
Synthesis, Characterization, and Antiproliferative Activity of Novel Chiral [QuinoxP*AuCl2]+ Complexes

 ,
,  , and
, and
Abstract
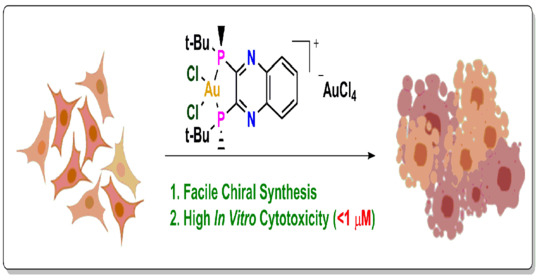
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
2.2. X-ray Crystallography
2.3. Biological Stability of 1 and 2
2.3.1. UV–Vis Stability in Biological Media
2.3.2. Reactivity of 1 and 2 with l-Glutathione (GSH)
2.4. Cytotoxicity of 1 and 2
2.5. Electrochemical Studies of 2
3. Materials and Methods
3.1. General Experimental Details
3.2. Synthesis of Compounds 1 and 2
3.3. Physical and Chemical Characterization
3.3.1. X-ray Crystallography
3.3.2. UV–Vis Stability
3.3.3. Cyclic Voltammetry of 2
3.4. In Vitro Biological Assays
3.4.1. Cell Culture
3.4.2. Cell Viability of 1 and 2
3.4.3. Reactivity of 1 and 2 with L-GSH
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rosenberg, B.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Franz, K.J.; Metzler-Nolte, N. Introduction: Metals in Medicine. Chem. Rev. 2019, 119, 727–729. [Google Scholar] [CrossRef] [Green Version]
- Farrell, N. Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Bierer, D.W. Bismuth subsalicylate: History, chemistry, and safety. Rev. Infect. Dis. 1990, 12 (Suppl. 1), S3–S8. [Google Scholar] [CrossRef]
- Mirabell, C.K.; Johnson, R.K.; Hill, D.T.; Faucette, L.F.; Girard, G.R.; Kuo, G.Y.; Sung, C.M.; Crooke, S.T. Correlation of the in vitro cytotoxic and in vivo antitumor activities of gold (I) coordination complexes. J. Med. Chem. 1986, 29, 218–223. [Google Scholar] [CrossRef]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arnér, E.S.; Linder, S. Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R.D. 2015, 15, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Islami, F.S.R.L.; Jemal, A. The changing landscape of cancer in the USA—Opportunities for advancing prevention and treatment. Nat. Rev. Clin. Oncol. 2020, 17, 631–649. [Google Scholar] [CrossRef]
- Fischer, J.; Ganellin, C.R.; Ganesan, A.; Proudfoot, J. ABDD; Wiley-VCH: Hoboken, NJ, USA, 2010. [Google Scholar]
- List, A.; Beran, M.; DiPersio, J.; Slack, J.; Vey, N.; Rosenfeld, C.; Greenberg, P. Opportunities for Trisenox®(arsenic trioxide) in the treatment of myelodysplastic syndromes. Leukemia 2003, 17, 1499–1507. [Google Scholar] [CrossRef]
- Gukathasan, S.; Parkin, S.; Awuah, S.G. Cyclometalated Gold (III) complexes bearing DACH ligands. Inorg. Chem. 2019, 58, 9326–9340. [Google Scholar] [CrossRef]
- Mertens, R.T.; Parkin, S.; Awuah, S.G. Cancer cell-selective modulation of mitochondrial respiration and metabolism by potent organogold (iii) dithiocarbamates. Chem. Sci. 2020, 11, 10465–10482. [Google Scholar] [CrossRef]
- Dennis, E.K.; Kim, J.H.; Parkin, S.; Awuah, S.G.; Garneau-Tsodikova, S. Distorted Gold (I)–Phosphine Complexes as Antifungal Agents. J. Med. Chem. 2019, 63, 2455–2469. [Google Scholar] [CrossRef]
- Kim, J.H.; Reeder, E.; Parkin, S.; Awuah, S.G. Gold(I/III)-Phosphine Complexes as Potent Antiproliferative Agents. Sci. Rep. 2019, 9, 12335. [Google Scholar] [CrossRef]
- Lopez, M.J.; Mohiuddin, S.S. Biochemistry, Essential Amino Acids. In StatPearls [Internet]; StatPearls Publishing: Tampa, FL, USA, 2020. [Google Scholar]
- Wang, Y.; Huang, H.; Zhang, Q.; Zhang, P. Chirality in metal-based anticancer agents. Dalton Trans. 2018, 47, 4017–4026. [Google Scholar] [CrossRef]
- Wilkinson, R.G.; Shepherd, R.G.; Thomas, J.P.; Baughn, C. Stereospecificity in a new type of synthetic antituberculous agent1,2. J. Am. Chem. Soc. 1961, 83, 2212–2213. [Google Scholar] [CrossRef]
- Kritsyn, A.M.; Likhosherstov, A.M.; Protopopova, T.V.; Skoldinov, A.P. In Ethambutol and related compounds. Synthesis and stereochemical relations. In Daklady Akademii Nawk; Russian Academy of Sciences: Moscow, Russia, 1962; Volume 145, pp. 332–335. [Google Scholar]
- Benedetti, M.; Malina, J.; Kasparkova, J.; Brabec, V.; Natile, G. Chiral discrimination in platinum anticancer drugs. Environ. Health Perspect. 2002, 110 (Suppl. 5), 779–782. [Google Scholar] [CrossRef] [Green Version]
- Von Zelewsky, A. Chiral complexes of platinum metals. Platin. Metals Rev. 1996, 40, 102–109. [Google Scholar]
- Coluccia, M.; Fanizzi, F.P.; Giannini, G.; Giordano, D. Synthesis, Mutagenicity, Binding to pBR 322 DNA and Antitumour Activity of Platinum (II) Complexes. Anticancer Res. 1991, 11, 281–288. [Google Scholar]
- Koch, J.H.; Gyarfas, E.C.; Dwyer, F. Biological Activity of Complex Ions Mechanism of Inhibition of Acetylcholinesterase. Aust. J. Biol. Sci. 1956, 9, 371–381. [Google Scholar] [CrossRef]
- Arnesano, F.; Pannunzio, A.; Coluccia, M.; Natile, G. Effect of chirality in platinum drugs. Coord. Chem. Rev. 2015, 284, 286–297. [Google Scholar] [CrossRef]
- Atilla-Gokcumen, G.E.; Williams, D.S.; Bregman, H.; Pagano, N.; Meggers, E. Organometallic compounds with biological activity: A very selective and highly potent cellular inhibitor for glycogen synthase kinase 3. ChemBioChem 2006, 7, 1443–1450. [Google Scholar] [CrossRef]
- Romero, M.J.; Sadler, P.J. Chirality in Organometallic Anticancer Complexes. In Bioorganometallic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp. 85–116. [Google Scholar]
- Fu, Y.; Soni, R.; Romero, M.J.; Pizarro, A.M.; Salassa, L.; Clarkson, G.J.; Hearn, J.M.; Habtemariam, A.; Wills, M.; Sadler, P.J. Mirror-Image Organometallic Osmium Arene Iminopyridine Halido Complexes Exhibit Similar Potent Anticancer Activity. Chem. Eur. J. 2013, 19, 15199–15209. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-A.; Ding, X.; Gong, L.; Meggers, E. Thioether-based anchimeric assistance for asymmetric coordination chemistry with ruthenium (II) and osmium (II). Dalton Trans. 2013, 42, 5623–5626. [Google Scholar] [CrossRef]
- Göbel, P.; Ritterbusch, F.; Helms, M.; Bischof, M.; Harms, K.; Jung, M.; Meggers, E. Probing chiral recognition of enzyme active sites with octahedral iridium (III) propeller complexes. Eur. J. Inorg. Chem. 2015, 2015, 1654–1659. [Google Scholar] [CrossRef]
- Kang, T.-S.; Mao, Z.; Ng, C.-T.; Wang, M.; Wang, W.; Wang, C.; Lee, S.M.-Y.; Wang, Y.; Leung, C.-H.; Ma, D.-L. Identification of an iridium (III)-based inhibitor of tumor necrosis factor-α. J. Med. Chem. 2016, 59, 4026–4031. [Google Scholar] [CrossRef]
- Rajaratnam, R.; Martin, E.K.; Dörr, M.; Harms, K.; Casini, A.; Meggers, E. Correlation between the Stereochemistry and Bioactivity in Octahedral Rhodium Prolinato Complexes. Inorg. Chem. 2015, 54, 8111–8120. [Google Scholar] [CrossRef]
- McGowan, M.A.; McGowan, P.C. A one-step synthesis of protected functionalised titanocene dichlorides. Inorg. Chem. Commun. 2000, 3, 337–340. [Google Scholar] [CrossRef]
- Potter, G.D.; Baird, M.C.; Cole, S.P. A new series of titanocene dichloride derivatives bearing chiral alkylammonium groups; assessment of their cytotoxic properties. Inorg. Chim. Acta 2010, 364, 16–22. [Google Scholar] [CrossRef]
- Kater, B.; Hunold, A.; Schmalz, H.-G.; Kater, L.; Bonitzki, B.; Jesse, P.; Prokop, A. Iron containing anti-tumoral agents: Unexpected apoptosis-inducing activity of a ferrocene amino acid derivative. J. Cancer Res. Clin. 2011, 137, 639–649. [Google Scholar] [CrossRef]
- Meléndez, E. Metallocenes as target specific drugs for cancer treatment. Inorg. Chim. Acta 2012, 393, 36–52. [Google Scholar] [CrossRef] [Green Version]
- Plazuk, D.; Zakrzewski, J.; Salmain, M.; Błauż, A.; Rychlik, B.E.; Strzelczyk, P.; Bujacz, A.; Bujacz, G. Ferrocene–biotin conjugates targeting cancer cells: Synthesis, interaction with avidin, cytotoxic properties and the crystal structure of the complex of avidin with a biotin–linker–ferrocene conjugate. Organometallics 2013, 32, 5774–5783. [Google Scholar] [CrossRef]
- Miklán, Z.; Szabo, R.; Zsoldos-Mády, V.; Reményi, J.; Banoczi, Z.; Hudecz, F. New ferrocene containing peptide conjugates: Synthesis and effect on human leukemia (HL-60) cells. Pept. Sci. Orig. Res. Biomol. 2007, 88, 108–114. [Google Scholar] [CrossRef]
- Mullick, A.B.; Chang, Y.M.; Ghiviriga, I.; Abboud, K.A.; Tan, W.; Veige, A.S. Human cancerous and healthy cell cytotoxicity studies of a chiral μ-dicarbene–digold (I) metallamacrocycle. Dalton Trans. 2013, 42, 7440–7446. [Google Scholar] [CrossRef]
- Li, B.-B.; Jia, Y.-X.; Zhu, P.-C.; Chew, R.J.; Li, Y.; Tan, N.S.; Leung, P.-H. Highly selective anti-cancer properties of ester functionalized enantiopure dinuclear gold (I)-diphosphine. Eur. J. Med. Chem. 2015, 98, 250–255. [Google Scholar] [CrossRef]
- Song, Y.; Vittal, J.J.; Srinivasan, N.; Chan, S.-H.; Leung, P.-H. Synthesis and anti-cancer activities of a pair of enantiomeric gold (I) complexes containing sulfanyl-substituted P-stereogenic phosphines. Tetrahedron Asymmetry 1999, 10, 1433–1436. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallog. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Messori, L.; Scaletti, F.; Massai, L.; Cinellu, M.A.; Krauss, I.R.; Di Martino, G.; Vergara, A.; Paduano, L.; Merlino, A. Interactions of gold-based drugs with proteins: Crystal structure of the adduct formed between ribonuclease A and a cytotoxic gold (III) compound. Metallomics 2014, 6, 233–236. [Google Scholar] [CrossRef]
- Messori, L.; Cinellu, M.A.; Merlino, A. Protein recognition of gold-based drugs: 3D structure of the complex formed when lysozyme reacts with Aubipyc. ACS Med. Chem. Lett. 2014, 5, 1110–1113. [Google Scholar] [CrossRef] [Green Version]
- Spell, S.R.; Farrell, N.P. [Au (dien)(N-heterocycle)] 3+: Reactivity with biomolecules and zinc finger peptides. Inorg. Chem. 2015, 54, 79–86. [Google Scholar] [CrossRef]
- Rajkumar, P.; Mathew, B.S.; Das, S.; Isaiah, R.; John, S.; Prabha, R.; Fleming, D.H. Cisplatin concentrations in long and short duration infusion: Implications for the optimal time of radiation delivery. J. Clin. Diag. Res. 2016, 10, XC01. [Google Scholar] [CrossRef]
- Rubbiani, R.; Schuh, E.; Meyer, A.; Lemke, J.; Wimberg, J.; Metzler-Nolte, N.; Meyer, F.; Mohr, F.; Ott, I. TrxR inhibition and antiproliferative activities of structurally diverse gold N-heterocyclic carbene complexes. MedChemComm 2013, 4, 942–948. [Google Scholar] [CrossRef]
- Maiore, L.; Cinellu, M.A.; Nobili, S.; Landini, I.; Mini, E.; Gabbiani, C.; Messori, L. Gold (III) complexes with 2-substituted pyridines as experimental anticancer agents: Solution behavior, reactions with model proteins, antiproliferative properties. J. Inorg. Biochem. 2012, 108, 123–127. [Google Scholar] [CrossRef]
- Bhabak, K.P.; Bhuyan, B.J.; Mugesh, G. Bioinorganic and medicinal chemistry: Aspects of gold (I)-protein complexes. Dalton Trans. 2011, 40, 2099–2111. [Google Scholar] [CrossRef]
- Boscutti, G.; Marchiò, L.; Ronconi, L.; Fregona, D. Insights into the reactivity of gold–dithiocarbamato anticancer agents toward model biomolecules by using multinuclear NMR spectroscopy. Chem. Eur. J. 2013, 19, 13428–13436. [Google Scholar] [CrossRef]
- Williams, M.R.; Bertrand, B.; Hughes, D.L.; Waller, Z.A.; Schmidt, C.; Ott, I.; O’Connell, M.; Searcey, M.; Bochmann, M. Cyclometallated Au (III) dithiocarbamate complexes: Synthesis, anticancer evaluation and mechanistic studies. Metallomics 2018, 10, 1655–1666. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Vanýsek, P. Electrochemical series. Handb. Chem. Phys. 2012, 93, 5–80. [Google Scholar]
- Parkin, S.; Hope, H. Macromolecular Cryocrystallography: Cooling, Mounting, Storage and Transportation of Crystals. J. Appl. Crystallog. 1998, 31, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Hope, H. X-ray crystallography—A fast, first-resort analytical tool. Prog. Inorg. Chem. 1994, 41, 1–19. [Google Scholar]
- Bruker. “APEX2” Bruker-AXS; Bruker: Madison, WI, USA, 2006. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SADABS, Program for Bruker Area Detector Absorption Correction; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sec. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Parkin, S. Expansion of scalar validation criteria to three dimensions: The R tensor. Erratum. Acta Crystallogr. A 2000, 56, 317. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Atom | Selected Atomic Distances (Å) and Angles (°) |
|---|---|
| Au1A-P2A | 2.292(6) Å |
| Au1A-P1A | 2.298(6) Å |
| Au1A-Cl2A | 2.320(6) Å |
| Au1A-Cl1A | 2.327(6) Å |
| P2A-Au1A-P1A | 89.4(2)° |
| Cl2A-Au1A-Cl1A | 91.8(2)° |
| X-ray Structural Data and Crystal Refinement | |
|---|---|
| Empirical Formula | C18H28Au2Cl6N2P2 |
| Molecular Weight (g/mol) | 941.00 |
| Temperature (K) | 90.0(2) |
| X-ray Radiation (Å) | Mo Kα (0.71073 Å) |
| Crystal System, Space Group | Triclinic, P1 |
| Unit Cell Dimensions (A), (o) | a = 11.0642(4) Å alpha = 80.985(1) |
| b = 11.2636(4) Å beta = 83.150(1) | |
| c = 12.5182(4) Å gamma = 63.674 (1) | |
| Volume | 1378.79(8) Å3 |
| Z | 2 |
| Absorption Coefficient | 11.336 mm−1 |
| F(000) | 880 |
| Crystal Size (mm) | 0.120 × 0.040 × 0.030 |
| Theta Range | 2.031 to 27.512 |
| Completeness to Theta = 25.242 | 100% |
| F2 | 1.022 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0518, wR2 = 0.1258 |
| IC50 (µM) | ||||||
|---|---|---|---|---|---|---|
| 1 | 2 | R,R QuinoxP* | S,S QuinoxP* | Cisplatin | Auranofin | |
| MDA-MB-468 | 1.51 ± 0.07 | 1.08 ± 0.13 | 14.8 0 ± 1.03 | 14.80 ± 1.03 | 2.09 ± 0.19 | 1.31 ± 0.09 |
| H460 | 4.53 ± 0.09 | 4.53 ± 0.06 | 11.75 ± 1.40 | 11.75 ± 1.40 | 1.41 ± 0.12 | 1.69 ± 0.21 |
| HCC1937 | 4.83 ± 0.1 | 3.85 ± 0.08 | - | - | 105.98 ± 0.97 a | - |
Sample Availability: Samples of the compounds are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arojojoye, A.S.; Mertens, R.T.; Ofori, S.; Parkin, S.R.; Awuah, S.G. Synthesis, Characterization, and Antiproliferative Activity of Novel Chiral [QuinoxP*AuCl2]+ Complexes. Molecules 2020, 25, 5735. https://doi.org/10.3390/molecules25235735
Arojojoye AS, Mertens RT, Ofori S, Parkin SR, Awuah SG. Synthesis, Characterization, and Antiproliferative Activity of Novel Chiral [QuinoxP*AuCl2]+ Complexes. Molecules. 2020; 25(23):5735. https://doi.org/10.3390/molecules25235735
Chicago/Turabian StyleArojojoye, Adedamola S., R. Tyler Mertens, Samuel Ofori, Sean R. Parkin, and Samuel G. Awuah. 2020. "Synthesis, Characterization, and Antiproliferative Activity of Novel Chiral [QuinoxP*AuCl2]+ Complexes" Molecules 25, no. 23: 5735. https://doi.org/10.3390/molecules25235735