
Comparative Assessment of Protein Kinase Inhibitors in Public Databases and in PKIDB

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results
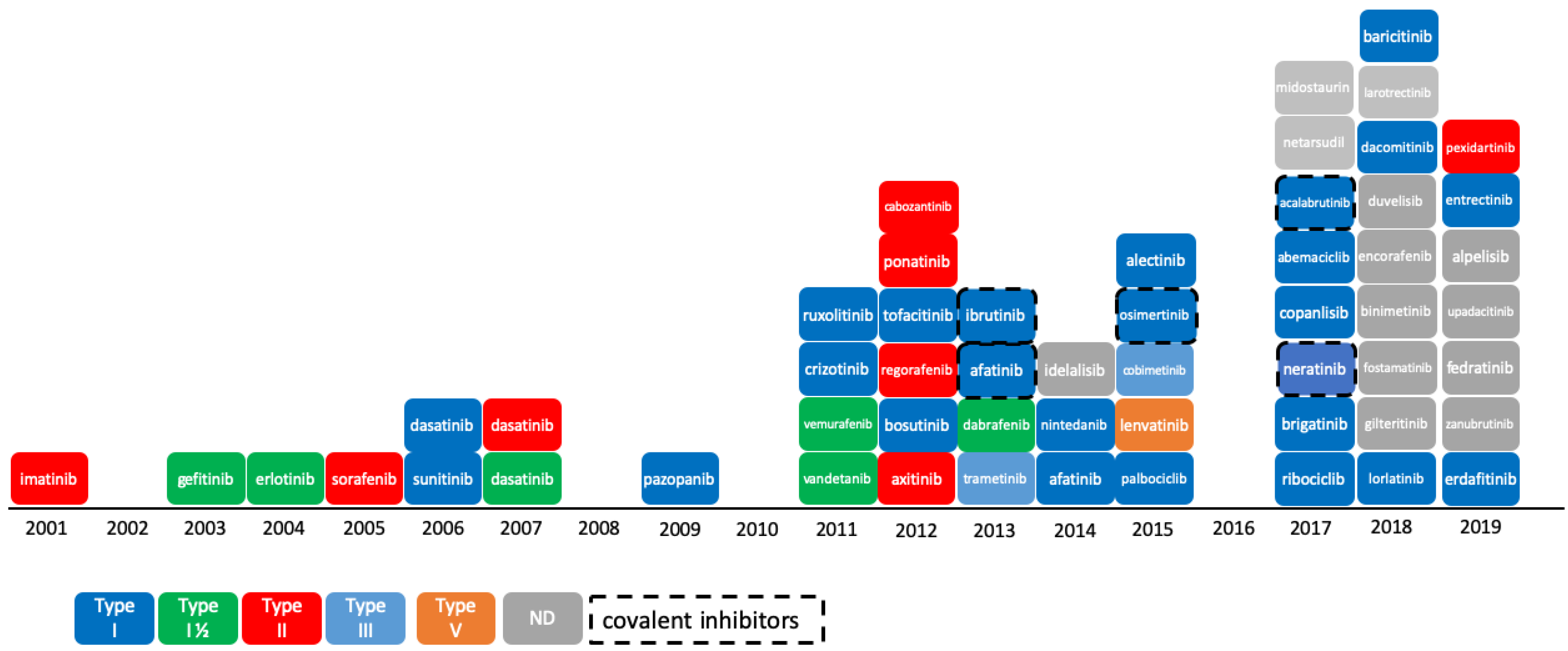
2.1. Update on PKIDB
2.2. Physicochemical Analysis of PKI Datasets
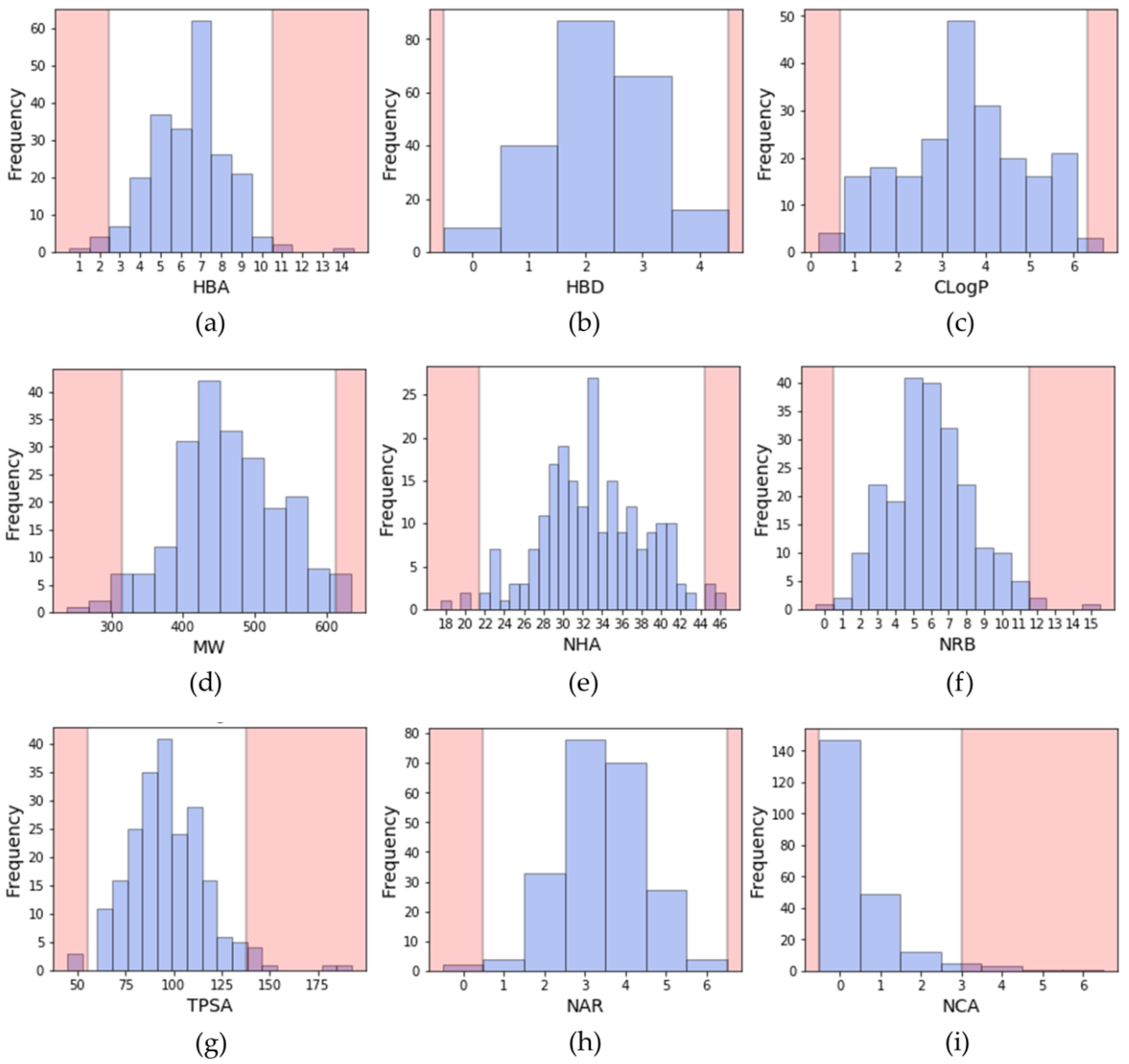
2.2.1. Distribution of Physicochemical Properties of PKIs
- A molecular weight (MW) between 314 and 613 Da (average of 463.4 Da);
- A ClogP (calculated with a Rational Discovery Kit (RDKit)) between 0.7 and 6.3 (average of 3.5);
- Between 0 and 4 hydrogen bond donors (HBD) (average of 2.2);
- Between 3 and 10 hydrogen bond acceptors (HBA) (average of 6.4);
- A topological polar surface area (TPSA) comprised between 55 and 138 Å2 (average of 96.6 Å2);
- Between 1 and 11 rotatable bonds (NRB) (average of 6.0);
- Number of aromatic rings (NAR) between 1 and 5 (average of 3.4);
- Number of chiral atoms (NCA) between 0 and 2 (average of 0.5).
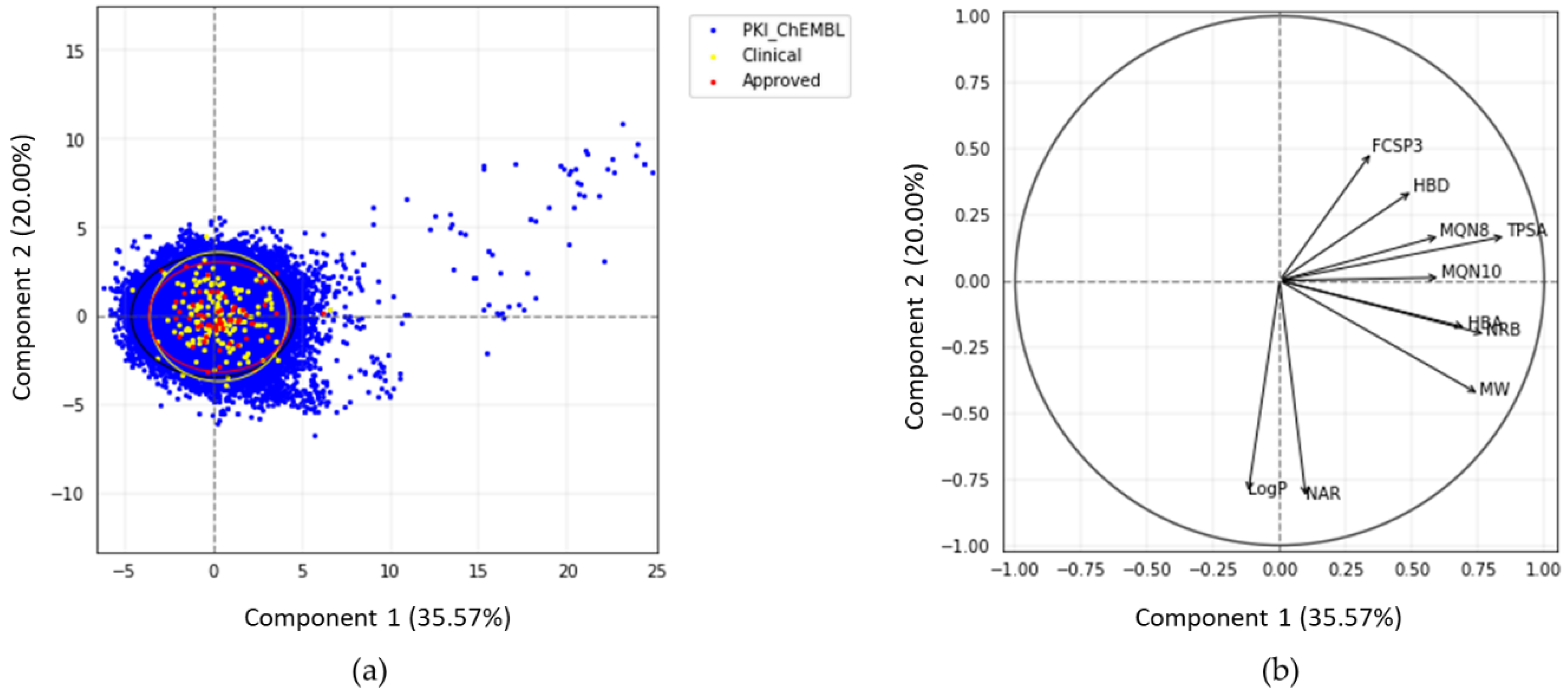
2.2.2. Statistical Analysis of Protein Kinase Inhibitors
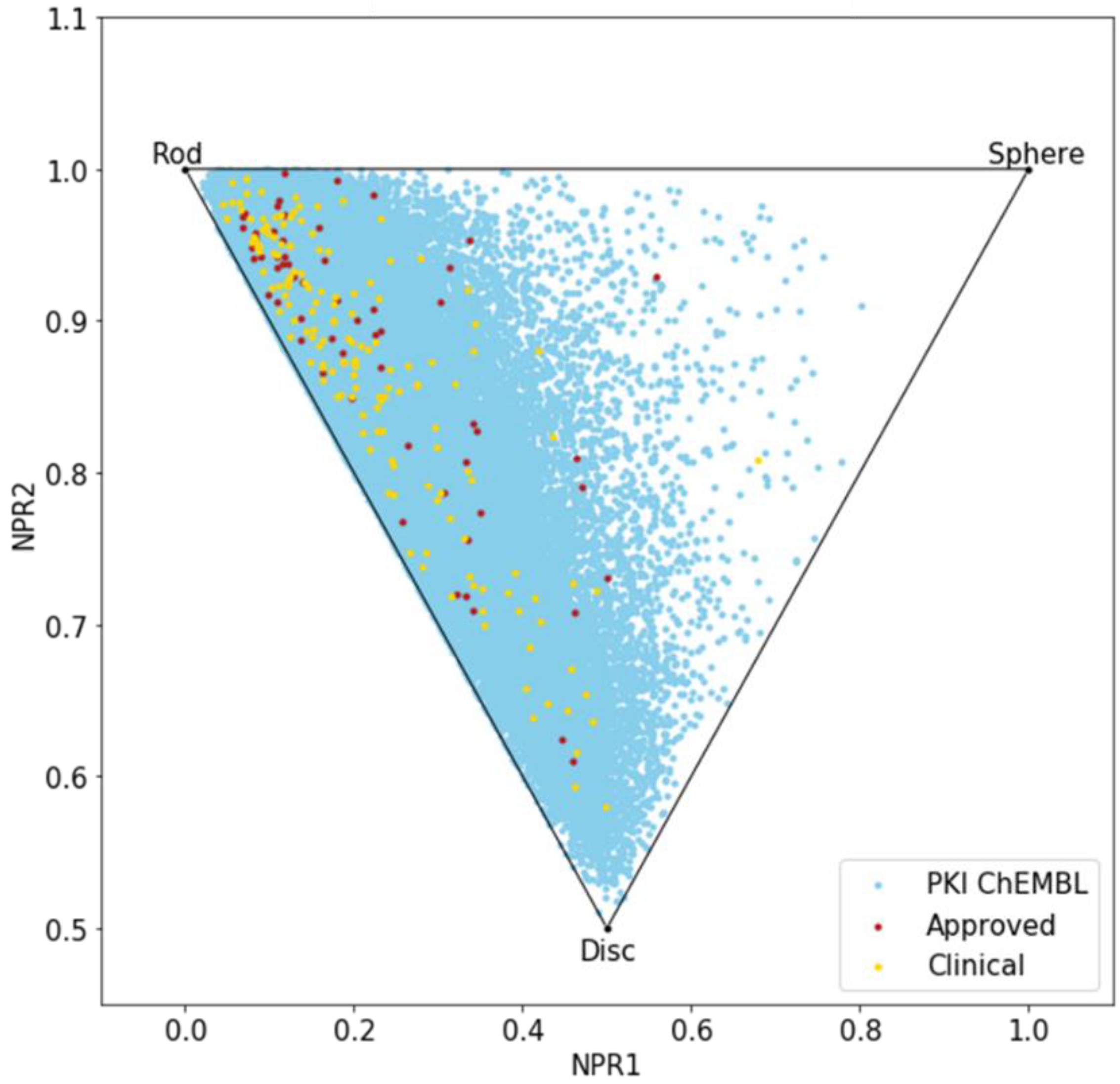
2.2.3. Principal Moments of Inertia
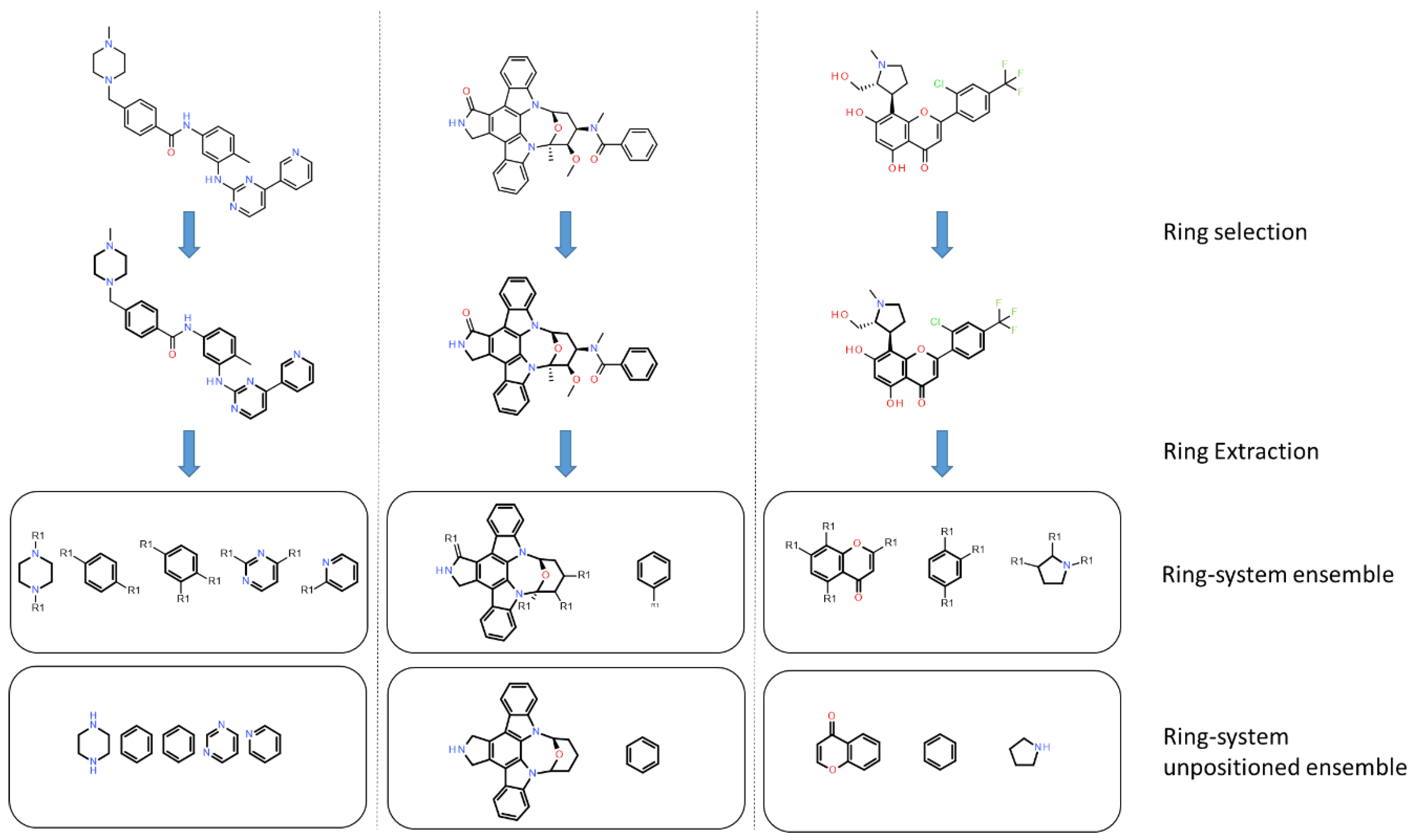
2.3. Scaffold Diversity Assessment
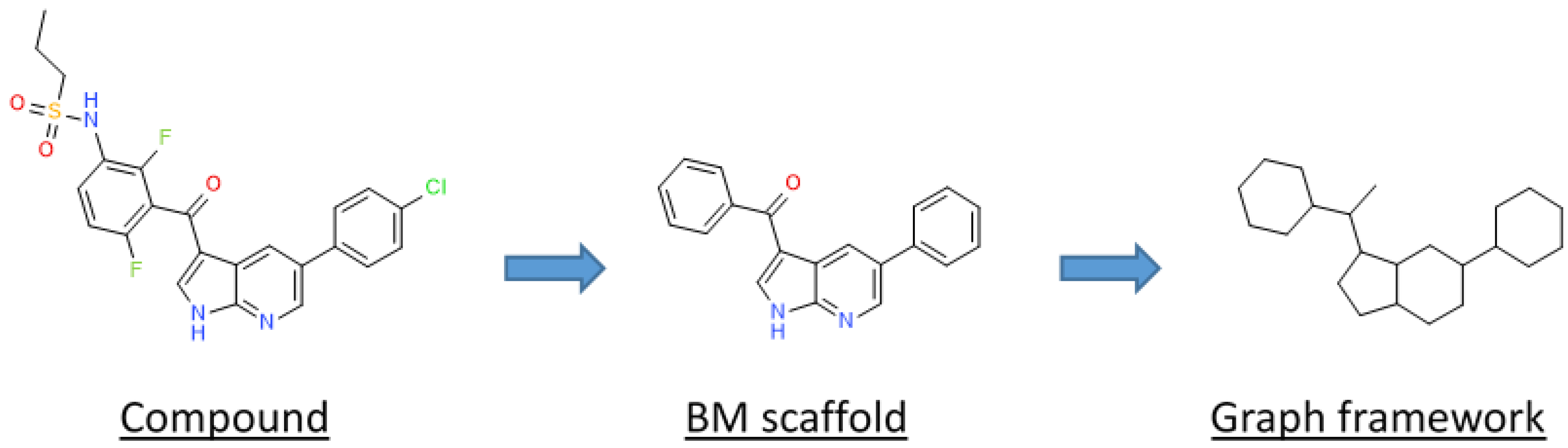
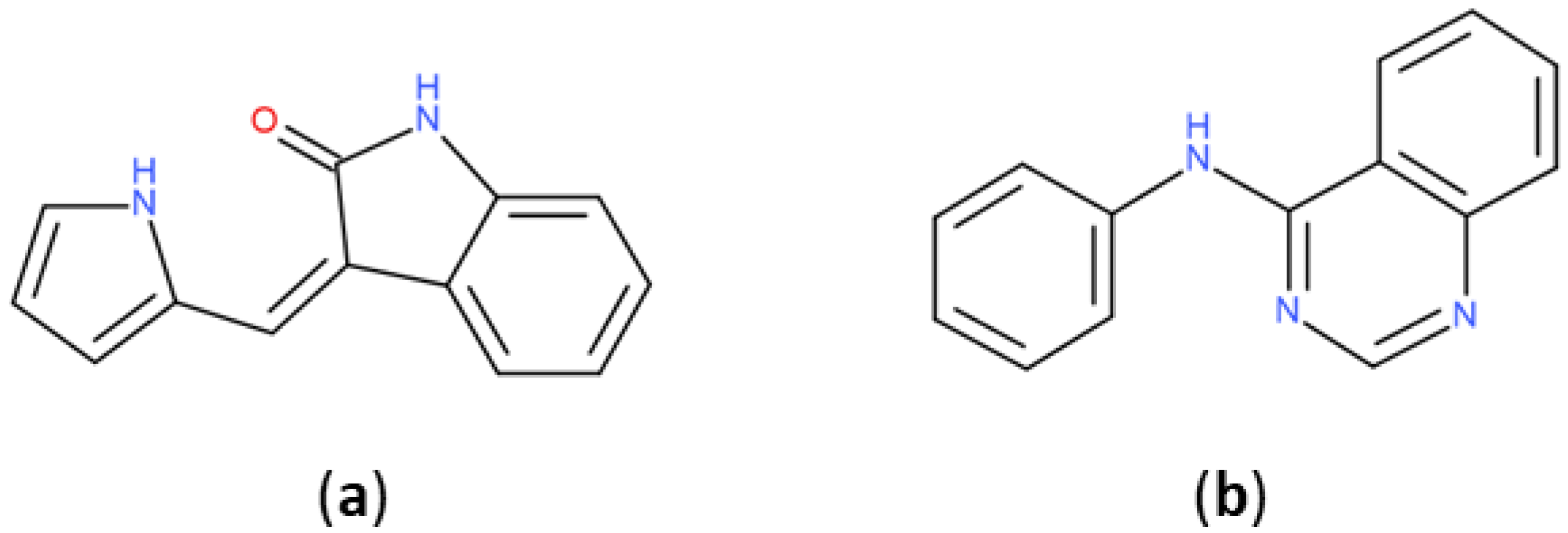
2.3.1. Analysis of Molecular Scaffolds
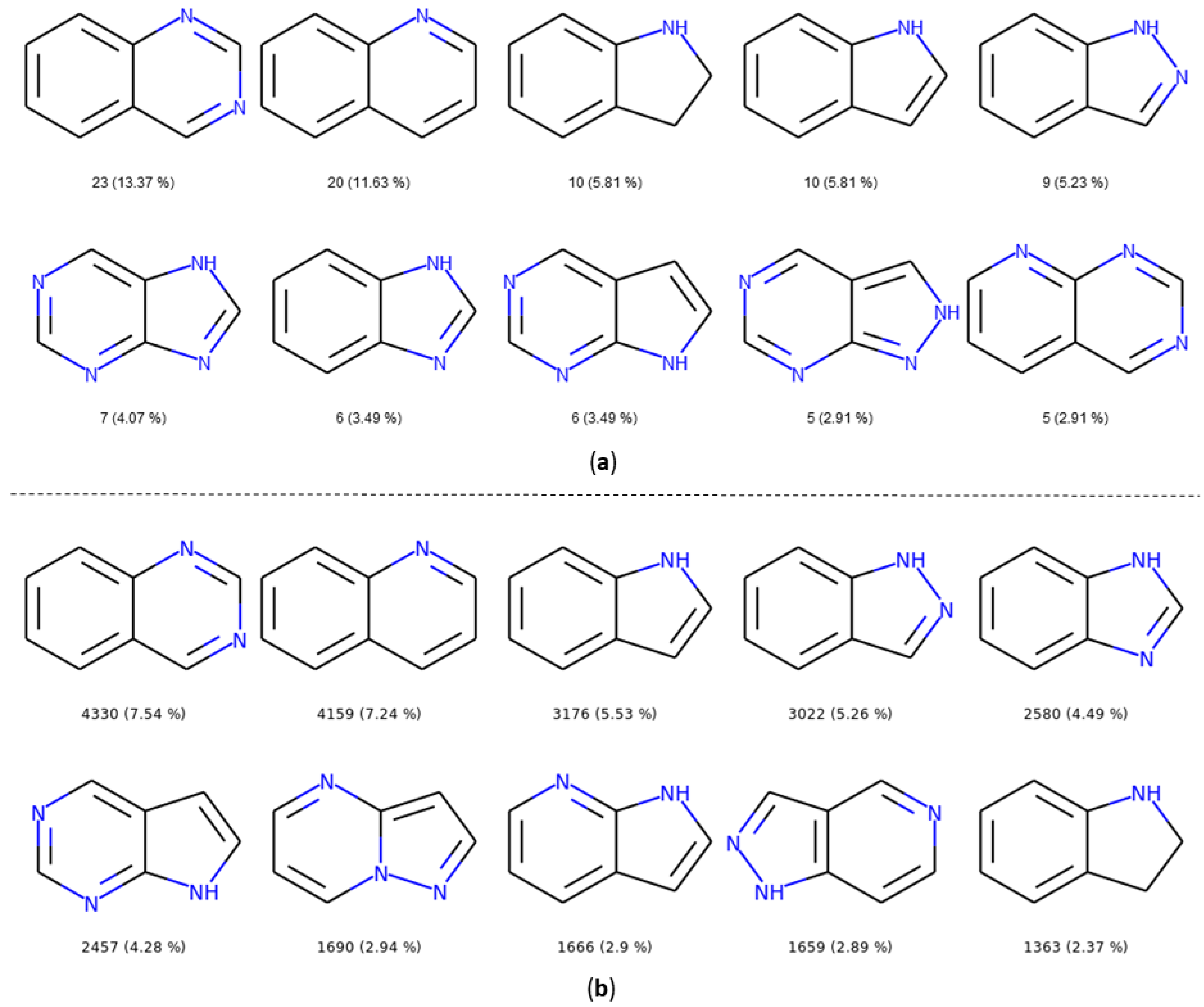
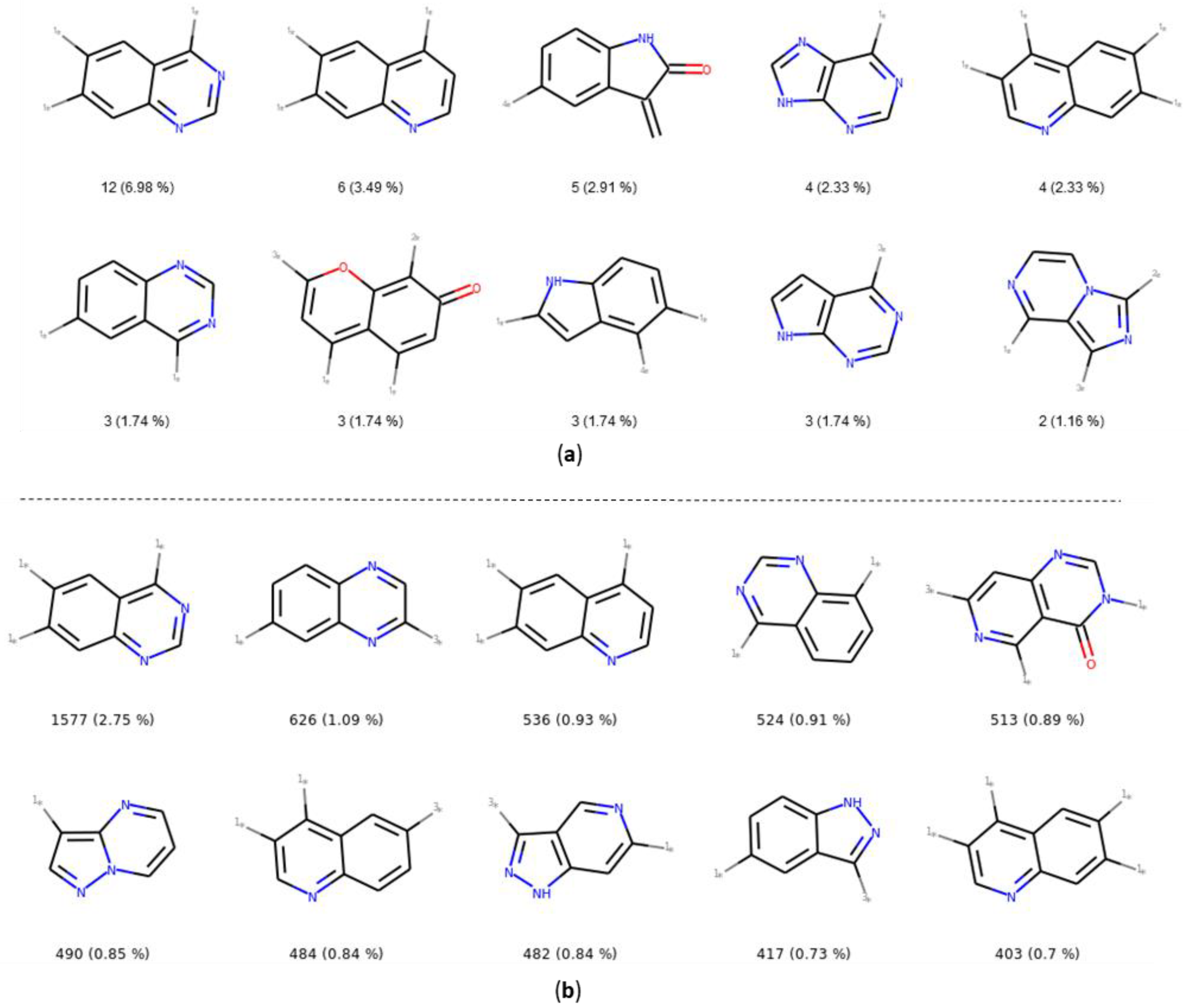
2.3.2. Ring Analysis
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giamas, G.; Stebbing, J.; Vorgias, C.E.; Knippschild, U. Protein kinases as targets for cancer treatment. Pharmacogenomics 2007, 8, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.K.; Mack, H.; Teusch, N. Rho kinase, a promising drug target for neurological disorders. Nat. Rev. Drug Discov. 2005, 4, 387–398. [Google Scholar] [CrossRef]
- Cohen, P. Immune diseases caused by mutations in kinases and components of the ubiquitin system. Nat. Immunol. 2014, 15, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Dimova, D.; Bajorath, J. Assessing scaffold diversity of kinase inhibitors using alternative scaffold concepts and estimating the scaffold hopping potential for different kinases. Molecules 2017, 22, 730. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Maximiano, S.; Magalhães, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the treatment of breast cancer. BioDrugs 2016, 30, 75–86. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.; Johnson, J.R.; Duan, J.; Gobburu, J.; Rahman, A.; Benson, K.; Leighton, J.; Kim, S.K.; Wood, R.; et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin. Cancer Res. 2002, 8, 935–942. [Google Scholar] [PubMed]
- WHO|INN Stems. Available online: http://www.who.int/medicines/services/inn/stembook/en/ (accessed on 20 March 2019).
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Schneider, P.; Schneider, G. Privileged structures revisited. Angew. Chem. Int. Ed. 2017, 56, 7971–7974. [Google Scholar] [CrossRef] [Green Version]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Scaffold hopping in medicinal chemistry. In Methods and Principles in Medicinal Chemistry; Brown, N., Ed.; Wiley-VCH-Verl: Weinheim, Germany, 2014; ISBN 978-3-527-33364-6. [Google Scholar]
- Dimova, D.; Stumpfe, D.; Bajorath, J. Computational design of new molecular scaffolds for medicinal chemistry, part II: Generalization of analog series-based scaffolds. Future Sci. OA 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Schuffenhauer, A.; Varin, T. Rule-based classification of chemical structures by scaffold. Mol. Inform. 2011, 30, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A curated, annotated and updated database of protein kinase inhibitors in clinical trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United States Adopted Names Approved Stems. Available online: https://www.ama-assn.org/about/united-states-adopted-names/united-states-adopted-names-approved-stems (accessed on 26 June 2019).
- Hiroshi, H.; Yoshikazu, I.; Megumu, O.; Tsuyoshi, A.; Toshihide, N.; Makiko, K.; Toshifumi, K.; Naoki, K.; Junko, O.; Kazunori, Y.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.E.; Beswick, P. What does the aromatic ring number mean for drug design? Expert Opin. Drug Discov. 2014, 9, 995–1003. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Sauer, W.H.B.; Schwarz, M.K. Molecular shape diversity of combinatorial libraries: A prerequisite for broad bioactivity. J. Chem. Inf. Comput. Sci. 2003, 43, 987–1003. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Dowling, R.J.O.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim. Biophys. Acta BBA-Proteins Proteom. 2010, 1804, 433–439. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Zhao, H.; Caflisch, A. Current kinase inhibitors cover a tiny fraction of fragment space. Bioorg. Med. Chem. Lett. 2015, 25, 2372–2376. [Google Scholar] [CrossRef] [PubMed]
- Conconi, M.T.; Marzaro, G.; Urbani, L.; Zanusso, I.; Di Liddo, R.; Castagliuolo, I.; Brun, P.; Tonus, F.; Ferrarese, A.; Guiotto, A.; et al. Quinazoline-based multi-tyrosine kinase inhibitors: Synthesis, modeling, antitumor and antiangiogenic properties. Eur. J. Med. Chem. 2013, 67, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Smaill, J.B.; Rewcastle, G.W.; Loo, J.A.; Greis, K.D.; Chan, O.H.; Reyner, E.L.; Lipka, E.; Showalter, H.D.; Vincent, P.W.; Elliott, W.L.; et al. Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino) quinazoline-and 4-(phenylamino) pyrido [3, 2-d] pyrimidine-6-acrylamides bearing additional solubilizing functions. J. Med. Chem. 2000, 43, 1380–1397. [Google Scholar] [CrossRef] [PubMed]
- Butina, D. Unsupervised data base clustering based on daylight’s fingerprint and tanimoto similarity: A fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- MaxHalford/Prince: Python Factor Analysis Library (PCA, CA, MCA, MFA, FAMD). Available online: https://github.com/MaxHalford/prince (accessed on 14 April 2020).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Riniker, S.; Landrum, G.A. Better informed distance geometry: Using what we know to improve conformation generation. J. Chem. Inf. Model. 2015, 55, 2562–2574. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Caswell, T.A.; Droettboom, M.; Hunter, J.; Firing, E.; Lee, A.; Stansby, D.; de Andrade, E.S.; Nielsen, J.H.; Klymak, J.; Varoquaux, N.; et al. Matplotlib/Matplotlib, version 3.0.1; Zenodo: Meyrin, Switzerland, 2018. [Google Scholar]
- Waskom, M.; Botvinnik, O.; O’Kane, D.; Hobson, P.; Ostblom, J.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; John, B.C.; et al. Mwaskom/Seaborn: v0.9.0 (July 2018); Zenodo: Meyrin, Switzerland, 2018. [Google Scholar]
- Gally, J.-M.; Bourg, S.; Do, Q.-T.; Aci-Sèche, S.; Bonnet, P. VSPrep: A general KNIME workflow for the preparation of molecules for virtual screening. Mol. Inform. 2017, 36, 1700023. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PKI | Unitprot ID | Gene Name |
|---|---|---|
| Alpelisib | P42336 | PI3KCA |
| Binimetinib | Q02750 | MAP2K1 |
| Dacomitinib | P00533 | EGFR |
| Duvelisib | O00329 | PI3KCD |
| P48736 | PI3KCG | |
| Encorafenib | P15056 | BRAF |
| Entrectinib | P04629 | NTRK1 |
| Q16620 | NTRK2 | |
| Q16288 | NTRK3 | |
| P08922 | ROS1 | |
| Q9UM73 | ALK | |
| Erdafitinib | P11362 | FGFR1 |
| Fedratinib | O60674 | JAK2 |
| P36888 | FLT3 | |
| O60885 | BRD4 | |
| Fostamatinib | P43405 | SYK |
| Gilteritinib | P36888 | FLT3 |
| P30530 | AXL | |
| Q9UM73 | ALK | |
| Larotractinib | P04629 | NTRK1 |
| Q16620 | NTRK2 | |
| Q16288 | NTRK3 | |
| Lorlatinib | Q9UM73 | ALK |
| P08922 | ROS1 | |
| Pexidartinib | P36888 | FLT3 |
| P10721 | KIT | |
| P07333 | CSF1R | |
| Upadacitinib | P23458 | JAK1 |
| Zanubrutinib | Q06187 | BTK |
| 1 | 0 Ro5 Violation | 1 Ro5 Violation | 2 Ro5 Violations | >2 Ro5 Violations |
|---|---|---|---|---|
| PKIs approved | 33/60 (55.0%) | 20/60 (33.0%) | 7/60 (12.0%) | 0/56 (0%) |
| PKIs in clinical trials | 101/158 (64.0%) | 41/158 (26.0%) | 16/158 (10.0%) | 0/158 (0%) |
| All PKIs | 134/218 (61.5%) | 61/218 (28.0%) | 23/218 (10.5%) | 0/218 (0%) |
| PKIs ChEMBL | 51,858/76,504 (67.8%) | 18,601/76,504 (24.3%) | 5876/76,504 (7.7%) | 169/76,504 (0.2%) |
| 1 | MW > 500 Da | ClogP > 5 | HBA > 10 | HBD > 5 | TPSA > 140 Å2 | NRB > 10 |
|---|---|---|---|---|---|---|
| PKIs approved | 20/60 (33.3%) | 12/60 (20.0%) | 2/60 (3.3%) | 0/60 (0%) | 2/60 (3.3%) | 2/60 (3.3%) |
| PKIs in clinical trials | 46/158 (29.1%) | 26/158 (16.5%) | 1/158 (0.6%) | 0/158 (0%) | 4/158 (2.5%) | 6/158 (3.8%) |
| All PKIs | 66/218 (30.3%) | 38/218 (17.4%) | 3/218 (1.4%) | 0/218 (0%) | 6/218 (2.8%) | 8/218 (3.7%) |
| PKIs ChEMBL | 18,892/76,504 (24.7%) | 10,897/76,504 (14.2%) | 924/76,504 (1.2%) | 208/76,504 (0.2%) | 3695/76,504 (4.8%) | 2051/76,504 (2.7%) |
| No. Molecules | No. Macrocycles | No. BM Scaffolds | No. Graph Frameworks | Molecular Similarity Mean a (SD) | |
|---|---|---|---|---|---|
| PKIDB | 218 | 4 (1.8%) | 207 (95.0%) | 195 (89.5%) | 0.51 (0.11) |
| PKI_ChEMBL | 76,504 | 487 (0.64%) | 28,732 (37.6%) | 13,331 (17.4%) | 0.49 (0.11) |
| Name Variable | Descriptor |
|---|---|
| MW | Molecular weight |
| LogP | Wildman-Crippen LogP value |
| TPSA | Topological polar surface area |
| HBA | Number of Hydrogen Bond Acceptors |
| HBD | Number of Hydrogen Bond Donors |
| NRB | Number of Rotatable Bonds |
| NAR | Number of aromatic rings |
| FCSP3 | Fraction of C atoms that are SP3 hybridized |
| MQN8 | Molecular Quantum Numbers |
| MQN10 | Molecular Quantum Numbers |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bournez, C.; Carles, F.; Peyrat, G.; Aci-Sèche, S.; Bourg, S.; Meyer, C.; Bonnet, P. Comparative Assessment of Protein Kinase Inhibitors in Public Databases and in PKIDB. Molecules 2020, 25, 3226. https://doi.org/10.3390/molecules25143226
Bournez C, Carles F, Peyrat G, Aci-Sèche S, Bourg S, Meyer C, Bonnet P. Comparative Assessment of Protein Kinase Inhibitors in Public Databases and in PKIDB. Molecules. 2020; 25(14):3226. https://doi.org/10.3390/molecules25143226
Chicago/Turabian StyleBournez, Colin, Fabrice Carles, Gautier Peyrat, Samia Aci-Sèche, Stéphane Bourg, Christophe Meyer, and Pascal Bonnet. 2020. "Comparative Assessment of Protein Kinase Inhibitors in Public Databases and in PKIDB" Molecules 25, no. 14: 3226. https://doi.org/10.3390/molecules25143226