
Synthesis, X-ray Characterization and Density Functional Theory (DFT) Studies of Two Polymorphs of the α,α,α,α, Isomer of Tetra-p-Iodophenyl Tetramethyl Calix[4]pyrrole: On the Importance of Halogen Bonds

 , , ,
, , , 

Abstract
:
1. Introduction
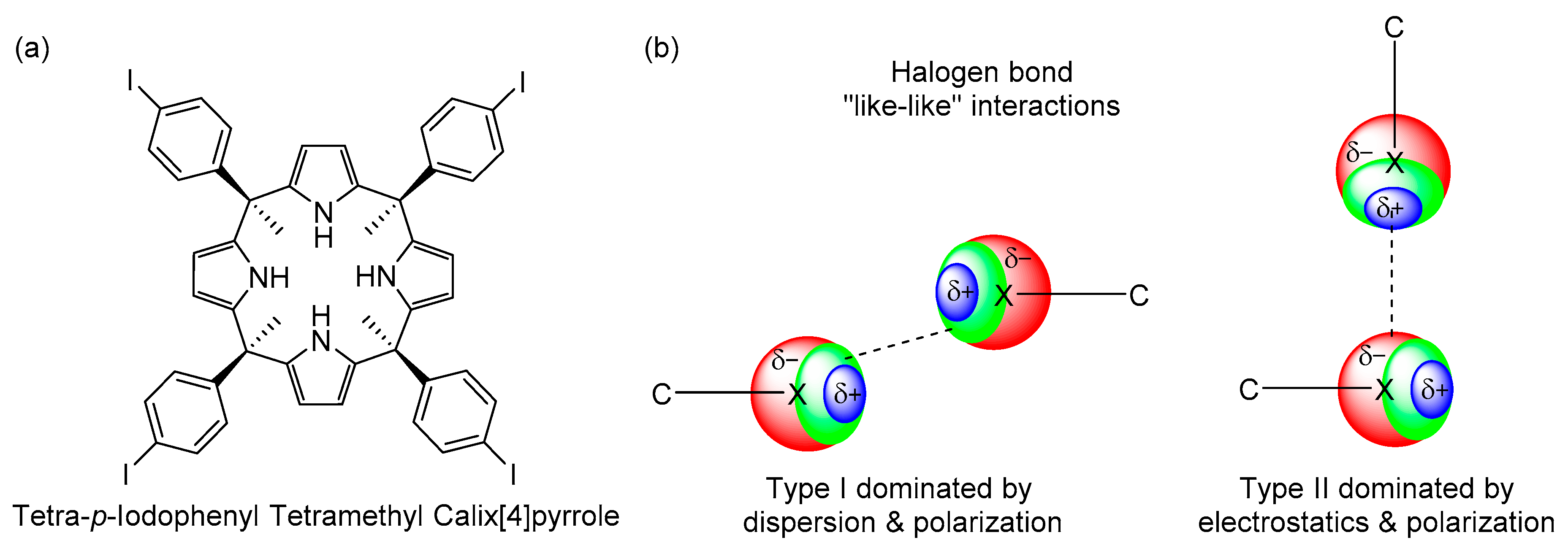
2. Results and Discussion
2.1. Synthesis
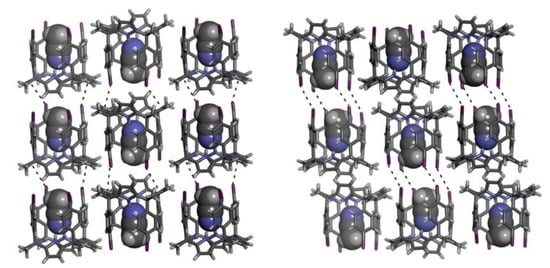
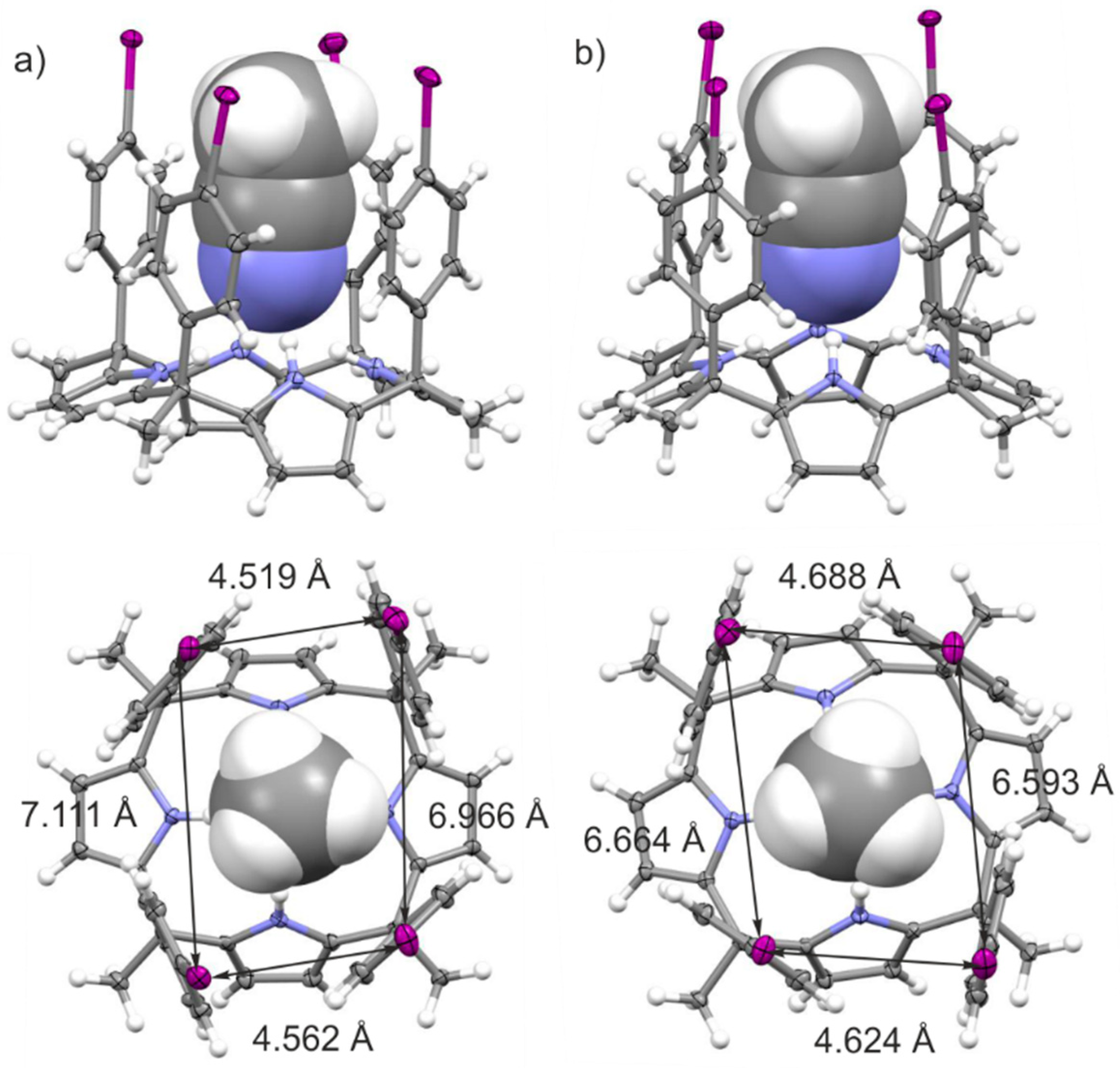
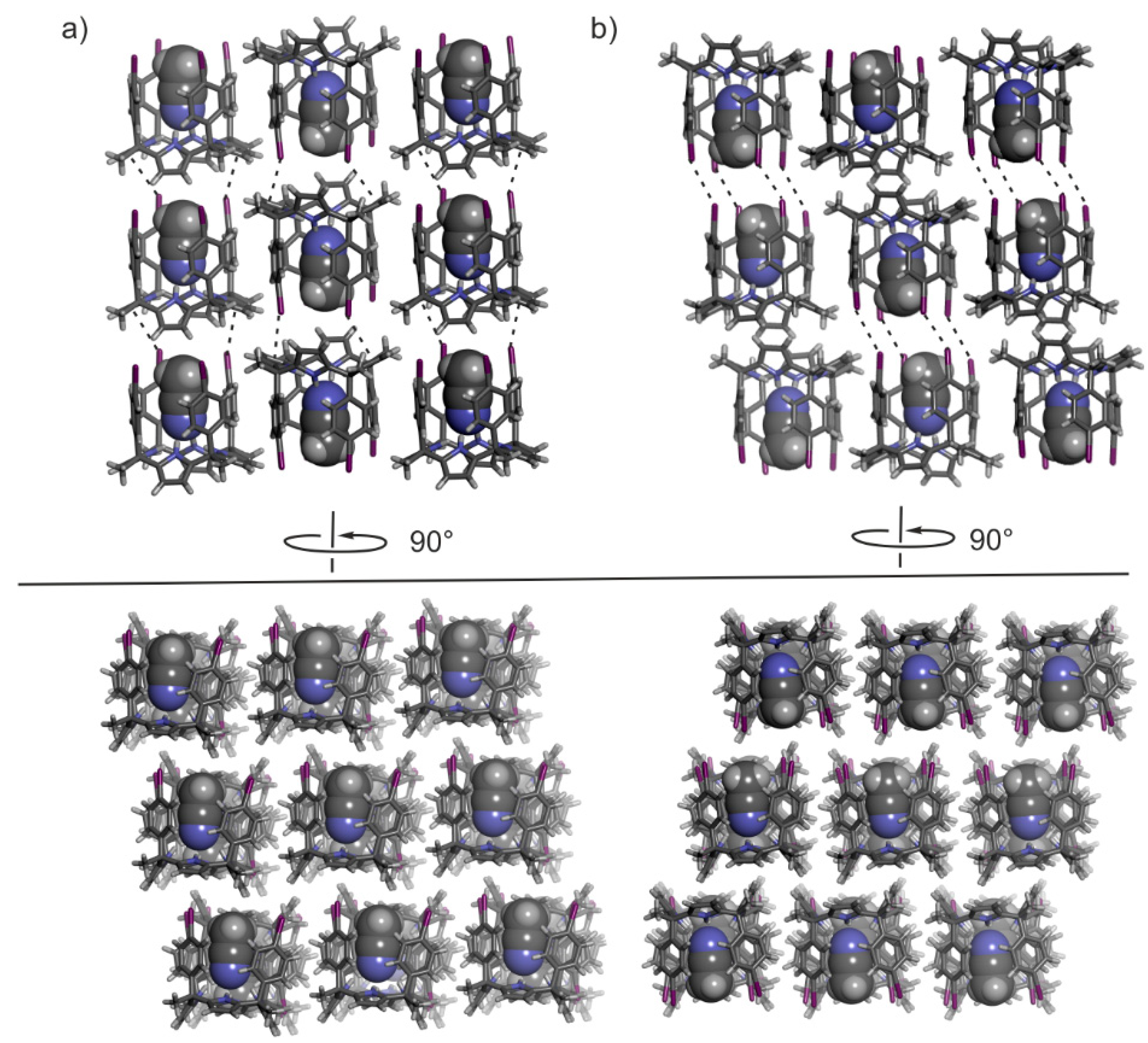
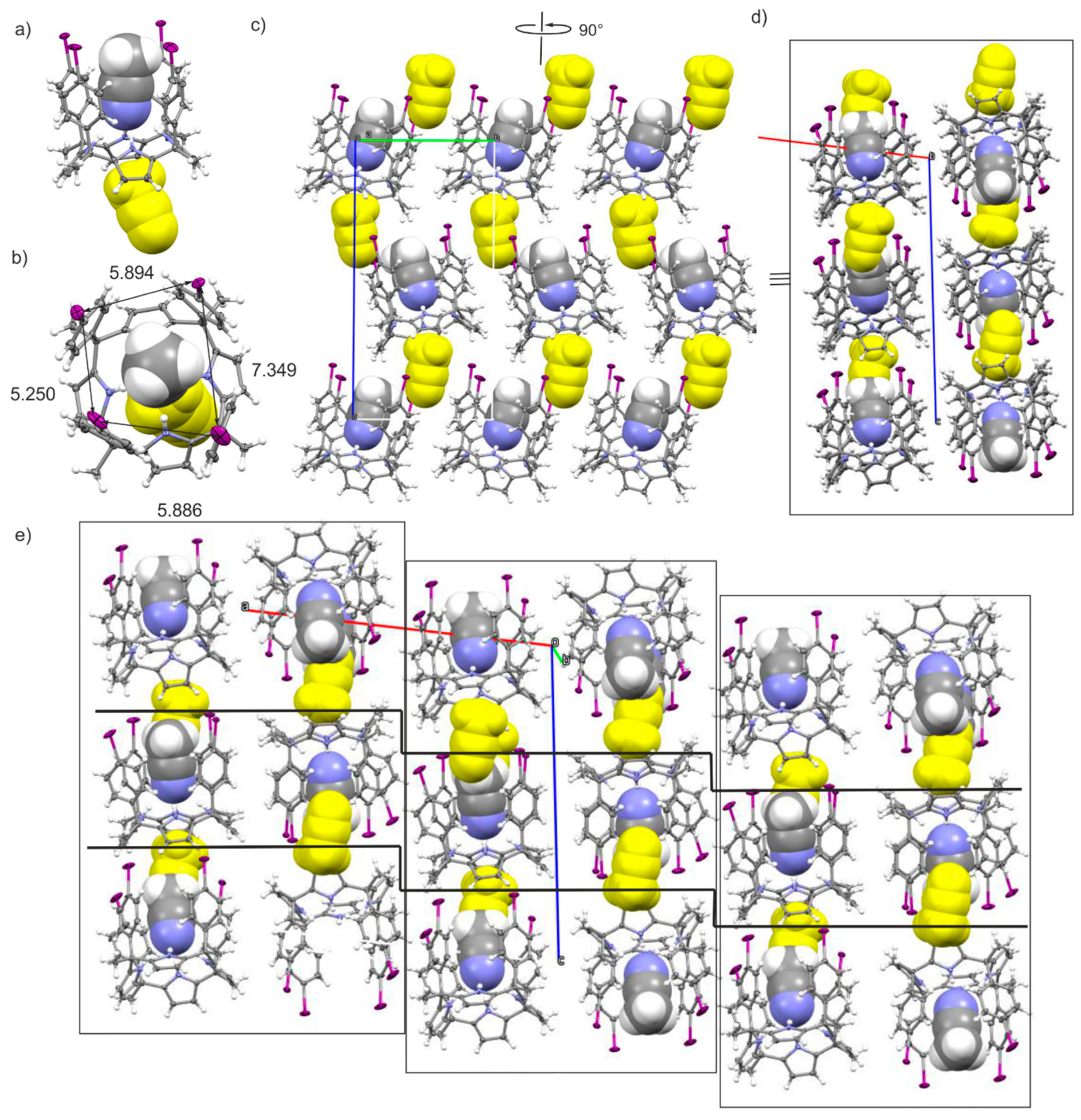
2.2. Structural Description of the Packing in the Single Crystals
2.3. Theoretical Study
2.3.1. Lattice Energies
2.3.2. MEP Surface Analysis
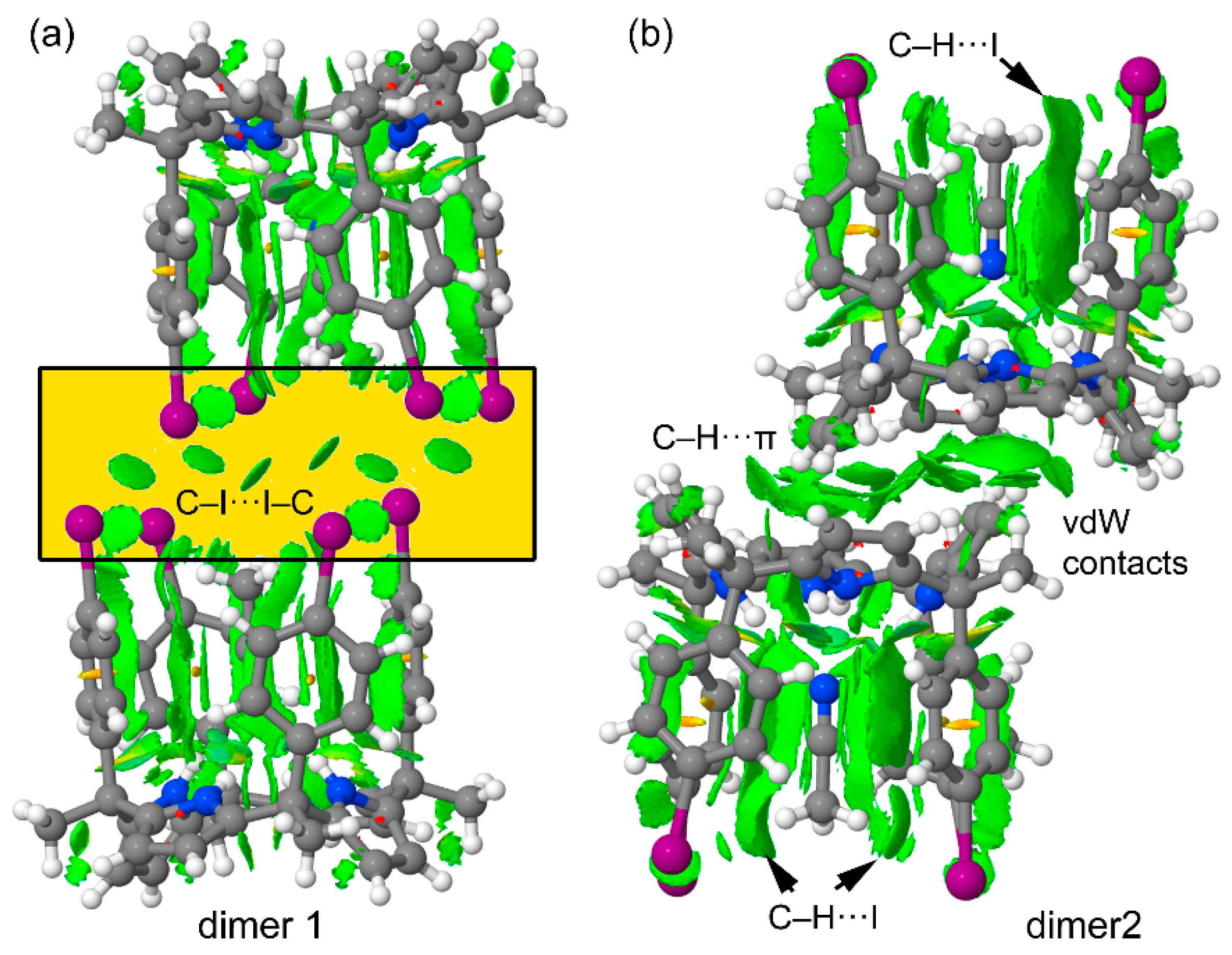
2.3.3. Energetic and Noncovalent Interaction Plot (NCIPLOT) Index Analyses
3. Materials and Methods
3.1. Materials and Techniques
3.2. Crystalization of Polymorphs A and B
3.3. Crystallographic Data Collection and Refinements
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shah, H.; Bhatt, K.D. Review on Calix[4]Pyrrole: A versatile receptor. Adv. Org. Chem. Lett. 2019, 6, 1–12. [Google Scholar]
- Kim, D.S.; Sessler, J.L. Calix[4]pyrroles: Versatile molecular containers with ion transport, recognition, and molecular switching functions. Chem. Soc. Rev. 2015, 44, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Park, I.W.; Yoo, J.; Adhikari, S.; Park, J.S.; Sessler, J.L.; Lee, C.-H. Calix[4]pyrrole-Based heteroditopic Ion-Pair receptor that displays Anion-Modulated, Cation-Binding behavior. Chem. Eur. J. 2012, 47, 15073–15078. [Google Scholar]
- Martinez-Crespo, L.; Sun-Wang, J.L.; Ferreira, P.; Mirabella, C.F.M.; Aragay, G.; Ballester, P. Influence of the insertion method of aryl-extended Calix[4]pyrroles into liposomal membranes on their properties as anion carriers. Chem. Eur. J. 2019, 25, 4775–4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adriaenssens, L.; Gil-Ramírez, G.; Frontera, A.; Quiñonero, D.; Eduardo, C.; Escudero-Adán, C.; Ballester, P. Thermodynamic characterization of Halide−π interactions in solution using “Two-Wall” aryl extended Calix[4]pyrroles as model system. J. Am. Chem. Soc. 2014, 136, 3208–3218. [Google Scholar] [CrossRef]
- Adriaenssens, L.; Estarellas, C.; Vargas-Jentzsch, A.; Martinez Belmonte, M.; Matile, S.; Ballester, P. Quantification of Nitrate−π Interactions and Selective Transport of Nitrate Using Calix[4]pyrroles with Two Aromatic Walls. J. Am. Chem. Soc. 2013, 135, 8324–8330. [Google Scholar]
- Lynch, V.M.; Gale, P.A.; Sessler, J.L.; Madeiros, D. Room-temperature monoclinic and low-temperature triclinic phase-transition structures of meso-octa-methylcalix[4]pyrrole±dimethyl sulfoxide (1/1). Acta Cryst. 2001, C57, 1426–1428. [Google Scholar]
- Mahanta, S.P.; Kumara, B.S.; Panda, P.K. Meso-diacylated calix[4]pyrrole: Structural diversities and enhanced binding towards dihydrogenphosphate ion. Chem. Commun. 2011, 47, 4496–4498. [Google Scholar] [CrossRef]
- He, Q.; Zhang, Z.; Brewster, J.T.; Lynch, V.M.; Kim, S.K.; Sessler, J.L. Hemispherand-Strapped Calix[4]pyrrole: An Ion-pair Receptor for the Recognition and Extraction of Lithium Nitrite. J. Am. Chem. Soc. 2016, 138, 9779–9782. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Proserpio, D.M.; Resnati, G. Dendrimeric Tectons in Halogen Bonding-Based Crystal Engineering. Cryst. Growth Des. 2008, 8, 654–659. [Google Scholar] [CrossRef]
- Yamamoto, H.M.; Maeda, R.; Yamaura, J.-I.; Kato, R. Structural and Physical Properties of Conducting Cation Radical Salts Containing Supramolecular Assemblies Based on p-Bis(iodoethynyl)benzene Derivatives. Annu. Rev. Synth. Met. 2001, 11, 101–102. [Google Scholar]
- Pang, X.; Zhao, X.R.; Wang, H.; Sun, H.L.; Jin, W.J. Modulating Crystal Packing and Magnetic Properties of Nitroxide Free Radicals by Halogen Bonding. Cryst. Growth Des. 2013, 13, 3739–3745. [Google Scholar] [CrossRef]
- He, W.; Ge, Y.C.; Tan, C.H. Halogen-Bonding-Induced Hydrogen Transfer to C═N Bond with Hantzsch Ester. Org. Lett. 2014, 16, 3244–3247. [Google Scholar] [CrossRef] [PubMed]
- Galan, A.; Aragay, G.; Ballester, P. A chiral “Siamese-Twin” calix[4]pyrrole tetramer. Chem. Sci. 2016, 7, 5976–5982. [Google Scholar] [CrossRef] [Green Version]
- Escobar, L.; Aragay, G.; Ballester, P. Super Aryl-Extended Calix[4]pyrroles: Synthesis, Binding Studies, and Attempts To Gain Water Solubility. Chem. Eur. J. 2016, 22, 13682–13689. [Google Scholar] [CrossRef]
- Escobar, L.; Villaron, D.; Escudero-Adan, E.C.; Ballester, P. A mono-metallic Pd(II)-cage featuring two different polar binding sites. Chem. Commun. 2019, 55, 604–607. [Google Scholar] [CrossRef]
- Perger, W.F.; Pandey, R.; Blanco, M.A.; Zhao, J.J. First-principles intermolecular binding energies in organic molecular crystals. Chem. Phys. Lett. 2004, 388, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theor. Comp. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A dispersion correction for density functionals, Hartree-Fock and semi-empirical quantum chemical methods DFT-D3. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bär, M.; Hacer, M.; Horn, H.; Kömel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grillo, M.E.; Andzelm, J.W.; Govind, N.; Fitzgerald, G.; Stark, K.B. Computational materials science with materials studio: Applications in catalysis. Lect. Notes Phys. 2004, 642, 214–220. [Google Scholar]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
Sample Availablity: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Percentage ACN in chloroform | Polymorph A (%) | Polymorph B (%) |
|---|---|---|
| 0.7% | 88 | 12 |
| 1% | 35 | 65 |
| 1.3% | 0 | 100 |
| Crystal | A | B | C |
|---|---|---|---|
| Empirical Formula | C50H43I4N5 | C50H43I4N5 | C52H46I4N6 |
| Formula weight | 1221.49 | 1221.49 | 1262.55 |
| Crystal system | Triclinic | Triclinic | Monoclinic |
| Space group | P ī | P ī | P21/c |
| a/Å | 10.66587(19) | 10.95660(10) | 21.4451(9) |
| b/Å | 11.1800(2) | 14.07090(10) | 10.8483(4) |
| c/Å | 19. 7496(3) | 15.34390(10) | 21.8771(6) |
| α/° | 76.8217(14) | 85.1190(10) | 90 |
| β/° | 88. 7021(14) | 89.8210(10) | 98.339(3) |
| γ/° | 80.1011(15) | 71.2270(10) | 90 |
| V/Å3 | 2258.53(27) | 2230.85(3) | 5035.8(3) |
| Z | 2 | 2 | 4 |
| Radiation type | Mo Kα | Mo Kα | Mo Kα |
| μ/mm−1 | 2.800 | 2.877 | 2.515 |
| Temperature/K | 100(2) | 100(2) | 100(2) |
| Crystal size/mm | 0.10 × 0.05 × 0.05 | 0.20 × 0.20 × 0.12 | 0.11 × 0.05 × 0.04 |
| Dcalc/g·cm−3 | 1.796 | 1.818 | 1.665 |
| Reflections collected | 64,252 | 113,257 | 76,454 |
| Independent Reflections | 11,647 [R(int) = 0.0637] | 12,126 [R(int) = 0.0241] | 10,380 [R(int) = 0.0503] |
| Completeness to theta = 25.242° | 99.9 % | 99.5 % | 99.9 % |
| F(000) | 1180 | 1180 | 2448 |
| Data/restraints/parameters | 11,647/0/537 | 12,126/0/537 | 10,838/0/537 |
| Goodness-of-fit | 1.013 | 1.396 | 1.396 |
| Final R indices [I < 2d(I)] | R1 = 0.0385, wR2 = 0.1066 | R1 = 0.0212, wR2 = 0.0503 | R1 = 0.0439, wR2 = 0.1222 |
| R indices (all data) | R1 = 0.0448, wR2 = 0.1107 | R1 = 0.0242, wR2 = 0.0513 | R1 = 0.0540, wR2 = 0.1259 |
| Largest diff. peak and hole/e·Å−3 | 1.828 and −1.097 | 0.872 and −0.645 | 1.217 and −1.483 |
| CCDC nº | 1971260 | 1971261 | 1971262 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dăbuleanu, D.; Bauzá, A.; Ortega-Castro, J.; Escudero-Adán, E.C.; Ballester, P.; Frontera, A. Synthesis, X-ray Characterization and Density Functional Theory (DFT) Studies of Two Polymorphs of the α,α,α,α, Isomer of Tetra-p-Iodophenyl Tetramethyl Calix[4]pyrrole: On the Importance of Halogen Bonds. Molecules 2020, 25, 285. https://doi.org/10.3390/molecules25020285
Dăbuleanu D, Bauzá A, Ortega-Castro J, Escudero-Adán EC, Ballester P, Frontera A. Synthesis, X-ray Characterization and Density Functional Theory (DFT) Studies of Two Polymorphs of the α,α,α,α, Isomer of Tetra-p-Iodophenyl Tetramethyl Calix[4]pyrrole: On the Importance of Halogen Bonds. Molecules. 2020; 25(2):285. https://doi.org/10.3390/molecules25020285
Chicago/Turabian StyleDăbuleanu, Dragoș, Antonio Bauzá, Joaquín Ortega-Castro, Eduardo C. Escudero-Adán, Pablo Ballester, and Antonio Frontera. 2020. "Synthesis, X-ray Characterization and Density Functional Theory (DFT) Studies of Two Polymorphs of the α,α,α,α, Isomer of Tetra-p-Iodophenyl Tetramethyl Calix[4]pyrrole: On the Importance of Halogen Bonds" Molecules 25, no. 2: 285. https://doi.org/10.3390/molecules25020285