
Phenylalanine Stereoisomers of CJ-15,208 and [d-Trp]CJ-15,208 Exhibit Distinctly Different Opioid Activity Profiles

 , , , and
, , , and
Abstract
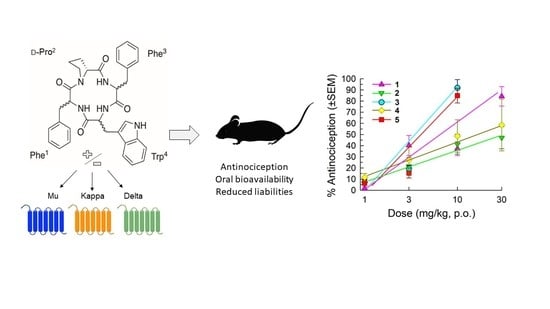
:
1. Introduction
2. Results
2.1. Synthesis
2.2. Metabolic Stability
2.3. In Vitro Pharmacological Evaluation
2.4. In Vivo Pharmacological Evaluation
2.4.1. Opioid Receptor Selectivity of Stereoisomer Antinociception
2.4.2. Determination of Stereoisomer Opioid-Receptor Mediated Selective Antagonist Activity
2.5. In Vivo Assessment of Opioid Related Liabilities
2.5.1. Assessment of Effects on Coordinated Locomotor Activity
2.5.2. Evaluation of Respiratory and Spontaneous Locomotor Effects
2.5.3. Assessment of Acute Antinociceptive Tolerance Development
2.5.4. Evaluation of Potential Reinforcing or Aversive Properties
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Instruments
4.3. Peptide Synthesis and Purification
4.3.1. [d-Phe1]CJ-15,208 (1)
4.3.2. [d-Phe3]CJ-15,208 (2)
4.3.3. [d-Phe1,3]CJ-15,208 (3)
4.3.4. [d-Phe1,d-Trp4]CJ-15,208 (4)
4.3.5. [d-Phe3,d-Trp4]CJ-15,208 (5)
4.4. Metabolism by Mouse Liver Microsomes
4.5. In Vitro Pharmacological Evaluation
4.6. In Vivo Testing
4.6.1. Animals and Drug Administration
4.6.2. Antinociceptive Testing
4.6.3. Acute Antinociceptive Tolerance Determination
4.6.4. Coordinated Locomotor Activity
4.6.5. Respiration and Ambulation
4.6.6. Evaluation of Potential Conditioned Place Preference and Conditioned Place Aversion
4.7. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and Exogenous Opioids in Pain. Annu. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; McLellan, A.T. Opioid Abuse in Chronic Pain—Misconceptions and Mitigation Strategies. N. Engl. J. Med. 2016, 374, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, A.; Brantl, V.; Herz, A.; Emrich, H.M. Psychotomimesis mediated by κ opiate receptors. Science 1986, 233, 774–776. [Google Scholar] [CrossRef] [PubMed]
- Jutkiewicz, E.M.; Baladi, M.G.; Folk, J.E.; Rice, K.C.; Woods, J.H. The Convulsive and Electroencephalographic Changes Produced by Nonpeptidic δ-Opioid Agonists in Rats: Comparison with Pentylenetetrazol. J. Pharmacol. Exp. Ther. 2006, 317, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Schiller, P.W. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010, 86, 598–603. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Husbands, S.M. Effects of atypical kappa-opioid receptor agonists on intrathecal morphine-induced itch and analgesia in primates. J. Pharmacol. Exp. Ther. 2008, 328, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negus, S.S.; Schrode, K.; Stevenson, G.W.; Schrode, K.K. Mu/kappa opioid interactions in rhesus monkeys: Implications for analgesia and abuse liability. Exp. Clin. Psychopharmacol. 2008, 16, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, G.W.; Folk, J.E.; Linsenmayer, D.C.; Rice, K.C.; Negus, S.S.; Litman, T.; Skovsgaard, T.; Stein, W.D. Opioid Interactions in Rhesus Monkeys: Effects of δ + μ and δ + κ Agonists on Schedule-Controlled Responding and Thermal Nociception. J. Pharmacol. Exp. Ther. 2003, 307, 1054–1064. [Google Scholar] [CrossRef]
- Brice-Tutt, A.C.; Wilson, L.L.; Eans, S.O.; Stacy, H.M.; Simons, C.A.; Simpson, G.G.; Coleman, J.S.; Ferracane, M.J.; Aldrich, J.V.; McLaughlin, J.P. Multifunctional opioid receptor agonism and antagonism by a novel macrocyclic tetrapeptide prevents reinstatement of morphine-seeking behaviour. Br. J. Pharmacol. 2020, 177, 4209–4222. [Google Scholar] [CrossRef]
- Anand, J.P.; Montgomery, D. Multifunctional Opioid Ligands. Handb. Exp. Pharmacol. 2018, 247, 21–51. [Google Scholar] [CrossRef]
- Bolaños, C.A.; Garmsen, G.M.; Clair, M.A.; McDougall, S.A. Effects of the κ-opioid receptor agonist U-50,488 on morphine-induced place preference conditioning in the developing rat. Eur. J. Pharmacol. 1996, 317, 1–8. [Google Scholar] [CrossRef]
- Dosaka-Akita, K.; Tortella, F.C.; Holaday, J.W.; Long, J.B. The Kappa Opioid Agonist U-50, 488H Antagonizes Respiratory Effects of Mu Opioid Receptor Agonists in Conscious. Rats 1993, 264, 631–637. [Google Scholar] [CrossRef]
- Hepburn, M.J.; Little, P.J.; Gingras, J.; Kuhn, C.M. Differential effects of naltrindole on morphine-induced tolerance and physical dependence in rats. J. Pharmacol. Exp. Ther. 1997, 281, 1350–1356. [Google Scholar]
- Daniels, D.J.; Kulkarni, A.; Xie, Z.; Bhushan, R.G.; Portoghese, P.S. A bivalent ligand (KDAN-18) containing delta-antagonist and kappa-agonist pharmacophores bridges delta2 and kappa1 opioid receptor phenotypes. J. Med. Chem. 2005, 48, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hirai, H.; Kim, Y.-J.; Kojima, Y.; Matsunaga, Y.; Nishida, H.; Sakakibara, T.; Suga, O.; Sujaku, T.; Kojima, N. CJ-15,208, a novel kappa opioid receptor antagonist from a fungus, Ctenomyces serratus ATCC15502. J. Antibiot. 2002, 55, 847–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, S.S.; Ross, N.C.; McLaughlin, J.P.; Aldrich, J.V. Synthesis of cyclic tetrapeptide CJ 15,208: A novel kappa opioid receptor antagonist. Adv. Exp. Med. Biol. 2009, 611, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Ross, N.C.; Kulkarni, S.S.; McLaughlin, J.P.; Aldrich, J.V. Synthesis of CJ-15,208, a novel κ-opioid receptor antagonist. Tetrahedron Lett. 2010, 51, 5020–5023. [Google Scholar] [CrossRef] [Green Version]
- Dolle, R.E.; Michaut, M.; Martinez-Teipel, B.; Seida, P.R.; Ajello, C.W.; Muller, A.L.; DeHaven, R.N.; Carroll, P.J. Nascent structure–activity relationship study of a diastereomeric series of kappa opioid receptor antagonists derived from CJ-15,208. Bioorg. Med. Chem. Lett. 2009, 19, 3647–3650. [Google Scholar] [CrossRef]
- Ross, N.C.; Reilley, K.J.; Murray, T.F.; Aldrich, J.V.; McLaughlin, J.P. Novel opioid cyclic tetrapeptides: Trp isomers of CJ-15,208 exhibit distinct opioid receptor agonism and short-acting κ opioid receptor antagonism. Br. J. Pharmacol. 2012, 165, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Aldrich, J.V.; Senadheera, S.N.; Ross, N.C.; Ganno, M.L.; Eans, S.O.; McLaughlin, J.P. The Macrocyclic Peptide Natural Product CJ-15,208 Is Orally Active and Prevents Reinstatement of Extinguished Cocaine-Seeking Behavior. J. Nat. Prod. 2013, 76, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Eans, S.; Ganno, M.L.; Reilley, K.J.; A Patkar, K.; Senadheera, S.N.; Aldrich, J.V.; McLaughlin, J.P. The macrocyclic tetrapeptide [d-Trp]CJ-15,208 produces short-acting κ opioid receptor antagonism in the CNS after oral administration. Br. J. Pharmacol. 2013, 169, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senadheera, S.N.; Kulkarni, S.S.; McLaughlin, J.P.; Aldrich, J.V. Improved Synthesis of CJ-15,208 Isomers and Their Pharmacological Activity at Opioid Receptors. In Peptides: Building Bridges; Lebl, M., Ed.; American Peptide Society: San Diego, CA, USA, 2011; pp. 346–347. [Google Scholar]
- Christians, U.; Sewing, K.-F. Cyclosporin metabolism in transplant patients. Pharmacol. Ther. 1993, 57, 291–345. [Google Scholar] [CrossRef]
- Delaforge, M.; André, F.; Jaouen, M.; Dolgos, H.; Benech, H.; Gomis, J.-M.; Noel, J.-P.; Cavelier, F.; Verducci, J.; Aubagnac, J.-L.; et al. Metabolism of Tentoxin by Hepatic Cytochrome P-450 3A Isozymes. JBIC J. Boil. Inorg. Chem. 1997, 250, 150–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoot, M.R.; Sypek, E.I.; Reilley, K.J.; Carey, A.N.; Bidlack, J.M.; McLaughlin, J.P. Inhibition of G??-subunit signaling potentiates morphine-induced antinociception but not respiratory depression, constipation, locomotion, and reward. Behav. Pharmacol. 2013, 24, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, J.V.; Kulkarni, S.S.; Senadheera, S.N.; Ross, N.C.; Reilley, K.J.; Eans, S.O.; Ganno, M.L.; Murray, T.F.; McLaughlin, J.P. Unexpected Opioid Activity Profiles of Analogues of the Novel Peptide Kappa Opioid Receptor Ligand CJ-15,208. ChemMedChem 2011, 6, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, J.V.; Senadheera, S.N.; Ross, N.C.; A Reilley, K.; Ganno, M.L.; E Eans, S.; Murray, T.F.; McLaughlin, J.P. Alanine analogues of [d-Trp]CJ-15,208: Novel opioid activity profiles and prevention of drug- and stress-induced reinstatement of cocaine-seeking behaviour. Br. J. Pharmacol. 2014, 171, 3212–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferracane, M.J.; Brice-Tutt, A.C.; Coleman, J.S.; Simpson, G.G.; Wilson, L.L.; Eans, S.O.; Stacy, H.M.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Design, Synthesis, and Characterization of the Macrocyclic Tetrapeptide cyclo[Pro-Sar-Phe-d-Phe]: A Mixed Opioid Receptor Agonist–Antagonist Following Oral Administration. ACS Chem. Neurosci. 2020, 11, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, G.W.; Luginbuhl, A.; Dunbar, C.; Lavigne, J.; Dutra, J.; Atherton, P.; Bell, B.; Cone, K.; Giuvelis, D.; Polt, R.; et al. The mixed-action delta/mu opioid agonist MMP-2200 does not produce conditioned place preference but does maintain drug self-administration in rats, and induces in vitro markers of tolerance and dependence. Pharmacol. Biochem. Behav. 2015, 132, 49–55. [Google Scholar] [CrossRef]
- Lenard, N.R.; Daniels, D.J.; Portoghese, P.S.; Roerig, S.C. Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and delta-opioid receptor antagonist pharmacophores. Eur. J. Pharmacol. 2007, 566, 75–82. [Google Scholar] [CrossRef]
- Anand, J.P.; E Kochan, K.; Nastase, A.F.; Montgomery, D.; Griggs, N.W.; Traynor, J.R.; I Mosberg, H.; Jutkiewicz, E.M. In vivo effects of μ-opioid receptor agonist/δ-opioid receptor antagonist peptidomimetics following acute and repeated administration. Br. J. Pharmacol. 2018, 175, 2013–2027. [Google Scholar] [CrossRef] [Green Version]
- Khaliq, T.; Williams, T.D.; Senadheera, S.N.; Aldrich, J.V. Development of a robust, sensitive and selective liquid chromatography-tandem mass spectrometry assay for the quantification of the novel macrocyclic peptide kappa opioid receptor antagonist [d-Trp]CJ-15,208 in plasma and application to an initial pharmacokinetic study. J. Chromatogr. B 2016, 1028, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Arttamangkul, S.; Ishmael, J.E.; Murray, T.F.; Grandy, D.K.; DeLander, G.E.; Kieffer, B.L.; Aldrich, J.V. Synthesis and Opioid Activity of Conformationally Constrained Dynorphin A Analogues. 2.1Conformational Constraint in the “Address” Sequence. J. Med. Chem. 1997, 40, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Siebenaller, J.F.; Murray, T.F. Hydrostatic Pressure Alters the Time Course of GTP[S] Binding to G Proteins in Brain Membranes from Two Congeneric Marine Fishes. Boil. Bull. 1999, 197, 388–394. [Google Scholar] [CrossRef]
- McGrath, J.; Drummond, G.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, J.P.; Hill, K.P.; Jiang, Q.; Sebastian, A.; Archer, S.; Bidlack, J.M. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: In vivo and in vitro characterization of mu-selective agonist and antagonist activity. J. Pharmacol. Exp. Ther. 1999, 289, 304–311. [Google Scholar]
- Jiang, Q.; Seyed-Mozaffari, A.; Sebastian, A.; Archer, S.; Bidlack, J.M. Preventing morphine antinociceptive tolerance by irreversible mu opioid antagonists before the onset of their antagonism. J. Pharmacol. Exp. Ther. 1995, 273, 680–688. [Google Scholar] [CrossRef]
- Mathews, J.L.; Smrcka, A.V.; Bidlack, J.M. A novel Gbetagamma-subunit inhibitor selectively modulates mu-opioid-dependent antinociception and attenuates acute morphine-induced antinociceptive tolerance and dependence. J. Neurosci. 2008, 28, 12183–12189. [Google Scholar] [CrossRef] [Green Version]
- Way, E.L.; Loh, H.H.; Shen, F.H. Simultaneous quantitative assessment of morphine tolerance and physical dependence. J. Pharmacol. Exp. Ther. 1969, 167, 1–8. [Google Scholar]
- Spetea, M.; O Eans, S.; Ganno, M.L.; Lantero, A.; Mairegger, M.; Toll, L.; Schmidhammer, H.; McLaughlin, J.P. Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br. J. Pharmacol. 2017, 174, 2444–2456. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds cyclo[d-Phe-d-Pro-Phe-Trp] (1), cyclo[Phe-d-Pro- d-Phe-Trp] (2), cyclo[d-Phe-d-Pro- d-Phe-Trp] (3), cyclo[d-Phe-d-Pro-Phe-d-Trp] (4) and cyclo[Phe-d-Pro- d-Phe-d-Trp] (5) are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stereoisomer | Ki (nM ± SEM) | Selectivity | |
|---|---|---|---|
| KOR | MOR | Ki (MOR) Ki (KOR) | |
| 1 (d-Phe1) | 5120 ± 690 | >10,000 | <2 |
| 2 (d-Phe3) | 362 ± 51 | 3920 ± 200 | 11 |
| 3 (d-Phe1,3) | 2560 ± 480 | 7780 ± 410 | 3 |
| CJ-15,208 | 27.4 ± 4.6 | 451 ± 114 | 16.5 |
| 4 (d-Phe1, d-Trp4) | >10,000 | >10,000 | - |
| 5 (d-Phe3, d-Trp4) | 353 ± 19 | 5800 ± 1450 | 16 |
| [d-Trp4]CJ-15,208 | 21.8 ± 4.8 | 259 ± 29 | 12 |
| Stereoisomer | ED50 (and 95% Confidence Interval (C.I.)) Values | ||
|---|---|---|---|
| i.c.v. (nmol) | p.o. (mg/kg) | Receptors Involved | |
| 1 | 0.75 (0.36–1.44) | 7.62 (5.12–12.2) | KOR, MOR, DOR |
| 2 | 20.4 (10–58.7) | ~ | KOR, MOR |
| 3 | 1.00 (0.64–1.60) | 4.12 (3.30–5.31) | KOR, DOR |
| CJ-15,208 b,c | 1.74 (0.62–4.82) | 3.49 (1.98–5.73) | KOR, MOR |
| 4 | 2.39 (1.40–4.56) | ~ | DOR, KOR |
| 5 | 0.56 (0.38–0.91) | 4.72 (3.70–6.39) | KOR, MOR, DOR |
| [d-Trp4]CJ-15,208 b,d | ~ | ~ | - |
| Stereoisomer | Naïve ED50 (95% C.I.) | ED50 (95% C.I.) Pretreated Mice | Fold-Shift, Naïve ED50 vs. Second ED50 |
|---|---|---|---|
| 1 | 0.65 (0.31–1.31) | 20.3 * (11.1–41.7) | 31.2 |
| 2 | 20.4 (10–58.7) | 125 (89.9–224) | 6.13 |
| 3 | 1.00 (0.64–1.60) | 5.19 * (2.57–11) | 5.19 |
| 4 | 2.39 (1.40–4.56) | 1.28 (0.96–1.66) | 0.54 |
| 5 | 0.45 (0.28–0.70) | 0.49 (0.09–1.36) | 1.09 |
| CJ-15,208 | 1.82 (1.22–2.74) | 5.23 (4.23–6.48) | 2.87 |
| Morphine | 2.91 (2.48–3.41) | 22.2 * (14.0–55.3) | 7.63 |
| Observed | TLC | HPLC, t (min) | ||
|---|---|---|---|---|
| Isomer | ES-MS, m/z a | Rf b | System A c | System B d |
| 1 | 600.2568 | 0.70 | 18.4 | 23.0 |
| 2 | 600.2565 | 0.10 | 18.9 | – |
| 3 | 600.2578 | 0.67 | 22.4 | 22.6 |
| 4 | 600.2584 | 0.49 | 18.6 e | 21.9 e |
| 5 | 600.2605 | 0.53 | 21.1 | 23.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brice-Tutt, A.C.; Senadheera, S.N.; Ganno, M.L.; Eans, S.O.; Khaliq, T.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Phenylalanine Stereoisomers of CJ-15,208 and [d-Trp]CJ-15,208 Exhibit Distinctly Different Opioid Activity Profiles. Molecules 2020, 25, 3999. https://doi.org/10.3390/molecules25173999
Brice-Tutt AC, Senadheera SN, Ganno ML, Eans SO, Khaliq T, Murray TF, McLaughlin JP, Aldrich JV. Phenylalanine Stereoisomers of CJ-15,208 and [d-Trp]CJ-15,208 Exhibit Distinctly Different Opioid Activity Profiles. Molecules. 2020; 25(17):3999. https://doi.org/10.3390/molecules25173999
Chicago/Turabian StyleBrice-Tutt, Ariana C., Sanjeewa N. Senadheera, Michelle L. Ganno, Shainnel O. Eans, Tanvir Khaliq, Thomas F. Murray, Jay P. McLaughlin, and Jane V. Aldrich. 2020. "Phenylalanine Stereoisomers of CJ-15,208 and [d-Trp]CJ-15,208 Exhibit Distinctly Different Opioid Activity Profiles" Molecules 25, no. 17: 3999. https://doi.org/10.3390/molecules25173999