4.1239 | 2.0620 | −3.8462 | 3.5872 |
| 38 | −5.6750 | −1.7682 | 3.9068 | 1.9534 | −3.7216 | 3.5452 |
| 24 | −7.1049 | −1.4487 | 5.6562 | 2.8281 | −4.2768 | 3.2339 |
| 25 | −7.2350 | −1.6602 | 5.5748 | 2.7874 | −4.4476 | 3.5483 |
| 26 | –7.2467 | –1.7339 | 5.5128 | 2.7564 | –4.4903 | 3.6575 |
| Compounda | Inhibition FXa (%)b | IC50 ± SD [μM]c |
|---|---|---|
| 6 | 19 | - |
| 7 | 18 | - |
| 8 | 20 | - |
| 10 | 15 | - |
| 11 | 17 | - |
| 12 | 14 | - |
| 13 | 25 | - |
| 14 | 24 | - |
| 17 | 25 | - |
| 19 | 27 | - |
| 20 | 15 | - |
| 27 | 52 | 102.10 ± 0.14 |
| 28 | 33 | - |
| 29 | 46 | - |
| 30 | 63 | 41.71 ± 0.07 |
| 31 | 73 | 29.73 ± 0.09 |
| 32 | 21 | - |
| 33 | 41 | - |
| 34 | 60 | 67.92 ± 0.08 |
| 35 | 28 | - |
| 36 | 34 | - |
| 37 | 34 | - |
| 38 | 39 | - |
| Compound | Anti-FXa [IU/mL] a |
|---|---|
| 27 | 0.29 ± 0.10 |
| 30 | 0.00 |
| 31 | 0.05 ± 0.09 |
| 34 | 0.42 ± 0.18 |
| Compound | Glide Emodel (kcal/mol) | Glide EVdW (Kcal/mol) | ∆Gbind (Kcal/mol) | ∆Gbind VdW (Kcal/mol) |
|---|---|---|---|---|
| 8 | −56.62 | −36.62 | −53.15 | −40.49 |
| 19 | −45.66 | −27.25 | −49.49 | −26.66 |
| 27 | −79.77 | −44.64 | −67.39 | −49.17 |
| 30 | −77.10 | −49.62 | −52.84 | −46.57 |
| 31 | −82.94 | −54.62 | −65.81 | −58.48 |
| 34 | −82.89 | −51.19 | −64.28 | −53.24 |
| Compound | logPo/w | Molecular Weight | logS | H-Bonds Donors | H-Bonds Acceptors | logBB | PHOA |
|---|---|---|---|---|---|---|---|
| 7 | 2.30 | 309.34 | −3.48 | 1.5 | 6.5 | 0.80 | 88.62 |
| 8 | 1.71 | 226.27 | −2.60 | 1.5 | 4.5 | 0.12 | 91.48 |
| 9 | 0.76 | 210.21 | −2.02 | 1.5 | 5.7 | 0.21 | 85.49 |
| 11 | 2.81 | 246.28 | −3.91 | 1.5 | 4.0 | 0.10 | 100.00 |
| 12 | 3.58 | 347.39 | −5.82 | 1.5 | 6.5 | 0.57 | 100.00 |
| 13 | 2.83 | 264.32 | −4.01 | 1.5 | 4.5 | 0.08 | 100.00 |
| 14 | 1.94 | 248.26 | −3.00 | 1.5 | 5.7 | 0.04 | 100.00 |
| 17 | 2.83 | 369.44 | −3.92 | 0.5 | 7.0 | 0.22 | 96.68 |
| 18 | 3.37 | 470.54 | −5.80 | 0.5 | 9.5 | 0.85 | 93.68 |
| 19 | 2.75 | 387.47 | −3.99 | 0.5 | 7.5 | 0.10 | 96.52 |
| 20 | 1.79 | 371.41 | −2.90 | 0.5 | 8.7 | 0.25 | 89.95 |
| 27 | 4.02 | 365.41 | −6.25 | 1.0 | 6.0 | 0.63 | 100.00 |
| 28 | 4.51 | 399.85 | −6.99 | 1.0 | 6.0 | −0.48 | 100.00 |
| 29 | 4.25 | 383.40 | −6.62 | 1.0 | 6.0 | −0.53 | 100.00 |
| 30 | 4.51 | 466.51 | −7.63 | 1.0 | 8.5 | −1.26 | 100.00 |
| 31 | 4.99 | 500.96 | −8.35 | 1.0 | 8.5 | −1.11 | 90.07 |
| 33 | 3.94 | 383.44 | −6.17 | 1.0 | 6.5 | −0.51 | 100.00 |
| 34 | 4.74 | 484.50 | −7.99 | 1.0 | 8.5 | −1.15 | 100.00 |
| 34 | 4.44 | 417.89 | −6.91 | 1.0 | 6.5 | −0.35 | 100.00 |
| 35 | 4.18 | 401.43 | −6.54 | 1.0 | 6.5 | −0.40 | 100.00 |
| 36 | 2.97 | 367.38 | −5.01 | 1.0 | 7.7 | −0.63 | 100.00 |
| 37 | 3.46 | 401.83 | −5.75 | 1.0 | 7.7 | −0.48 | 100.00 |
| 38 | 3.21 | 385.37 | −5.43 | 1.0 | 7.7 | −0.54 | 100.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana-Romo, F.; Lagos, C.F.; Duarte, Y.; Castillo, F.; Moglie, Y.; Maestro, M.A.; Charbe, N.; Zacconi, F.C. Innovative Three-Step Microwave-Promoted Synthesis of N-Propargyltetrahydroquinoline and 1,2,3-Triazole Derivatives as a Potential Factor Xa (FXa) Inhibitors: Drug Design, Synthesis, and Biological Evaluation. Molecules 2020, 25, 491. https://doi.org/10.3390/molecules25030491
Santana-Romo F, Lagos CF, Duarte Y, Castillo F, Moglie Y, Maestro MA, Charbe N, Zacconi FC. Innovative Three-Step Microwave-Promoted Synthesis of N-Propargyltetrahydroquinoline and 1,2,3-Triazole Derivatives as a Potential Factor Xa (FXa) Inhibitors: Drug Design, Synthesis, and Biological Evaluation. Molecules. 2020; 25(3):491. https://doi.org/10.3390/molecules25030491
Chicago/Turabian StyleSantana-Romo, Fabián, Carlos F. Lagos, Yorley Duarte, Francisco Castillo, Yanina Moglie, Miguel A. Maestro, Nitin Charbe, and Flavia C. Zacconi. 2020. "Innovative Three-Step Microwave-Promoted Synthesis of N-Propargyltetrahydroquinoline and 1,2,3-Triazole Derivatives as a Potential Factor Xa (FXa) Inhibitors: Drug Design, Synthesis, and Biological Evaluation" Molecules 25, no. 3: 491. https://doi.org/10.3390/molecules25030491