
Synthesis of a Fluorous-Tagged Hexasaccharide and Interaction with Growth Factors Using Sugar-Coated Microplates


Glycosystems Laboratory, Instituto de Investigaciones Químicas (IIQ), cicCartuja, CSIC and Universidad de Sevilla, Americo Vespucio, 49, 41092 Sevilla, Spain
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(8), 1591; https://doi.org/10.3390/molecules24081591
Submission received: 20 February 2019
/
Revised: 17 April 2019
/
Accepted: 18 April 2019
/
Published: 22 April 2019
(This article belongs to the Special Issue Carbohydrates in Synthesis)
Abstract
:Here, we report the synthesis of a sulfated, fully protected hexasaccharide as a glycosaminoglycan mimetic and the study of its interactions with different growth factors: midkine, basic fibroblast growth factor (FGF-2) and nerve growth factor (NGF). Following a fluorous-assisted approach, monosaccharide building blocks were successfully assembled and the target oligosaccharide was prepared in excellent yield. The use of more acid stable 4,6-O-silylidene protected glucosamine units was crucial for the efficiency of this strategy because harsh reaction conditions were needed in the glycosylations to avoid the formation of orthoester side products. Fluorescence polarization experiments demonstrated the strong interactions between the synthesized hexamer, and midkine and FGF-2. In addition, we have developed an alternative assay to analyse these molecular recognition events. The prepared oligosaccharide was non-covalently attached to a fluorous-functionalized microplate and the direct binding of the protein to the sugar-immobilized surface was measured, affording the corresponding KD,surf value.
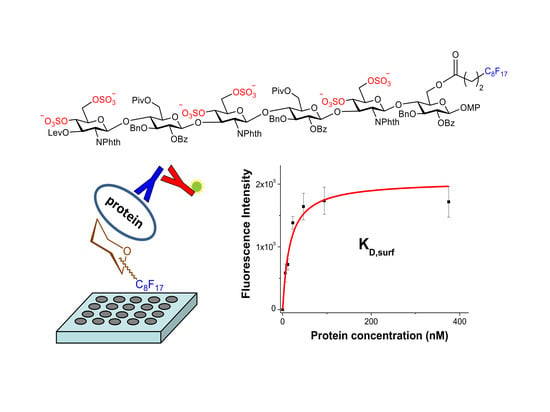
1. Introduction
Growth factors are a family of secreted, low molecular weight proteins that regulate cell proliferation and differentiation. Many of these growth factors bind to glycosaminoglycans (GAGs), including heparin and chondroitin sulfate (CS), which are heterogeneous polysaccharides found ubiquitously in mammalian tissues. This binding is essential for the biological activities of growth factors, and usually involves particular GAG sequences with well-defined structures. For instance, midkine is a heparin-binding growth factor that promotes growth, survival and differentiation of target cells [1,2]. This cytokine is implicated in many diseases, such as cancer and multiple sclerosis. It has been demonstrated that midkine interacts with chondroitin sulfate E (CS-E), a subtype of this polysaccharide characterized by the disulfated repeating unit d-glucuronic acid (GlcA)-β(1→3)-N-acetyl-d-galactosamine (GalNAc)(4,6-di-OSO3)-β(1→4) [3,4]. The interaction of midkine with CS is sensitive to the location of the sulfate groups along the sugar backbone [5]. Basic fibroblast growth factor (FGF-2) is another protein that plays a key role in cell growth, mediating the formation of new blood vessels [6]. FGF-2 also binds with nanomolar affinity to heparin and CS-E [3]. On the other hand, nerve growth factor (NGF) is a neurotrophin involved in the development, maintenance and survival of the vertebrate nervous system [7]. Hsieh-Wilson and coworkers showed for the first time that CS-E is able to modulate NGF signaling pathways [8], although in this case the interaction GAG-protein is weaker, in the micromolar range [9].
The preparation of molecules that mimic the properties of GAGs and bind to growth factors with high affinity is of great interest [10,11,12]. Such compounds could potentially control the biological functions of these proteins and have applications in tissue repair and anticancer therapies. In fact, the synthesis of different types of GAG mimetics has been reported, including GAG multivalent systems [13,14,15,16,17,18,19,20,21,22,23]. In this context, we previously found that sulfated oligosaccharides displaying hydrophobic protecting groups can be considered as GAG mimetics, strongly interacting with growth factors such as midkine and FGF-2 [24]. Interestingly, the binding affinities of these protected oligomers were much higher than those corresponding to the fully deprotected compounds [24]. A tetrasaccharide derivative following the sequence d-glucosamine(GlcN)(4,6-di-OSO3)-β(1→4)-d-glucose(Glc)-β(1→3) was synthesized using a fluorous-tag assisted approach and its interaction with midkine and FGF-2 was demonstrated by fluorescence polarization measurements [25]. In this work, we present the synthesis of the analogous hexasaccharide chain in order to study the influence of sugar length on the growth factor binding affinity. The synthetic strategy was optimized for the efficient preparation of this complex oligosaccharide. Moreover, we developed an experimental protocol for the interaction study between this sugar derivative and several growth factors based on the immobilization of the fluorinated carbohydrate on standard microtiter plates. These binding data were compared with the results obtained from fluorescence polarization assays.
2. Results and Discussion
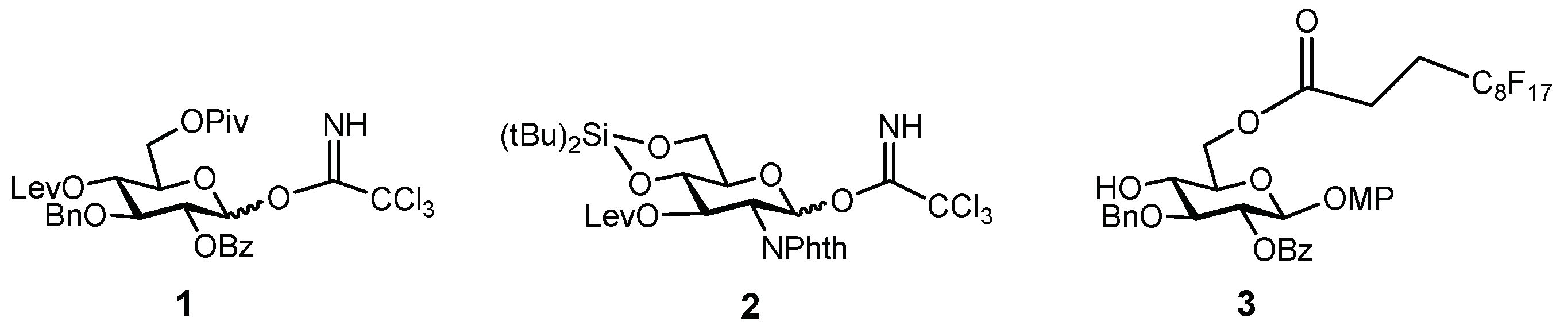
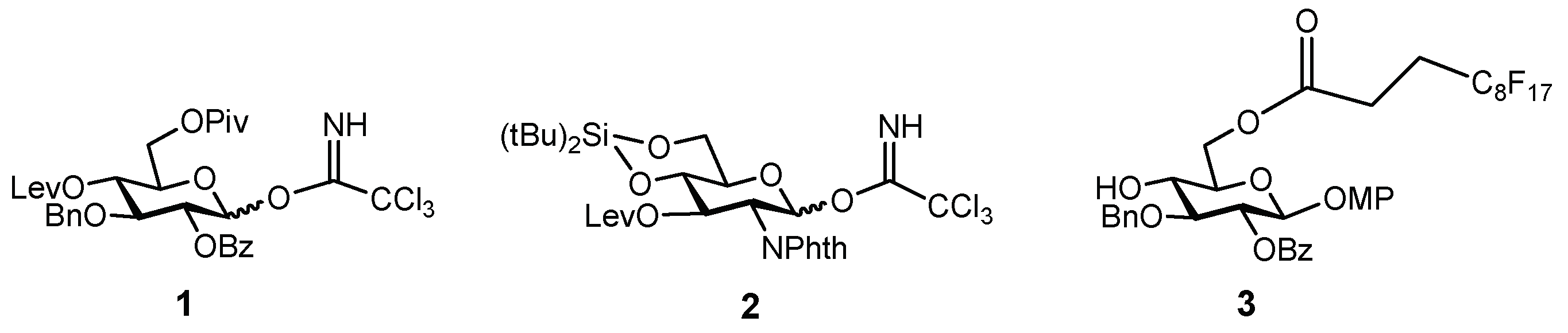
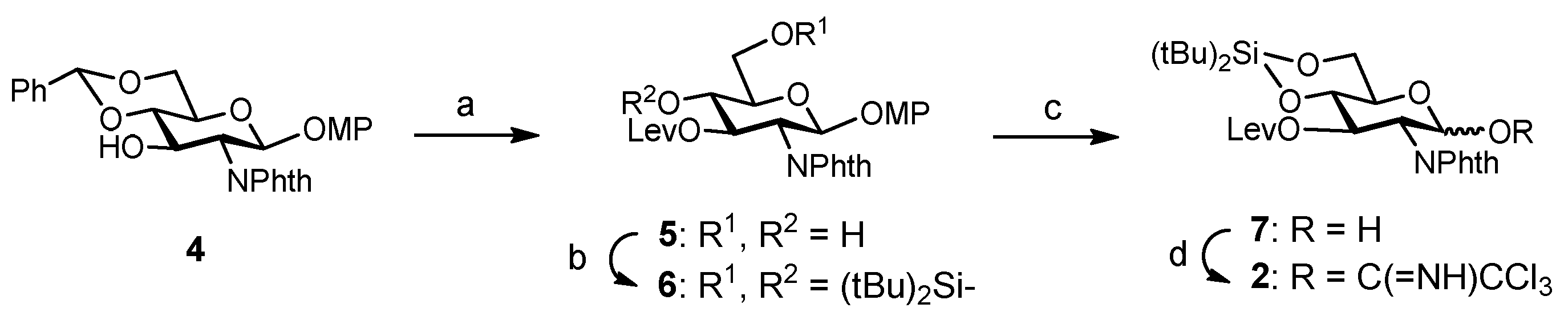
The synthesis of the target hexasaccharide was accomplished by assembling the monosaccharide building blocks 1, 2 and 3 shown in Scheme 1. The trichloroacetimidate method was chosen to construct the glycosidic bonds [26,27]. Participating protecting groups at position 2 in glycosyl donors 1 and 2 (benzoyl and N-phthalimido) favoured the selective formation of the desired β linkages. Glucose derivative 3 constituted the reducing end of the oligosaccharide chain. The C8F17 tail at the 6-OH position allowed the separation of fluorinated intermediates by fluorous solid-phase extraction (F-SPE) [28,29,30,31], avoiding time-consuming chromatographic purifications. Both glucose units 1 and 3 were prepared as previously reported, starting from diacetone d-glucose [25,32]. On the other hand, glucosamine trichloroacetimidate 2 was synthesized from commercially available 4 (Scheme 2). Treatment with levulinic anhydride and catalytic DMAP (4-dimethylamino pyridine) followed by trifluoroacetic acid (TFA)-mediated hydrolysis of the benzylidene acetal afforded diol 5 in excellent yield. Hydroxyl functions at positions 4 and 6 were then masked with a cyclic silylidene group by using di-tert-butylsilyl bis(trifluoromethanesulfonate) in dry pyridine. Thus, compound 6 was obtained in 93% yield. This silyl group can be selectively removed at the end of the synthesis in order to install the sulfates at the desired positions. Notably, in our previous synthesis of a tetrasaccharide GAG mimetic, we used 4,6-O-benzylidene glucosamine derivatives [25]. However, unwanted cleavage of the benzylidene moiety can occur under the acidic conditions of glycosylation reactions. For this reason, we decided to employ the more acid stable di-tert-butylsilylidene group. The advantages of this function over the alternative benzylidene acetals were shown by Codee and coworkers during the synthesis of hyaluronic acid oligomers [33]. Oxidative removal of the 4-methoxyphenyl moiety at the anomeric position using cerium (IV) ammonium nitrate (CAN) and trichloroacetimidate activation with trichloroacetonitrile and catalytic DBU (1,8-diazabicycloundec-7-ene) finally gave glycosyl donor 2.
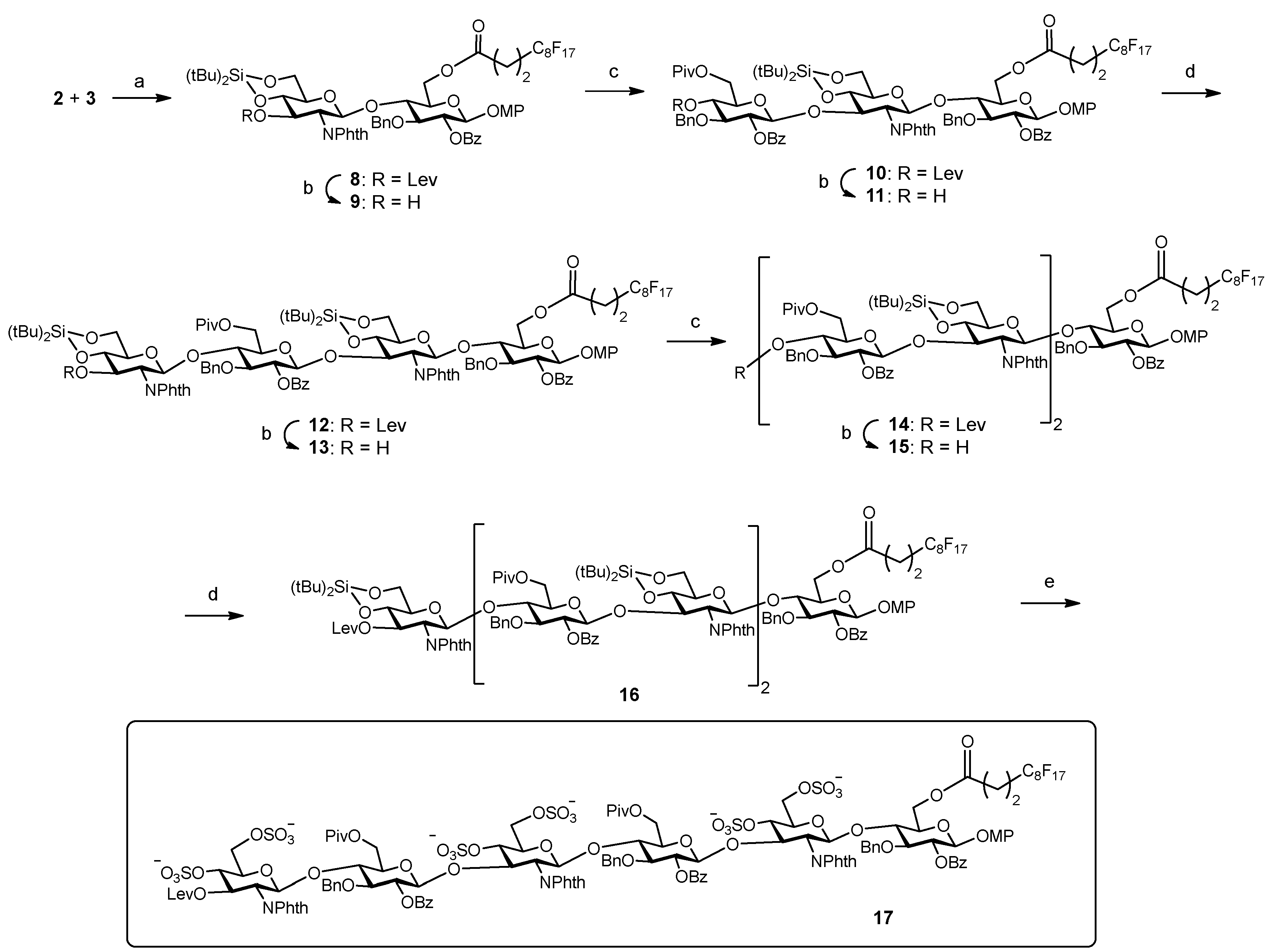
Next, we proceeded with the fluorous-tag assisted coupling of the building blocks to generate the target hexasaccharide sequence (Scheme 3). Glycosylation between fluorinated glucose 3 and glucosamine 2 (2 equiv.) was performed at 0 °C using TBSOTf (tert-butyldimethylsilyl trifluoromethanesulfonate) instead of TMSOTf (trimethylsilyl trifluoromethanesulfonate) in order to avoid the formation of the trimethylsilylated acceptor byproduct. After F-SPE purification, NMR, TLC and mass spectrometry analysis indicated the clean formation of disaccharide 8. Treatment with hydrazine monohydrate in pyridine/acetic acid buffer selectively removed the levulinoyl group providing disaccharide acceptor 9 ready for subsequent chain elongation. Synthesis of trisaccharide 10 was then accomplished. In previous work [25], we observed the formation of an orthoester side product [34] in the condensation between donor 1 and a 3-OH GlcN-Glc derivative, closely related to disaccharide acceptor 9. Increased amounts of Lewis acid catalyst were required to prevent the formation of the orthoester. Similarly, the first glycosylation trial of building blocks 1 and 9 using 10 mol% of TBSOTf with respect to the donor at 0 °C gave the orthoester trisaccharide as the major product. Taking advantage of the high acid stability of the silylidene, harsher reaction conditions (20 mol% of TBSOTf with respect to the donor at room temperature) were tested. Thus, compound 9 was coupled with 2.5 equiv. of trichloroacetimidate 1 to afford the desired trimer 10. The crude mixture was quickly purified by F-SPE and the study of the fluorinated fraction by NMR, TLC and mass spectrometry revealed the generation of 10 as the main reaction product (see NMR spectra, Supplementary Materials). A small amount of unreacted 9 (<10%) was also identified. Therefore, conventional acetylation (Ac2O, Py, DMAP, overnight) was carried out in order to block the remaining acceptor 9 and prevent deletion sequence problems [35]. Iterative delevulination and glycosylation with glucosamine 2 (2.8 equiv.) smoothly led to tetramer 12. Subsequent deprotection of the temporary levulinoyl group gave acceptor 13 that was treated with donor 1 (3 equiv.) to obtain pentasaccharide 14. After this first glycosylation cycle, F-SPE was performed and a significant amount (around 25%) of unreacted tetrasaccharide 13 was detected in the fluorous elution. In order to complete the reaction, a second glycosylation cycle with additional 1 (2 equiv.) was run. Interestingly, our results suggest that the construction of the Glc-GlcN bonds is less efficient in terms of consumption of donor building blocks than the formation of the GlcN-Glc linkages. Then, levulinoyl deprotection and condensation with glucosamine 2 (3 equiv.) afforded the desired compound 16. After F-SPE and standard silica gel column chromatography, pure hexasaccharide 16 was isolated in an excellent 39% yield, which means a 90% average yield per step. Finally, the three silylidene groups in 16 were selectively removed with (HF)n·Py complex in THF at 0 °C and the resulting hexaol was extensively sulfated with SO3·NMe3 at 100 ºC using microwave heating [36] to give sulfated derivative 17. Three sulfation cycles were required to complete the reaction and minimize partially sulfated intermediates.
As previously reported [25], treatment of compounds, such as 17, with ethylene diamine followed by N-acetylation and hydrogenolysis could afford a GAG-like deprotected sequence. However, in previous papers [24,25], we found that the binding affinities of these fully deprotected oligosaccharides are much lower than those corresponding to the sulfated, protected intermediates. For this reason, in this work we focus on the binding properties of hexasaccharide 17.
Thus, we initiated the study of the interactions between 17 and midkine, FGF-2 and NGF growth factors. First, we estimated the solution-phase binding affinities using our previously reported fluorescence polarization (FP) competition experiment (see Supplementary Materials) [37]. Briefly, we measured the ability of hexasaccharide 17 to compete with a fluorescent probe for protein binding. Thus, we calculated IC50 values as 17 concentration required for 50% inhibition. As shown in Table 1, our results indicated that derivative 17 interacted with midkine and FGF-2 in the nano- and micromolar range, respectively. These relative binding affinities were similar to those previously found for the analogous tetrasaccharide, although slightly lower (IC50 = 270 nM and 2.4 µM for the tetramer/midkine and tetramer/FGF-2 interactions, respectively) [25]. Our results indicated that the increase in oligosaccharide length, from tetra- to hexasaccharide, does not enhance growth factor binding. We also attempted to measure the binding of 17 to NGF. However, we could not apply our FP competition assay to the study of this interaction because NGF did not significantly bind to the probe, a fluorescently labelled heparin sequence, even at 3.7 µM concentration. This fact suggested that NGF displays less affinity for GAGs compared to midkine and FGF-2, in agreement with literature data (see also below) [3,9].
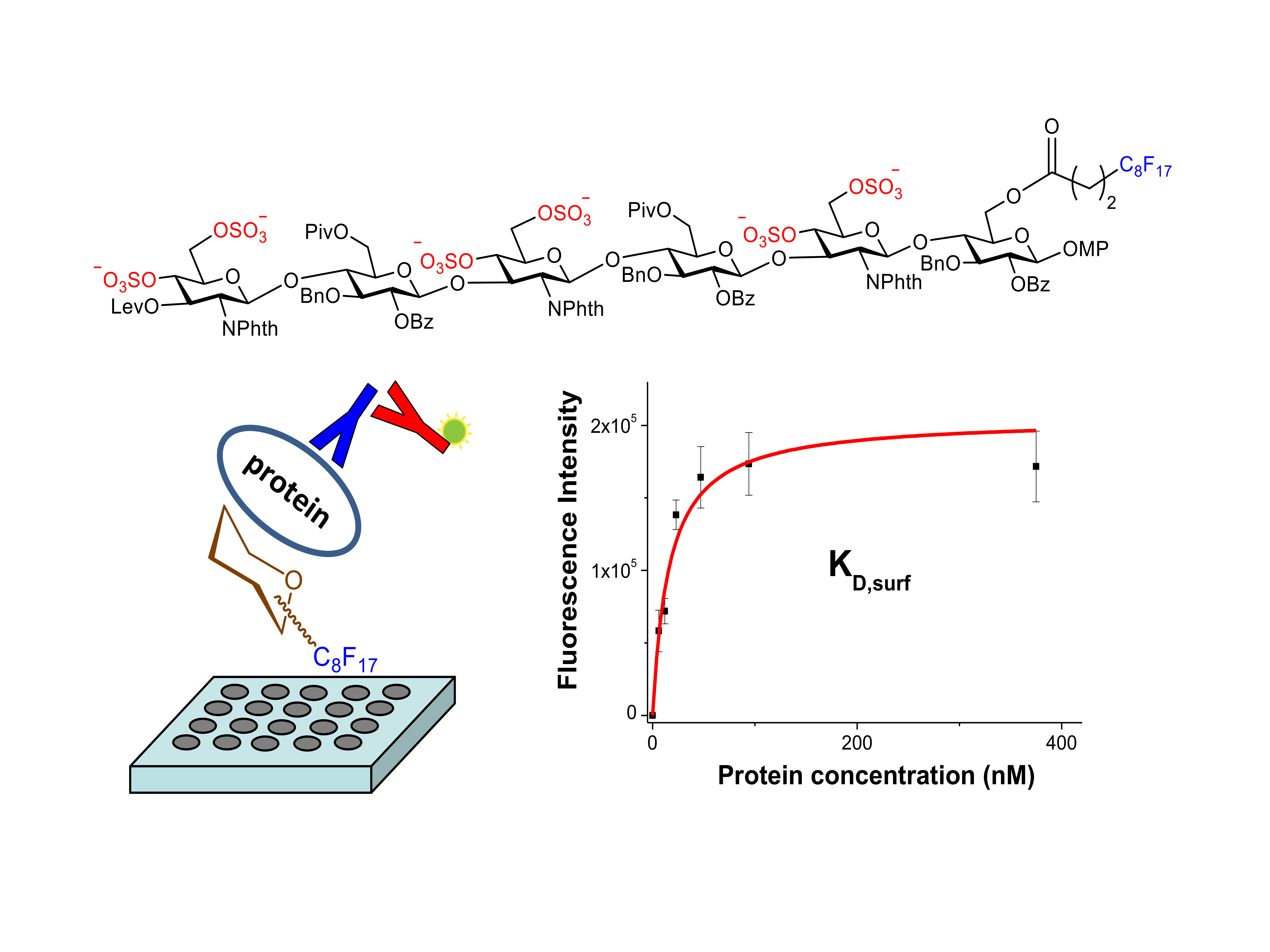
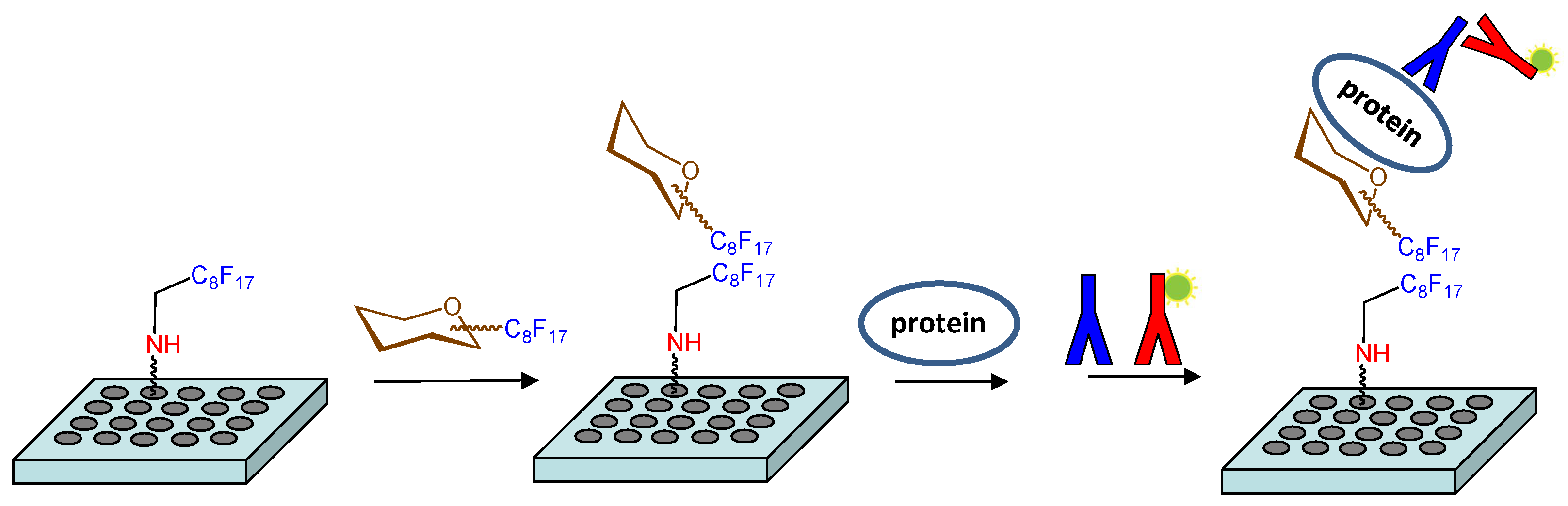
An alternative strategy was also considered to study these molecular recognition events. This approach involved the non-covalent immobilization of the fluorinated hexamer 17 on microtiter plates (Figure 1). Fluorous-based microarrays for the investigation of carbohydrate-protein interactions have been previously developed. A C8F17 tail was employed for the noncovalent immobilization of sugar derivatives on fluorous coated glass slides [38,39,40,41]. However, to the best of our knowledge, an experimental protocol to study the interactions between fluorous sugar-coated conventional microplates and proteins has not been published. For this purpose, we first treated an amino reactive 384-well plate (Nunc Immobilizer AminoTM) with a 1 mM solution of heptadecafluorononylamine (C8F17CH2NH2) in DMSO. Thus, we created a fluorinated surface ready for the immobilization of compound 17 through noncovalent fluorous-fluorous interactions. After quenching with ethanolamine, microplate wells were filled with a 250 µM solution of 17 in water/DMSO 9:1, incubated at room temperature for 6 h, and extensively washed with water. Some wells were not treated with 17 and were used as blank samples. Then, the binding of the attached sugar 17 to growth factors was analyzed. Incubation with different concentrations of midkine, FGF-2 and NGF was carried out and the bound protein was detected by further incubations with the corresponding primary and fluorescently labelled secondary antibodies (see Materials and Methods). Finally, the fluorescence was recorded using a standard microplate reader. The observation of fluorescence signals confirmed the interaction between immobilized 17 and the protein at a particular concentration. Fluorescence was observed neither from wells incubated only with the antibodies, in the absence of the protein, nor from blank wells coated with the fluorous C8F17CH2NH2 linker alone, in the absence of sugar 17, ruling out the non-specific binding of the proteins on the fluorous linker-coated wells.
Importantly, we performed a control experiment in which sugar 3, instead of hexamer 17, was immobilized on the fluorous-functionalized plate (see Supplementary Materials). No fluorescence signals above background were detected from these wells, discarding the non-specific interaction between any fluorous-tagged sugar and the growth factors. In addition, wells coated with hexamer 17 were incubated with two fluorescently labelled non-GAG binding proteins, Concanavalin A and bovine serum albumin, at 11 µM concentration (see Supplementary Materials). Again, we observed no fluorescence signals from these wells. These results indicated that compound 17 did not interact with Concanavalin A and bovine serum albumin and confirmed the specificity of the interactions between the growth factors and 17.
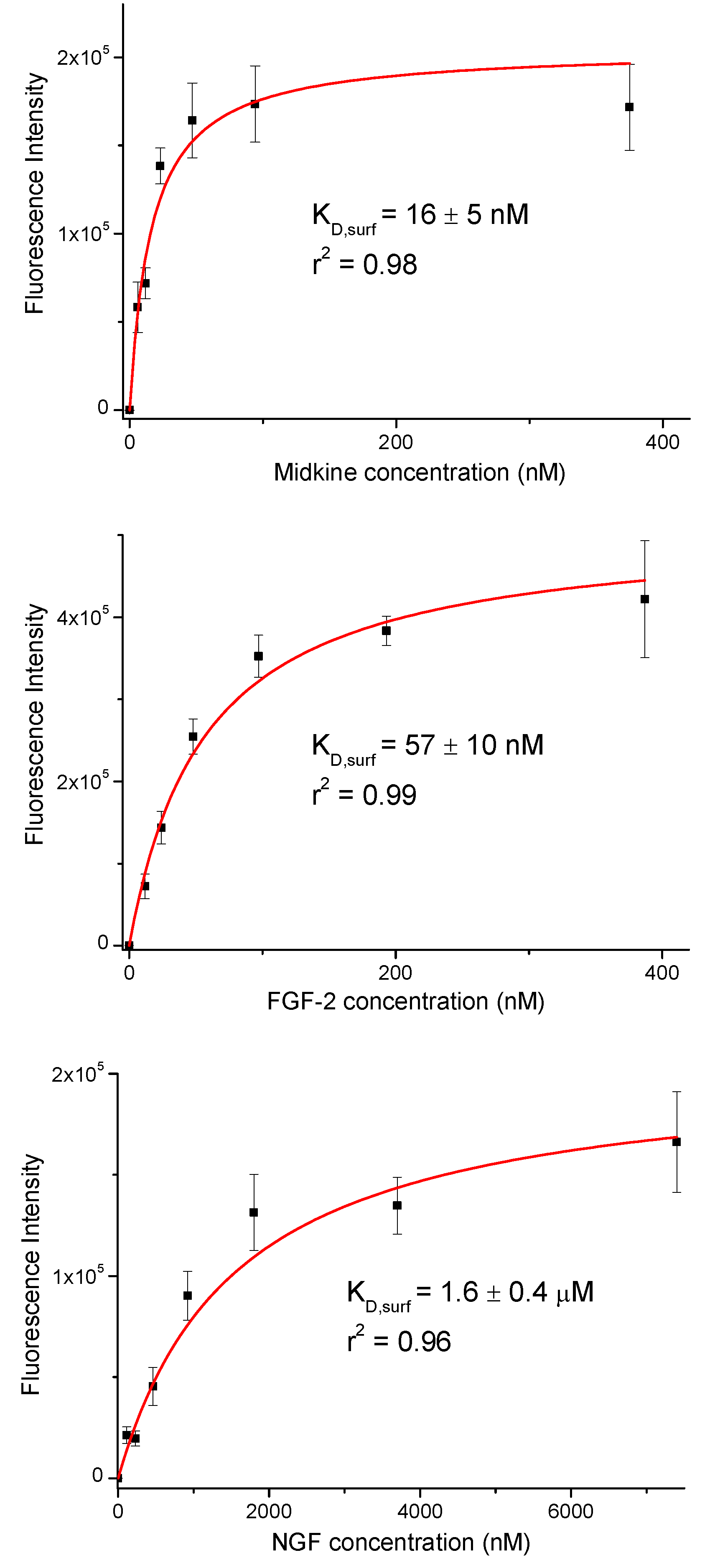
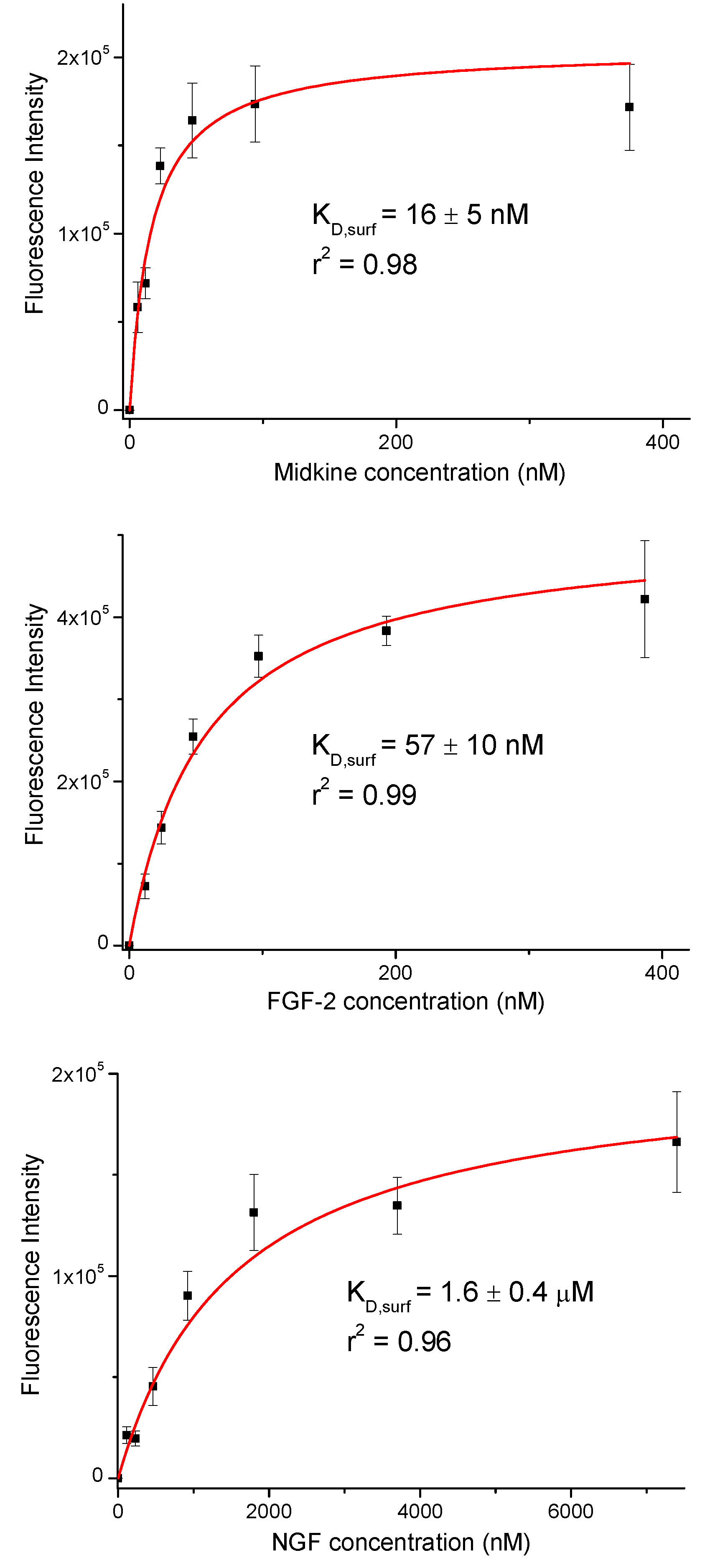
Fluorescence intensities were plotted against growth factor concentration (Figure 2). We observed a concentration dependent increase in the fluorescence signal. The resulting curves were fitted to the equation for a one-site binding model and surface dissociation constants (KD,surf) were obtained (Figure 2 and Table 1) [42,43]. The KD,surf values for the interaction between immobilized 17 and midkine/FGF-2 were 16 and 57 nM, respectively, confirming the strong carbohydrate-protein binding. A KD,surf value of 1.6 µM was determined for the 17/NGF interaction, indicating that the hexasaccharide recognizes NGF with less affinity. The obtained dissociation constants followed the same trend as literature data for GAG-growth factor interactions (GAG-midkine/FGF-2 and GAG-NGF bindings in the nano- and micromolar ranges, respectively) [3,9]. On the other hand, these results also showed clear differences between the relative binding affinities obtained from the FP experiment and the sugar-coated surface assay (Table 1). This fact can be easily explained by the differences in the two experimental setups. In the FP competition method, we measured the binding of hexasaccharide 17 and the protein in solution, while in the sugar immobilized experiment, we observed the direct interaction between the growth factor and a multivalent functionalized surface. As previously shown, the format of the interaction experiment can strongly influence the calculated affinity values [4,43,44]. Thus, the multivalent presentation of the fluorinated oligosaccharide on the microplate provided much higher binding affinities, in comparison with the solution-phase results.
3. Materials and Methods
3.1. General Synthetic Procedures
Thin-layer chromatography (TLC) analyses were performed on silica gel 60F-254 precoated aluminium plates from Merck (Darmstadt, Germany). The compounds were detected by UV visualization (λ = 254 nm) and by staining with 5% v/v anisaldehyde-5% v/v H2SO4-0.2% v/v AcOH in EtOH or 0.2% w/v cerium (IV) sufate-5% w/v ammonium molybdate tetrahydrate in 1 M H2SO4 followed by heating at over 200 °C. Column chromatography was carried out on silica gel 60 (0.2–0.063 mm or 0.040–0.015 mm; Merck). Optical rotations were determined with a Perkin-Elmer 341 polarimeter (Perkin Elmer, Waltham, MA, USA). 1H- and 13C-NMR spectra were acquired on an Avance III-400 spectrometer (Bruker, Billerica, MA, USA). 2-D COSY and HSQC experiments were routinely carried out to assist in signal assignment. Unit A refers to the reducing end monosaccharide in the NMR data. Electrospray mass spectra (ESI MS) were carried out with an Esquire 6000 ESI-Ion Trap from Bruker Daltonics (Billerica, MA, USA). High resolution mass spectra (HR MS) were carried out by CITIUS (Universidad de Sevilla, Sevilla, Spain). Microwave-based sulfation reactions were performed using a Biotage Initiator Eight synthesizer in sealed reaction vessels (Biotage, Uppsala, Sweden). Compound 4 was purchased from Carbosynth. FluoroFlash silica gel was purchased from Sigma-Aldrich (Merck, Darmstadt, Germany).
3.2. General Procedure for F-SPE
We followed our previously reported experimental procedure [25,30,45]. Briefly, FluoroFlash silica gel (5 g., Fluorous Technologies, Inc., Ambridge, PA, USA) was placed in a glass chromatography column (1.7 cm diameter). The F-SPE column was washed with DMF (2 mL) and then preconditioned with MeOH/H2O 80:20 (15 mL). Next, the crude sample (50–300 mg) was dissolved in DMF/H2O 9:1 (0.8 mL) and loaded on the column. The fluorophobic elution was carried out with 15 mL of MeOH/H2O 80:20. The fluorous compounds were then eluted using 100% acetone (20 mL). To regenerate the F-SPE column, we washed with additional acetone (20 mL). The initial DMF washing step can be omitted when reusing the F-SPE column.
3.3. Immobilization of Fluorous-Tagged Sugar on Microtiter Plates and Determination of Surface Dissociation Constants (KD, surf)
A 1 mM solution of 2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,9-heptadecafluorononylamine (C8F17CH2NH2) in DMSO was placed in the wells of Nunc Immobilizer AminoTM 384 microtiter plates (from Thermo Scientific, Waltham, MA, USA) (20 µL/well). The plate was shaken overnight at room temperature and the wells were emptied and washed twice with water. Then, the wells were incubated with 100 mM ethanolamine in sodium bicarbonate buffer (50 mM, pH 9.6) (80 µL/well). After shaking for 1 h, the wells were washed twice with water. Next, fluorous-coated wells were filled with a 250 µM solution of 17 in water/DMSO 9:1 (20 µL/well). After 6 h, the plate was washed with water and was ready to perform protein interaction studies. Blank samples were wells not incubated with 17, and coated with the fluorous linker alone.
Recombinant human FGF-2, midkine and NGF (from Peprotech) were reconstituted in PBS buffer (10 mM, pH 7.4) containing 1% BSA. Wells were incubated with 20 µL of protein solutions at different concentrations. All samples were performed at least in three replicates. After shaking at room temperature for 1 h, the microplate was washed with PBS containing 1% Tween 20 and 0.1% BSA, and water. Rabbit anti-human FGF-2, midkine and NGF antibodies (from Peprotech) were reconstituted in PBS containing 1% BSA. 20 µL of primary antibody solution at 5 µg/mL concentration was added to the wells in order to detect the bound protein. The plate was shaken for 1 h and washed with PBS containing 1% Tween 20 and 0.1% BSA, and water. Finally, the primary rabbit antibodies were detected by using Alexa Fluor 488 labelled anti-rabbit IgG (from Invitrogen, 20 µg/mL in PBS containing 1% BSA, 20 µL/well). The plate was incubated with shaking in the dark for 1 h and then washed with PBS containing 1% Tween 20 and 0.1% BSA, and water.
The fluorescence was read at 535 nm using a TRIAD multimode microplate reader (from Dynex). Blank wells, coated only with heptadecafluorononylamine, and not treated with 17, were also incubated with each protein and the corresponding antibodies. The residual fluorescence intensities obtained from these blank samples were subtracted from all values. The average fluorescence intensity values of at least three replicates were plotted against protein concentration. The resulting curve was fitted to the equation for a one-site binding model: y = Fmaxx/(x + KD,surf) where Fmax is the maximum fluorescence intensity approached with increasing protein concentrations and KD,surf is the dissociation constant of the surface sugar/protein interaction.
3.4. Procedure for Synthesis of Hexasaccharide 17
3.4.1. Synthesis of 4-Methoxyphenyl 2-deoxy-3-O-levulinoyl-2-phthalimido-β-d-glucopyranoside (5)
Lev2O preparation: LevOH (3.48 mL, 33.4 mmol) was added at 0 °C to a solution of 1,3-dicyclohexylcarbodiimide (3.48 g, 16.7 mmol) in CH2Cl2 (32 mL). After stirring for 5 min at room temperature, the mixture was cooled and filtered, and the urea precipitate was washed with additional CH2Cl2 (6 mL) to give 38 mL of a 0.44 M Lev2O solution.
Lev2O (38 mL of a 0.44 M solution in CH2Cl2) was added at room temperature to a mixture of 4 (2.8 g, 5.6 mmol) and DMAP (103 mg, 0.834 mmol). The mixture was stirred for 1.5 h, diluted with CH2Cl2, and washed with saturated aqueous NaHCO3, and brine. The organic phase was dried (MgSO4), filtered and concentrated to dryness. The residue was suspended in hexane/EtOAc (1:1, 8 mL) and the mixture was filtered. The solid was washed with additional hexane/EtOAc to give the 3-O-levulinated monosaccharide as a white amorphous solid (3.21 g, 96%) that was used without further purification. TLC (toluene-EtOAc 4:1) Rf 0.33.
TFA (6.2 mL) and H2O (0.93 mL) were added to a solution of the levulinated compound (3.21 g, 5.34 mmol) in CH2Cl2 (62 mL) at 0 °C. The solution was stirred for 2 h at room temperature, then it was diluted with CH2Cl2, and washed with saturated NaHCO3 aqueous solution and brine. The organic layer was dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by column chromatography (toluene/EtOAc 1:1 → toluene/EtOAc 1:3) to give 5 (2.5 g, 91%) as a white foam. TLC (toluene/EtOAc 1:2): Rf = 0.15. [α]20D + 35° (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3): δ 7.85 (m, 2H, Ar), 7.73 (m, 2H, Ar), 6.84 (m, 2H, Ar), 6.74 (m, 2H, Ar), 5.92 (d, 1H, J1,2 = 8.4 Hz, H-1), 5.78 (dd, 1H, J2,3 = 10.8 Hz, J3,4 = 8.9 Hz, H-3), 4.48 (dd, 1H, H-2), 4.01 (m, 1H, H-6a), 3.93–3.87 (m, 2H, H-6b, H-4), 3.77–3.72 (m, 4H, H-5, Me (OMP)), 3.52 (d, 1H, J4,OH = 4.0 Hz, OH), 2.68–2.33 (m, 4H, CH2(Lev)), 2.24 (br t, 1H, OH), 2.03 (s, 3H, CH3(Lev)); 13C-NMR (100 MHz, CDCl3): δ 207.5, 172.9, 168.0 (CO), 155.5–114.6 (Ar), 97.2 (C-1), 75.8 (C-5), 73.7 (C-3), 69.8 (C-4), 62.1 (C-6), 55.6 (Me (OMP)), 54.5 (C-2), 38.0 (CH2(Lev)), 29.5 (CH3(Lev)), 28.0 (CH2(Lev)); HR MS: m/z: calcd for C26H27NO10Na: 536.1527; found: 536.1529 [M + Na]+.
3.4.2. Synthesis of 4-Methoxyphenyl 4,6-O-di-tert-butylsilylidene-2-deoxy-3-O-levulinoyl-2-phthalimido-β-d-glucopyranoside (6)
Compound 5 (2.0 g, 3.89 mmol) was dissolved in dry Py (60 mL) and cooled (0 °C). di-tert-Butylsilyl bis(trifluoromethanesulfonate) (1.57 mL, 4.67 mmol) was added and the mixture was stirred at room temperature for 1.5 h. The reaction was quenched with MeOH (17 mL), diluted with EtOAc (300 mL), and washed with 1 M HCl, saturated aqueous NaHCO3, and brine. The organic phase was dried (MgSO4), filtered and concentrated to dryness. The residue was purified by column chromatography (toluene-EtOAc 10:1) to afford 6 as a white foam (2.37 g, 93%). TLC (toluene-EtOAc 5:1) Rf 0.43; [α]20D + 22° (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3): δ 7.86 (m, 2H, Ar), 7.73 (m, 2H, Ar), 6.82 (m, 2H, Ar), 6.73 (m, 2H, Ar), 5.90 (d, 1H, J1,2 = 8.4 Hz, H-1), 5.75 (dd, 1H, J2,3 = 10.8 Hz, J3,4 = 9.0 Hz, H-3), 4.48 (dd, 1H, H-2), 4.25 (dd, 1H, J5,6a = 5.1 Hz, J6a,6b = 10.3 Hz, H-6a), 4.03 (t, 1H, J4,5 = 9.4 Hz, H-4), 4.00 (t, 1H, J5,6b = 10.3 Hz, H-6b), 3.74 (td, 1H, H-5), 3.71 (s, 3H, Me (OMP)), 2.64–2.41 (m, 4H, CH2(Lev)), 1.96 (s, 3H, CH3(Lev)), 1.06, 0.99 (2s, 18H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3): δ 205.7, 172.0, 168.3, 167.7 (4 × CO), 155.8-114.6 (Ar), 97.9 (C-1), 75.6 (C-4), 72.5 (C-3), 70.8 (C-5), 66.3 (C-6), 55.6 (Me (OMP)), 54.6 (C-2), 37.9 (CH2(Lev)), 29.5 (CH3(Lev)), 28.0 (CH2(Lev)), 27.5, 27.0 (Si(C(CH3)3)2), 22.8, 20.0 (Si(C(CH3)3)2); HR MS: m/z: calcd for C34H43NO10SiNa: 676.2548; found: 676.2555 [M + Na]+.
3.4.3. Synthesis of 4,6-O-di-tert-Butylsilylidene-2-deoxy-3-O-levulinoyl-2-phthalimido-α,β-d-glucopyranose (7)
CAN (20 mL of a 0.73 M solution in H2O) was added to a cooled solution of 6 (2.37 g, 3.62 mmol) in CH2Cl2/MeCN (1:2; 180 mL). After stirring for 50 min at 0 °C, the reaction mixture was diluted with EtOAc, washed with H2O, saturated aqueous NaHCO3, and brine. The organic phase was dried (MgSO4), filtered and concentrated to dryness. The residue was purified by column chromatography (toluene-EtOAc 4:1) to afford 7 as light-yellow foam (1.81 g, 91%, mixture of α/β anomers). TLC (toluene/EtOAc 4:1) Rf 0.23; 1H-NMR (400 MHz, CDCl3) (data for β anomer): δ 7.85 (m, 2H, Ar), 7.72 (m, 2H, Ar), 5.74 (dd, 1H, J2,3 = 10.7 Hz, J3,4 = 8.9 Hz, H-3), 5.62 (br t, 1H, H-1), 4.25 (dd, 1H, J5,6a = 5.0 Hz, J6a,6b = 10.3 Hz, H-6a), 4.17 (dd, 1H, J1,2 = 8.4 Hz, H-2), 3.99–3.94 (m, 2H, H-4, H-6b), 3.72 (td, 1H, J4,5 = J5,6b = 9.9 Hz, H-5), 3.24 (d, 1H, J1,OH = 7.6 Hz, OH), 2.63–2.39 (m, 4H, CH2(Lev)), 1.95 (s, 3H, CH3(Lev)), 1.05, 0.99 (2s, 18H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3) (data for β anomer): δ 206.0, 172.0, 168.3, 168.1 (4 × CO), 134.6–123.6 (Ar), 93.1 (C-1), 75.8 (C-4), 72.4 (C-3), 70.8 (C-5), 66.3 (C-6), 56.2 (C-2), 37.9 (CH2(Lev)), 29.5 (CH3(Lev)), 28.0 (CH2(Lev)), 27.4, 27.0 (Si(C(CH3)3)2), 22.7, 20.0 (Si(C(CH3)3)2); HR MS: m/z: calcd for C27H37NO9SiNa: 570.2130; found: 570.2132 [M + Na]+.
3.4.4. Synthesis of O-(4,6-O-di-tert-Butylsilylidene-2-deoxy-3-O-levulinoyl-2-phthalimido-α,β-d-glucopyranosyl) trichloroacetimidate (2)
Trichloroacetonitrile (1.95 mL, 19.2 mmol) and catalytic DBU (19 µL, 0.13 mmol) were added to a solution of 7 (700 mg, 1.28 mmol) in dry CH2Cl2 (13 mL) under an argon atmosphere. After stirring for 4 h at room temperature, the reaction mixture was concentrated to dryness. The residue was purified by a short silica gel column (toluene-EtOAc 8:1 + 1% Et3N) to afford 2 as a white foam (659 mg, 75%, mixture of α/β anomers). TLC (toluene-EtOAc 7:1) Rf 0.41; 1H-NMR (400 MHz, CDCl3) (data for β anomer): δ 8.62 (s, 1H, NH), 7.83 (m, 2H, Ar), 7.71 (m, 2H, Ar), 6.65 (d, 1H, J1,2 = 8.8 Hz, H-1), 5.81 (dd, 1H, J2,3 = 10.8 Hz, J3,4 = 8.9 Hz, H-3), 4.54 (dd, 1H, H-2), 4.32 (dd, 1H, J5,6a = 5.0 Hz, J6a,6b = 10.1 Hz, H-6a), 4.05 (t, 1H, H-4), 4.01 (t, 1H, J5,6b = 10.1 Hz, H-6b), 3.87 (td, 1H, J4,5 = 9.8 Hz, H-5), 2.65–2.41 (m, 4H, CH2(Lev)), 1.95 (s, 3H, CH3(Lev)), 1.05, 1.00 (2s, 18H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3) (data for β anomer): δ 205.6, 171.8, 167.6 (4 × CO), 160.7 (C=NH), 134.2-123.6 (Ar), 93.9 (C-1), 90.2 (CCl3), 75.4 (C-4), 72.0 (C-3), 71.4 (C-5), 66.2 (C-6), 53.6 (C-2), 37.9 (CH2(Lev)), 29.5 (CH3(Lev)), 27.9 (CH2(Lev)), 27.4, 26.9 (Si(C(CH3)3)2), 22.7, 20.0 (Si(C(CH3)3)2); ESI MS: m/z: calcd for C29H37Cl3N2O9SiNa: 713.1; found: 713.1 [M + Na]+.
3.4.5. Synthesis of 4-Methoxyphenyl O-(4,6-O-di-tert-butylsilylidene-2-deoxy-3-O-levulinoyl-2-phthalimido-β-d-glucopyranosyl)-(1→4)-O-(2-O-benzoyl-3-O-benzyl-6-O-pivaloyl-β-d-glucopyranosyl)-(1→3)-O-(4,6-O-di-tert-butylsilylidene-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1→4)-O-(2-O-benzoyl-3-O-benzyl-6-O-pivaloyl-β-d-glucopyranosyl)-(1→3)-O-(4,6-O-di-tert-butylsilylidene-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1→4)-2-O-benzoyl-3-O-benzyl-6-O-4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecanoyl-β-d-glucopyranoside (16)
Donor 2 (55 mg, 0.08 mmol) and acceptor 3 (38 mg, 0.04 mmol) were dissolved in dry CH2Cl2 (1.1 mL) in the presence of activated 4Å molecular sieves (MS, 90 mg). The reaction mixture was stirred, under an argon atmosphere, for 10 min at 0 °C and TBSOTf (65 µL of a 0.085 M solution in dry CH2Cl2) was added. After stirring for 25 min at 0 °C, the reaction mixture was quenched with triethylamine, filtered, and then concentrated under reduced pressure. The crude product was purified using a fluorous solid-phase extraction (F-SPE) column to give disaccharide 8 as a white foam (59 mg). TLC (hexane-EtOAc 2:1) Rf 0.35; 1H-NMR (400 MHz, CDCl3): δ 7.98 (m, 2H, Ar), 7.85 (m, 2H, Ar), 7.73 (m, 2H, Ar), 7.57 (m, 1H, Ar), 7.43 (m, 2H, Ar), 7.28-7.17 (m, 5H, Ar), 6.79 (m, 2H, Ar), 6.68 (m, 2H, Ar), 5.62 (m, 2H, H-3′, H-1′), 5.41 (t, 1H, J2,3 = 6.8 Hz, H-2), 5.00 (d, 1H, J1,2 = 6.6 Hz, H-1), 4.77 (m, 2H, CH2(Bn)), 4.43 (dd, 1H, J5,6a = 2.1 Hz, J6a,6b = 11.8 Hz, H-6a), 4.21 (dd, 1H, J1,2 = 8.4 Hz, J2,3 = 10.7 Hz, H-2′), 4.03 (dd, 1H, J3,4 = 8.0 Hz, J4,5 = 9.4 Hz, H-4), 3.92 (dd, 1H, J5,6a = 5.0 Hz, J6a,6b = 10.2 Hz, H-6′a), 3.86 (br t, 1H, H-3), 3.82 (t, 1H, J3,4 = J4,5 = 9.3 Hz, H-4′), 3.77 (dd, 1H, J5,6b = 4.5 Hz, H-6b), 3.68–3.63 (m, 4H, H-5, Me (OMP)), 3.60 (t, 1H, J5,6b = 10.2 Hz, H-6’b), 3.42 (td, 1H, H-5’), 2.63-2.08 (m, 8H, -CH2-CH2-, CH2(Lev)), 1.92 (s, 3H, CH3(Lev)), 0.99, 0.89 (2s, 18H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3, selected data from HSQC experiment): δ 99.4 (C-1), 98.5 (C-1’), 80.5 (C-3), 76.7 (C-4), 75.1 (C-4’), 73.4 (CH2(Bn)), 72.2 (C-2), 72.1 (C-5), 72.0 (C-3’), 70.3 (C-5’), 65.6 (C-6’), 62.3 (C-6), 55.0 (C-2’, Me (OMP)); ESI MS: m/z: calcd for C65H66F17NO17SiNa: 1506.4; found: 1506.3 [M + Na]+.
Compound 8 (59 mg, 0.04 mmol) was dissolved in CH2Cl2 (1.0 mL) and hydrazine monohydrate (160 µL of a 0.5 M solution in Py/AcOH 3:2) was added. After stirring at room temperature for 1 h, the reaction mixture was quenched with acetone (0.2 mL). The mixture was diluted with CH2Cl2 and washed with 1 M HCl aqueous solution, saturated NaHCO3 aqueous solution and H2O. The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified using a F-SPE column to give disaccharide 9 as a white foam (51 mg). TLC (hexane-EtOAc 2:1) Rf 0.54; 1H-NMR (400 MHz, CDCl3): δ 7.99–7.73 (m, 6H, Ar), 7.57 (m, 1H, Ar), 7.43 (m, 2H, Ar), 7.28–7.16 (m, 5H, Ar), 6.79 (m, 2H, Ar), 6.68 (m, 2H, Ar), 5.50 (d, 1H, J1,2 = 8.3 Hz, H-1′), 5.41 (br t, 1H, H-2), 5.00 (d, 1H, J1,2 = 6.6 Hz, H-1), 4.77 (m, 2H, CH2(Bn)), 4.45 (dd, 1H, J5,6a = 2.1 Hz, J6a,6b = 11.8 Hz, H-6a), 4.30 (m, 1H, H-3′), 4.17 (dd, 1H, J2,3 = 10.8 Hz, H-2′), 4.03 (dd, 1H, J3,4 = 8.0 Hz, J4,5 = 9.4 Hz, H-4), 3.93 (dd, 1H, J5,6a = 5.1 Hz, J6a,6b = 10.2 Hz, H-6′a), 3.87 (t, 1H, J2,3 = 7.4 Hz, H-3), 3.82 (dd, 1H, J5,6b = 4.7 Hz, H-6b), 3.68–3.63 (m, 5H, H-5, H-4′, Me (OMP)), 3.61 (t, 1H, J5,6b = 10.2 Hz, H-6′b), 3.35 (td, 1H, J4,5 = 9.8 Hz, H-5′), 2.51 (br d, 1H, OH), 2.29–2.12 (m, 4H, -CH2-CH2-), 1.01, 0.92 (2s, 18H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3, selected data from HSQC experiment): δ 99.5 (C-1), 98.8 (C-1′), 80.6 (C-3), 77.9 (C-4′), 76.8 (C-4), 73.5 (CH2(Bn)), 72.5 (C-2, C-5), 71.0 (C-3′), 70.3 (C-5′), 65.9 (C-6′), 62.7 (C-6), 56.3 (C-2′), 55.3 (Me (OMP)); ESI MS: m/z: calcd for C60H60F17NO15SiNa: 1408.3; found: 1408.1 [M + Na]+.
Donor 1 (65 mg, 0.093 mmol) and acceptor 9 (51 mg, 0.037 mmol) were dissolved in dry CH2Cl2 (1.0 mL) in the presence of activated 4Å MS (90 mg). The reaction mixture was stirred, under an argon atmosphere, for 10 min at rt and TBSOTf (143 µL of a 0.13 M solution in dry CH2Cl2) was added. After stirring for 25 min at rt, the reaction mixture was quenched with triethylamine, filtered, and then concentrated under reduced pressure. The crude product was purified using a F-SPE column to give a fluorophilic fraction (70 mg) containing trisaccharide 10 as main product together with a small amount (<10%) of unreacted acceptor 9 that was capped by standard acetylation. Data for trisaccharide 10: TLC (hexane-EtOAc 2:1) Rf 0.27; 1H-NMR (400 MHz, CDCl3): δ 7.95 (m, 2H, Ar), 7.72–7.54 (m, 8H, Ar), 7.44–7.36 (m, 4H, Ar), 7.24–6.92 (m, 10H, Ar), 6.74 (m, 2H, Ar), 6.65 (m, 2H, Ar), 5.36 (t, 1H, J1,2 = J2,3 = 6.7 Hz, H-2A), 5.27 (d, 1H, J1,2 = 8.4 Hz, H-1B), 5.10 (t, 1H, J3,4 = J4,5 = 9.4 Hz, H-4C), 4.99–4.94 (m, 3H, H-1C, H-2C, H-1A), 4.71 (m, 2H, CH2(Bn)), 4.54 (dd, 1H, J2,3 = 10.5, J3,4 = 8.6 Hz, H-3B), 4.34 (m, 2H, CH2(Bn)), 4.28 (dd, 1H, J5,6a = 2.0 Hz, J6a,6b = 11.8 Hz, H-6aA), 4.23 (dd, 1H, H-2B), 4.20 (dd, 1H, J5,6a = 2.1 Hz, J6a,6b = 12.2 Hz, H-6aC), 4.14 (dd, 1H, J5,6b = 4.6 Hz, H-6bC), 3.97–3.90 (m, 3H, H-4B, H-4A, H-6aB), 3.78 (t, 1H, J3,4 = 7.5 Hz, H-3A), 3.69-3.51 (m, 8H, Me (OMP), H-6bA, H-6bB, H-3C, H-5C, H-5A), 3.35 (td, 1H, J5,6 = 4.9 Hz, J5,6 = J4,5 = 9.7 Hz, H-5B), 2.65-2.13 (m, 8H, -CH2-CH2-, CH2(Lev)), 2.11 (s, 3H, CH3(Lev)), 1.23 (s, 9H, C(CH3)3), 1.05, 0.95 (2s, 18H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3, selected data from HSQC experiment): δ 99.3 (C-1C, C-1A), 98.7 (C-1B), 80.4 (C-3A), 80.1 (C-3C), 76.7 (C-3B), 76.6 (C-4B, C-4A), 74.2 (C-2C), 73.4 (2 × CH2(Bn)), 72.5 (C-5A, C-5C), 72.4 (C-2A), 70.4 (C-5B), 69.3 (C-4C), 65.9 (C-6B), 62.5 (C-6A), 62.2 (C-6C), 55.8 (C-2B), 55.3 (Me (OMP)); ESI MS: m/z: calcd for C90H94F17NO24SiNa: 1946.6; found: 1946.5 [M + Na]+.
Compound 10 (70 mg, 0.036 mmol) was dissolved in CH2Cl2 (1.0 mL) and hydrazine monohydrate (144 µL of a 0.5 M solution in Py/AcOH 3:2) was added. After stirring at room temperature for 45 min, the reaction mixture was quenched with acetone (0.2 mL). The mixture was diluted with CH2Cl2 and washed with 1 M HCl aqueous solution, saturated NaHCO3 aqueous solution and H2O. The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified by F-SPE to give trisaccharide 11 as a white amorphous solid (63 mg). TLC (hexane-EtOAc 2:1) Rf 0.39; 1H-NMR (400 MHz, CDCl3): δ 7.95 (m, 2H, Ar), 7.71–7.54 (m, 8H, Ar), 7.44–7.36 (m, 4H, Ar), 7.24–6.97 (m, 10H, Ar), 6.74 (m, 2H, Ar), 6.65 (m, 2H, Ar), 5.36 (t, 1H, J1,2 = J2,3 = 6.8 Hz, H-2A), 5.28 (d, 1H, J1,2 = 8.5 Hz, H-1B), 4.97 (d, 1H, J1,2 = 7.6 Hz, H-1C), 4.94 (d, 1H, J1,2 = 6.6 Hz, H-1A), 4.90 (br t, 1H, H-2C), 4.71 (m, 2H, CH2(Bn)), 4.55–4.50 (m, 3H, CH2(Bn), H-3B, H-6aC), 4.41 (m, 1H, CH2(Bn)), 4.29–4.21 (m, 3H, H-6aA, H-6bC, H-2B), 4.00-3.90 (m, 3H, H-4B, H-4A, H-6aB), 3.77 (t, 1H, J3,4 = 7.5 Hz, H-3A), 3.67–3.57 (m, 5H, Me (OMP)), H-6bA, H-6bB), 3.53 (m, 1H, H-5A), 3.49 (m, 1H, H-4C), 3.44–3.32 (m, 3H, H-3C, H-5C, H-5B), 2.95 (br s, 1H, OH), 2.23–2.02 (m, 4H, -CH2-CH2-), 1.24 (s, 9H, C(CH3)3), 1.06, 0.96 (2s, 18H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3, selected data from HSQC experiment): δ 99.4 (C-1C, C-1A), 98.8 (C-1B), 82.2 (C-3C), 80.5 (C-3A), 77.0 (C-3B), 76.8 (C-4B, C-4A), 74.6 (CH2(Bn), C-5C), 74.4 (C-2C), 73.4 (CH2(Bn)), 72.4 (C-2A, C-5A), 70.4 (C-5B), 69.9 (C-4C), 65.9 (C-6B), 63.2 (C-6C), 62.6 (C-6A), 56.0 (C-2B), 55.4 (Me (OMP)); ESI MS: m/z: calcd for C85H88F17NO22SiNa: 1848.5; found: 1848.2 [M + Na]+.
Donor 2 (67 mg, 0.097 mmol) and acceptor 11 (63 mg, 0.034 mmol) were dissolved in dry CH2Cl2 (1.0 mL) in the presence of activated 4Å MS (75 mg). The reaction mixture was stirred, under an argon atmosphere, for 10 min at rt and TBSOTf (114 µL of a 0.085 M solution in dry CH2Cl2) was added. After stirring for 40 min at rt, the reaction mixture was quenched with triethylamine, filtered, and then concentrated under reduced pressure. The crude product was purified by F-SPE to give a fluorophilic fraction (77 mg) containing tetrasaccharide 12 as main product. A small amount (<10%) of unreacted acceptor 11 was also detected and capped by standard acetylation. Data for tetrasaccharide 12: TLC (hexane-EtOAc 2:1) Rf 0.22; 1H-NMR (400 MHz, CDCl3): δ 7.95–7.33 (m, 18H, Ar), 7.20–6.98 (m, 10H, Ar), 6.73 (m, 2H, Ar), 6.64 (m, 2H, Ar), 5.59 (dd, 1H, J2,3 = 10.6 Hz, J3,4 = 9.0 Hz, H-3D), 5.35–5.32 (m, 2H, H-2A, H-1D), 5.22 (d, 1H, J1,2 = 8.5 Hz, H-1B), 4.92 (d, 1H, J1,2 = 6.6 Hz, H-1A), 4.85 (t, 1H, J2,3 = 7.9 Hz, H-2C), 4.72 (d, 1H, J1,2 = 7.2 Hz, H-1C), 4.68 (m, 2H, CH2(Bn)), 4.62 (m, 1H, CH2(Bn)), 4.38 (dd, 1H, J2,3 = 10.5 Hz, J3,4 = 8.5 Hz, H-3B), 4.34 (m, 1H, CH2(Bn)), 4.29–4.22 (m, 2H, H-6aC, H-6aA), 4.16–4.09 (m, 2H, H-2B, H-2D), 3.90–3.81 (m, 5H, H-4A, H-4B, H-4C, H-6aB, H-6aD), 3.75 (t, 1H, J3,4 = 7.5 Hz, H-3A), 3.69–3.66 (m, 4H, H-4D, Me (OMP)), 3.63–3.56 (m, 2H, H-6bA, H-6bC), 3.53–3.46 (m, 3H, H-6bB, H-3C, H-5A), 3.40 (td, 1H, J5,6 = 5.0 Hz, J5,6 = J4,5 = 9.9 Hz, H-5D), 3.31–3.23 (m, 3H, H-5B, H-6bD, H-5C), 2.60–1.99 (m, 8H, -CH2-CH2-, CH2(Lev)), 1.92 (s, 3H, CH3(Lev)), 1.17 (s, 9H, C(CH3)3), 0.95, 0.91, 0.90, 0.86 (4s, 36H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3, selected data from HSQC experiment): δ 99.4 (C-1C), 99.3 (C-1A), 98.4 (C-1B), 97.9 (C-1D), 80.2 (C-3A), 79.6 (C-3C), 77.3 (C-3B), 76.3 (C-4A, C-4B, C-4C), 75.4 (C-4D), 73.9 (C-2C), 73.8 (CH2(Bn)), 73.6 (C-5C), 73.3 (CH2(Bn)), 72.1 (C-3D, C-2A, C-5A), 70.2 (C-5B), 70.1 (C-5D), 65.5 (C-6B, C-6D), 62.3 (C-6A, C-6C), 55.1 (C-2B, C-2D, Me (OMP)); ESI MS: m/z: calcd for C112H123F17N2O30Si2Na: 2377.7; found: 2377.4 [M + Na]+.
Compound 12 (77 mg, 0.033 mmol) was dissolved in CH2Cl2 (1.0 mL) and hydrazine monohydrate (132 µL of a 0.5 M solution in Py/AcOH 3:2) was added. After stirring at room temperature for 50 min, the reaction mixture was quenched with acetone (0.2 mL). The mixture was diluted with CH2Cl2 and washed with 1 M HCl aqueous solution, saturated NaHCO3 aqueous solution and H2O. The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified by F-SPE to give tetrasaccharide 13 as a white amorphous solid (64 mg). TLC (hexane-EtOAc 2:1) Rf 0.43; 1H-NMR (400 MHz, CDCl3): δ 7.95–7.74 (m, 6H, Ar), 7.64–7.33 (m, 12H, Ar), 7.21–6.97 (m, 10H, Ar), 6.73 (m, 2H, Ar), 6.64 (m, 2H, Ar), 5.33 (t, 1H, J2,3 = 6.9 Hz, H-2A), 5.22 (d, 1H, J1,2 = 8.5 Hz, H-1B), 5.21 (d, 1H, J1,2 = 8.3 Hz, H-1D), 4.91 (d, 1H, J1,2 = 6.6 Hz, H-1A), 4.86 (br t, 1H, H-2C), 4.72 (d, 1H, J1,2 = 7.3 Hz, H-1C), 4.68 (m, 2H, CH2(Bn)), 4.63 (m, 1H, CH2(Bn)), 4.39 (dd, 1H, J2,3 = 10.5 Hz, J3,4 = 8.4 Hz, H-3B), 4.34 (m, 1H, CH2(Bn)), 4.30–4.22 (m, 3H, H-6aC, H-3D, H-6aA), 4.13 (dd, 1H, H-2B), 4.08 (dd, 1H, J2,3 = 10.8 Hz, H-2D), 3.90–3.81 (m, 5H, H-4A, H-4B, H-4C, H-6aB, H-6aD), 3.75 (m, 1H, H-3A), 3.66–3.58 (m, 5H, Me (OMP), H-6bA, H-6bC), 3.56–3.46 (m, 4H, H-6bB, H-5A, H-4D, H-3C), 3.37–3.23 (m, 4H, H-5D, H-6bD, H-5B, H-5C), 2.46 (br s, 1H, OH), 2.22–1.99 (m, 4H, -CH2-CH2-), 1.17 (s, 9H, C(CH3)3), 0.97, 0.94, 0.91, 0.86 (4s, 36H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3, selected data from HSQC experiment): δ 99.5 (C-1C), 99.3 (C-1A), 98.2 (C-1B, C-1D), 80.4 (C-3A), 80.1 (C-3C), 78.0 (C-4D), 77.2 (C-3B), 76.3 (C-4A, C-4B, C-4C), 74.0 (C-2C), 73.9 (CH2(Bn)), 73.7 (C-5C), 73.5 (CH2(Bn)), 72.5 (C-2A), 72.3 (C-5A), 71.3 (C-3D), 70.7 (C-5B), 70.1 (C-5D), 65.7 (C-6B, C-6D), 62.5 (C-6A, C-6C), 56.2 (C-2D), 55.8 (C-2B), 55.3 (Me (OMP)); ESI MS: m/z: calcd for C107H117F17N2O28Si2Na: 2279.7; found: 2279.4 [M + Na]+.
Donor 1 (60 mg, 0.085 mmol) and acceptor 13 (64 mg, 0.028 mmol) were dissolved in dry CH2Cl2 (1.2 mL) in the presence of activated 4Å MS (90 mg). The reaction mixture was stirred, under an argon atmosphere, for 10 min at rt and TBSOTf (132 µL of a 0.13 M solution in dry CH2Cl2) was added. After stirring for 25 min at rt, the reaction mixture was quenched with triethylamine, filtered, and then concentrated under reduced pressure. The crude product was purified by F-SPE to give pentasaccharide 14 (75 mg) as the main product. A significant amount (around 25%) of unreacted acceptor 13 was also detected in the fluorous fraction. For this reason, we ran a second glycosylation cycle with additional donor 1 (38 mg, 0.054 mmol). F-SPE purification then gave a fluorous fraction (71 mg) containing pentasaccharide 14 as the main product and a small amount (<10%) of unreacted acceptor 13 that was capped by standard acetylation. Data for pentasaccharide 14: TLC (hexane-EtOAc 3:2) Rf 0.43; 1H-NMR (400 MHz, CDCl3): δ 7.94–7.30 (m, 23H, Ar), 7.19–6.91 (m, 15H, Ar), 6.74–6.62 (m, 4H, Ar), 5.32 (t, 1H, J1,2 = J2,3 = 6.8 Hz, H-2A), 5.19 (d, 1H, J1,2 = 8.5 Hz, H-1B or D), 5.10 (t, 1H, J3,4 = J4,5 = 9.4 Hz, H-4E), 5.00 (br t, 1H, H-2E), 4.98 (d, 1H, J1,2 = 8.3 Hz, H-1B or D), 4.90 (d, 1H, J1,2 = 6.6 Hz, H-1A), 4.84 (d, 1H, J1,2 = 7.4 Hz, H-1E), 4.80 (br t, 1H, H-2C), 4.67–4.64 (m, 3H, CH2(Bn), H-1C), 4.57 (d, 1H, CH2(Bn)), 4.51 (dd, 1H, J2,3 = 10.4 Hz, J3,4 = 8.5 Hz, H-3B or D), 4.38-4.29 (m, 3H, CH2(Bn), H-3B or D), 4.25–4.20 (m, 2H, CH2(Bn), H-6aA), 4.16–4.06 (m, 5H, H-6aE, H-6bE, H-6aC, H-2B, H-2D), 3.88–3.76 (m, 6H, H-6aB, H-6aD, H-4A, H-4B, H-4C, H-4D), 3.73 (t, 1H, J3,4 = 7.4 Hz, H-3A), 3.65 (s, 3H, Me (OMP)), 3.63–3.57 (m, 2H, H-3E, H-6bA), 3.55–3.45 (m, 3H, H-5E, H-6bB or D, H-5A), 3.37–3.22 (m, 5H, H-6bC, H-6bB or D, H-3C, H-5B, H-5D), 3.03 (m, 1H, H-5C), 2.64-2.11 (m, 8H, -CH2-CH2-, CH2(Lev)), 2.10 (s, 3H, CH3(Lev)), 1.21, 1.15 (2s, 18H, C(CH3)3), 1.01, 0.98, 0.83, 0.82 (4s, 36H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3, selected data from HSQC experiment): δ 99.8 (C-1E), 99.6 (C-1C), 99.4 (C-1A), 98.6, 97.6 (C-1B, C-1D), 80.5 (C-3C), 80.4 (C-3A), 80.3 (C-3E), 77.5, 77.4 (C-3B, C-3D), 77.4–75.7 (C-4A, C-4B, C-4C, C-4D), 74.5 (C-2E, CH2(Bn)), 74.1 (C-2C), 73.6 (2 × CH2(Bn)), 73.5 (C-5C), 72.8 (C-5E), 72.5 (C-2A, C-5A), 70.5 (C-5B, C-5D), 69.6 (C-4E), 66.0, 65.8 (C-6B, C-6D), 62.7 (C-6A), 62.4 (C-6C, C-6E), 55.9 (C-2B, C-2D), 55.6 (Me (OMP)); ESI MS: m/z: calcd for C137H151F17N2O37Si2Na: 2817.9; found: 2817.5 [M + Na]+.
Compound 14 (71 mg, 0.025 mmol) was dissolved in CH2Cl2 (1.0 mL) and hydrazine monohydrate (100 µL of a 0.5 M solution in Py/AcOH 3:2) was added. After stirring at room temperature for 1 h, the reaction mixture was quenched with acetone (0.15 mL). The mixture was diluted with CH2Cl2 and washed with 1 M HCl aqueous solution, saturated NaHCO3 aqueous solution and H2O. The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified by F-SPE to give pentasaccharide 15 as a white amorphous solid (61 mg). TLC (hexane-EtOAc 3:2) Rf 0.35; 1H-NMR (400 MHz, CDCl3): δ 7.94-7.30 (m, 23H, Ar), 7.19–6.92 (m, 15H, Ar), 6.74–6.62 (m, 4H, Ar), 5.32 (t, 1H, J1,2 = J2,3 = 6.8 Hz, H-2A), 5.19 (d, 1H, J1,2 = 8.4 Hz, H-1B or D), 4.99 (d, 1H, J1,2 = 8.5 Hz, H-1B or D), 4.92 (br t, 1H, H-2E), 4.90 (d, 1H, J1,2 = 6.6 Hz, H-1A), 4.85 (d, 1H, J1,2 = 7.6 Hz, H-1E), 4.80 (br t, 1H, H-2C), 4.67–4.63 (m, 3H, CH2(Bn), H-1C), 4.59–4.47 (m, 4H, CH2(Bn), H-6aE, H-3B or D), 4.40 (d, 1H, CH2(Bn)), 4.33 (dd, 1H, J2,3 = 10.5 Hz, J3,4 = 8.4 Hz, H-3B or D), 4.25–4.17 (m, 3H, CH2(Bn), H-6aA, H-6bE), 4.12–4.07 (m, 3H, H-6aC, H-2B, H-2D), 3.88–3.78 (m, 6H, H-6aB, H-6aD, H-4A, H-4B, H-4C, H-4D), 3.73 (t, 1H, J3,4 = 7.4 Hz, H-3A), 3.65 (s, 3H, Me (OMP)), 3.59 (dd, 1H, J5,6b = 4.6 Hz, J6a,6b = 11.8 Hz, H-6bA), 3.52-3.44 (m, 3H, H-6bB or D, H-5A, H-4E), 3.40 (t, 1H, J2,3 = J3,4 = 8.8 Hz, H-3E), 3.37–3.22 (m, 6H, H-6bC, H-5E, H-3C, H-6bB or D, H-5B, H-5D), 3.03 (m, 1H, H-5C), 2.98 (br d, 1H, OH), 2.23–1.89 (m, 4H, -CH2-CH2-), 1.23, 1.15 (2s, 18H, C(CH3)3), 1.02, 0.99, 0.83 (4s, 36H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3, selected data from HSQC experiment): δ 99.8 (C-1E), 99.5 (C-1C), 99.4 (C-1A), 98.5, 97.4 (C-1B, C-1D), 81.9 (C-3E), 80.3 (C-3A), 80.2 (C-3C), 77.4 (C-3B, C-3D), 77.4–75.4 (C-4A, C-4B, C-4C, C-4D), 74.6 (C-5E), 74.5 (CH2(Bn)), 74.4 (C-2E, CH2(Bn)), 74.0 (C-2C), 73.5 (CH2(Bn)), 73.3 (C-5C), 72.3 (C-5A), 72.2 (C-2A), 70.4 (C-5B, C-5D), 69.9 (C-4E), 65.9, 65.7 (C-6B, C-6D), 63.1 (C-6E), 62.6 (C-6A), 62.3 (C-6C), 55.7 (C-2B, C-2D), 55.4 (Me (OMP)); ESI MS: m/z: calcd for C132H145F17N2O35Si2Na: 2719.9; found: 2719.7 [M + Na]+.
Donor 2 (48 mg, 0.069 mmol) and acceptor 15 (61 mg, 0.023 mmol) were dissolved in dry CH2Cl2 (1.0 mL) in the presence of activated 4Å MS (75 mg). The reaction mixture was stirred, under an argon atmosphere, for 10 min at rt and TBSOTf (108 µL of a 0.064 M solution in dry CH2Cl2) was added. After stirring for 25 min at rt, the reaction mixture was quenched with triethylamine, filtered, and then concentrated under reduced pressure. The crude product was first purified by F-SPE. The fluorous fraction was then purified by standard silica gel column chromatography (hexane-EtOAc 2:1) to afford pure 16 (50 mg, 39% from 3; 9 steps) as a white amorphous solid. TLC (toluene-EtOAc 4:1) Rf 0.59; [α]20D −5° (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3): δ 7.93-6.88 (m, 42H, Ar), 6.73–6.61 (m, 4H, Ar), 5.59 (dd, 1H, J2,3 = 10.6 Hz, J3,4 = 9.0 Hz, H-3F), 5.33–5.30 (m, 2H, H-1F, H-2A), 5.18 (d, 1H, J1,2 = 8.4 Hz, H-1B or D), 4.93–4.86 (m, 3H, H-1B or D, H-1A, H-2C or E), 4.77 (t, 1H, J1,2 = J2,3 = 8.1 Hz, H-2C or E), 4.66–4.52 (m, 6H, H-1C, H-1E, CH2(Bn)), 4.38–4.19 (m, 6H, H-3B, H-3D, CH2(Bn), H-6aA or C or E), 4.13–4.06 (m, 3H, H-2F, H-2B or D, H-6aA or C or E), 4.00 (dd, 1H, J1,2 = 8.4 Hz, J2,3 = 10.3 Hz, H-2B or D), 3.87–3.66 (m, 10H, H-6aB, H-6aD, H-6aF, H-4A, H-4C, H-4E, H-4F, H-4B, H-4D, H-3A), 3.65 (s, 3H, Me (OMP)), 3.60-3.53 (m, 2H, H-6bA or C or E), 3.51–3.37 (m, 4H, H-6bB or D or F, H-5A, H-3C or E, H-5B or D or F), 3.33-3.20 (m, 7H, H-6bA or C or E, H-3C or E, H-6bB or D or F, H-5B or D or F, H-5C or E), 2.99 (m, 1H, H-5C or E), 2.63–2.01 (m, 8H, -CH2-CH2-, CH2(Lev)), 1.92 (s, 3H, CH3(Lev)), 1.16, 1.13 (2s, 18H, C(CH3)3), 0.95, 0.91, 0.88, 0.81 (5s, 54H, Si(C(CH3)3)2); 13C-NMR (100 MHz, CDCl3): δ 205.7, 177.4, 177.2, 172.0, 170.2 (5 x CO), 168.5-167.0 (6 × CO), 165.1, 165.0, 164.9 (3 × CO), 155.5-114.4 (Ar), 100.1, 99.7 (C-1C, C-1E), 99.5 (C-1A), 98.6 (C-1B or D), 97.8 (C-1F), 97.5 (C-1B or D), 80.4, 80.3, 80.1 (C-3A, C-3C, C-3E), 77.8 (C-3B, C-3D), 77.0-75.3 (C-4A, C-4C, C-4E, C-4B, C-4D, C-4F), 74.5 (CH2(Bn)), 74.2, 74.1 (CH2(Bn), C-2C, C-2E), 73.8, 73.6, 73.5 (CH2(Bn), C-5C, C-5E), 72.5 (C-2A, C-5A, C-3F), 70.4 (C-5B, C-5D, C-5F), 65.9, 65.7 (C-6B, C-6D, C-6F), 62.7, 62.6, 62.4 (C-6A, C-6C, C-6E), 56.0, 55.9 (C-2B, C-2D), 55.5 (Me (OMP)), 55.2 (C-2F), 38.8 (C(CH3)3), 37.9 (CH2(Lev)), 29.6 (CH3(Lev)), 27.9 (CH2(Lev)), 27.4, 27.3, 27.1, 27.0, 26.9 (C(CH3)3, Si(C(CH3)3)2), 26.2 (t, JC,F = 21.1 Hz, -CH2-CF2-), 24.7 (-CH2-), 22.6, 22.5, 20.0, 19.9, 19.8 (Si(C(CH3)3)2); HR MS: m/z: calcd for C159H180F17N3O43Si3Na: 3249.0919; found: 3249.0895 [M + Na]+.
3.4.6. Synthesis of 4-Methoxyphenyl O-(2-deoxy-3-O-levulinoyl-2-phthalimido-4,6-di-O-sulfo-β-d-glucopyranosyl)-(1→4)-O-(2-O-benzoyl-3-O-benzyl-6-O-pivaloyl-β-d-glucopyranosyl)-(1→3)-O-(2-deoxy-2-phthalimido-4,6-di-O-sulfo-β-d-glucopyranosyl)-(1→4)-O-(2-O-benzoyl-3-O-benzyl-6-O-pivaloyl-β-d-glucopyranosyl)-(1→3)-O-(2-deoxy-2-phthalimido-4,6-di-O-sulfo-β-d-glucopyranosyl)-(1→4)-2-O-benzoyl-3-O-benzyl-6-O-4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecanoyl-β-d-glucopyranoside (17)
An excess of (HF)n·Py (79 µL, 3.0 mmol) was added at 0 °C under an argon atmosphere to a solution of 16 (49 mg, 15 µmol) in dry THF (1.6 mL). After 18 h at 0 °C the mixture was diluted with CH2Cl2 and washed with H2O, saturated NaHCO3 solution and H2O. The organic layers were dried (MgSO4), filtered and concentrated in vacuo to give the corresponding hexaol (42 mg) as a white amorphous solid that was used without further purification. TLC (toluene-EtOAc 1:3) Rf 0.35; ESI MS: m/z: calcd for C135H132F17N3O43Cl: 2840.8; found: 2841.2 [M + Cl]−.
The desilylated compound (15 µmol) and sulfur trioxide–trimethylamine complex (125 mg, 0.9 mmol) were dissolved in dry DMF (4.0 mL) and heated at 100 °C for 30 min using microwave radiation (40 W average power). The reaction vessel was cooled and Et3N (350 µL), MeOH (2.0 mL) and CH2Cl2 (2.0 mL) were added. The solution was purified by Sephadex LH 20 chromatography (CH2Cl2-MeOH 1:1). Partially sulfated intermediates were still detected by TLC and, for this reason, the residue was submitted to a second and third sulfation cycle with new sulfating reagent using the same reaction conditions. After the third sulfation cycle, the reaction was completed and the crude mixture was purified by Sephadex LH 20 chromatography (CH2Cl2-MeOH 1:1) and silica gel column chromatography (EtOAc-MeOH-H2O 24:5:3 → EtOAc-MeOH-H2O 16:5:3). The residue was finally eluted from a Dowex 50WX2-Na+ column (MeOH as eluent) to obtain 17 as sodium salt (29 mg, 56%, 2 steps, white amorphous solid). TLC (EtOAc-MeOH-H2O 16:5:3) Rf 0.34; 1H-NMR (400 MHz, CD3OD): δ 7.96–6.86 (m, 42H, Ar), 6.78–6.66 (m, 4H, Ar), 5.83 (dd, 1H, J2,3 = 10.8 Hz, J3,4 = 8.8 Hz, H-3F), 5.44 (d, 1H, J1,2 = 8.3 Hz, H-1F), 5.33 (d, 1H, J1,2 = 8.3 Hz, H-1B or D), 5.25 (t, 1H, J2,3 = 8.3 Hz, H-2A), 5.16 (d, 1H, J1,2 = 7.7 Hz, H-1A), 5.12 (d, 1H, J1,2 = 8.4 Hz, H-1B or D), 4.90 (t, 1H, J1,2 = J2,3 = 8.3 Hz, H-2C or E), 4.83–4.78 (m, 2H, H-2C or E, CH2(Bn)), 4.76–4.54 (m, 7H, H-3B, H-3D, H-6aB, H-6aD, H-6aF, CH2(Bn), H-1C or E), 4.52–4.47 (m, 4H, H-1C or E, CH2(Bn)), 4.42 (d, 1H, CH2(Bn)), 4.36–4.22 (m, 5H, H-6aA, H-6aC or E, H-4F, H-4B, H-4D), 4.19–3.98 (m, 11H, H-2F, H-3A, H-2B, H-2D, H-6aC or E, H-6bA, H-6bB, H-6bD, H-6bF, H-5A, H-4C or E), 3.97–3.89 (m, 3H, H-4C or E, H-4A, H-5B or D), 3.83–3.73 (m, 3H, H-5B or D, H-5F, H-6bC or E), 3.65 (m, 4H, Me (OMP), H-6bC or E), 3.55 (t, 1H, J2,3 = J3,4 = 8.7 Hz, H-3C or E), 3.44–3.39 (m, 2H, H-3C or E, H-5C or E), 3.28 (m, 1H, H-5C or E), 2.59-2.32 (m, 8H, -CH2-CH2-, CH2(Lev)), 1.87 (s, 3H, CH3(Lev)), 1.19, 1.16 (2s, 18H, C(CH3)3); 13C-NMR (100 MHz, CD3OD): δ 208.8, 179.7, 179.6, 173.9, 172.2 (5 x CO), 169.9–168.8 (6 × CO), 167.2, 166.9 (3 × CO), 156.7–115.4 (Ar), 100.6, 100.5, 100.4 (C-1A C-1C, C-1E), 98.3 (C-1F), 98.0, 96.7 (C-1B, C-1D), 81.0, 80.8 (C-3C, C-3E), 78.9 (C-3A), 77.7, 77.4 (C-4B, C-4D), 77.1, 76.7, 76.4, 76.0 (C-4A, C-4C, C-4E, C-4F, C-3B, C-3D), 75.1 (C-2C, C-2E, C-5B, C-5D, C-5F, 2 x CH2(Bn)), 74.6 (C-2A), 74.3 (C-5C, C-5E), 73.9 (C-5A), 73.2 (CH2(Bn)), 71.8 (C-3F), 68.8, 68.3, 68.0 (C-6B, C-6D, C-6F), 64.5 (C-6A), 63.9 (C-6C, C-6E), 57.4, 57.3 (C-2B, C-2D), 56.4 (C-2F), 55.8 (Me (OMP)), 39.9, 39.8 (C(CH3)3), 38.5 (CH2(Lev)), 29.2 (CH3(Lev)), 29.1 (CH2(Lev)), 27.8 (C(CH3)3), 27.1 (t, JC,F = 22.0 Hz, -CH2-CF2-), 25.9 (-CH2-); ESI MS: m/z: calcd for C135H126F17N3O61S6Na33−: 1116.2; found: 1116.2 [M + 3Na]3−; HR MS: m/z: calcd for C135H126F17N3O61S6Na42−: 1685.7252; found: 1685.7295 [M + 4Na]2−.
4. Conclusions
The synthesis of a hexasaccharide with the sequence GlcN(4,6-di-OSO3)-β(1→4)-Glc-β(1→3), related to the CS-E structure, was successfully accomplished. Starting monosaccharide building blocks 1, 2 and 3 were efficiently assembled for the preparation of the target oligosaccharide and only two standard chromatographic purifications were required due to the assistance of a fluorinated tag. Using solution-phase FP competition experiments, we showed that the synthesized hexasaccharide 17 strongly interacted with midkine and FGF-2, although the IC50 values were slightly higher than those corresponding to the analogous tetramer. In addition, we also measured the direct binding of the growth factors to a microplate surface that was functionalized with the fluorous-tagged sugar. The calculated surface dissociation constants indicated a nanomolar interaction for midkine and FGF-2 (KD,surf = 16 and 57 nM, respectively). On the contrary, NGF bound to the sugar-coated surface with less affinity (KD,surf = 1.6 µM), suggesting that derivative 17 is more selective (≥28 fold) for midkine and FGF-2.
Supplementary Materials
The supplementary materials are available online.
Author Contributions
S.M., J.L.P. and P.M.N. designed the experiments; S.M. and J.L.P. performed the experiments and analyzed the data; S.M. and J.L.P. prepared and wrote the original draft; J.L.P. and P.M.N. reviewed and edited the article, and supervised the project and funding acquisition.
Funding
This research was funded by the Spanish Ministry of Economy and Competitiveness, grant number CTQ2015-70134-P, Junta de Andalucía, grant number P12-BIO-1938 and the European Union (ERDF).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Muramatsu, T. Midkine: A Promising Molecule for Drug Development to Treat Diseases of the Central Nervous System. Curr. Pharm. Des. 2011, 17, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, K.; Kishida, S.; Tsubota, S. The heparin-binding growth factor midkine: The biological activities and candidate receptors. J. Biochem. 2013, 153, 511–521. [Google Scholar] [CrossRef]
- Deepa, S.S.; Umehara, Y.; Higashiyama, S.; Itoh, N.; Sugahara, K. Specific molecular interactions of oversulfated chondroitin sulfate E with various heparin-binding growth factors—Implications as a physiological binding partner in the brain and other tissues. J. Biol. Chem. 2002, 277, 43707–43716. [Google Scholar] [CrossRef]
- Solera, C.; Macchione, G.; Maza, S.; Kayser, M.M.; Corzana, F.; de Paz, J.L.; Nieto, P.M. Chondroitin Sulfate Tetrasaccharides: Synthesis, Three-Dimensional Structure and Interaction with Midkine. Chem. Eur. J. 2016, 22, 2356–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gama, C.I.; Tully, S.E.; Sotogaku, N.; Clark, P.M.; Rawat, M.; Vaidehi, N.; Goddard, W.A.; Nishi, A.; Hsieh-Wilson, L.C. Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol. 2006, 2, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Rogers, C.J.; Clark, P.M.; Tully, S.E.; Abrol, R.; Garcia, K.C.; Goddard, W.A., III; Hsieh-Wilson, L.C. Elucidating glycosaminoglycan-protein-protein interactions using carbohydrate microarray and computational approaches. Proc. Natl. Acad. Sci. USA 2011, 108, 9747–9752. [Google Scholar] [CrossRef]
- Liu, P.; Chen, L.; Toh, J.K.C.; Ang, Y.L.; Jee, J.-E.; Lim, J.; Lee, S.S.; Lee, S.-G. Tailored chondroitin sulfate glycomimetics via a tunable multivalent scaffold for potentiating NG/TrkA-induced neurogenesis. Chem. Sci. 2015, 6, 450–456. [Google Scholar] [CrossRef]
- Desai, U.R. The promise of sulfated synthetic small molecules as modulators of glycosaminoglycan function. Future Med. Chem. 2013, 5, 1363–1366. [Google Scholar] [CrossRef]
- Weiss, R.J.; Esko, J.D.; Tor, Y. Targeting heparin and heparan sulfate protein interactions. Org. Biomol. Chem. 2017, 15, 5656–5668. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. Heparin/heparan sulphate-based drugs. Drug Discovery Today 2010, 15, 1058–1069. [Google Scholar] [CrossRef]
- Dominguez-Rodriguez, P.; Reina, J.J.; Gil-Caballero, S.; Nieto, P.M.; de Paz, J.L.; Rojo, J. Glycodendrimers as Chondroitin Sulfate Mimetics: Synthesis and Binding to Growth Factor Midkine. Chem. Eur. J. 2017, 23, 11338–11345. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, W.; Xu, X.J.; Sun, H.F.; Zhao, J.H.; Meng, X.B.; Wu, M.Y.; Li, Z.J. Synthesis of Fucosylated Chondroitin Sulfate Glycoclusters: A Robust Route to New Anticoagulant Agents. Chem. Eur. J. 2018, 24, 1694–1700. [Google Scholar] [CrossRef]
- Loka, R.S.; Yu, F.; Sletten, E.T.; Nguyen, H.M. Design, synthesis, and evaluation of heparan sulfate mimicking glycopolymers for inhibiting heparanase activity. Chem. Commun. 2017, 53, 9163–9166. [Google Scholar] [CrossRef]
- Tyler, P.C.; Guimond, S.E.; Turnbull, J.E.; Zubkova, O.V. Single-Entity Heparan Sulfate Glycomimetic Clusters for Therapeutic Applications. Angew. Chem. Int. Ed. 2015, 54, 2718–2723. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.I.; Sheng, G.J.; Chang, S.-K.; Hsieh-Wilson, L.C. Tailored Glycopolymers as Anticoagulant Heparin Mimetics. Angew. Chem. Int. Ed. 2013, 52, 11796–11799. [Google Scholar] [CrossRef] [Green Version]
- Sheng, G.J.; Oh, Y.I.; Chang, S.-K.; Hsieh-Wilson, L.C. Tunable Heparan Sulfate Mimetics for Modulating Chemokine Activity. J. Am. Chem. Soc. 2013, 135, 10898–10901. [Google Scholar] [CrossRef] [PubMed]
- Kuhnast, B.; El Hadri, A.; Boisgard, R.; Hinnen, F.; Richard, S.; Caravano, A.; Nancy-Portebois, V.; Petitou, M.; Tavitian, B.; Dolle, F. Synthesis, radiolabeling with fluorine-18 and preliminary in vivo evaluation of a heparan sulphate mimetic as potent angiogenesis and heparanase inhibitor for cancer applications. Org. Biomol. Chem. 2016, 14, 1915–1920. [Google Scholar] [CrossRef]
- Lee, S.S.; Fyrner, T.; Chen, F.; Alvarez, Z.; Sleep, E.; Chun, D.S.; Weiner, J.A.; Cook, R.W.; Freshman, R.D.; Schallmo, M.S.; et al. Sulfated glycopeptide nanostructures for multipotent protein activation. Nat. Nanotechnol. 2017, 12, 821–829. [Google Scholar] [CrossRef]
- Johnstone, K.D.; Karoli, T.; Liu, L.; Dredge, K.; Copeman, E.; Li, C.P.; Davis, K.; Hammond, E.; Bytheway, I.; Kostewicz, E.; et al. Synthesis and Biological Evaluation of Polysulfated Oligosaccharide Glycosides as Inhibitors of Angiogenesis and Tumor Growth. J. Med. Chem. 2010, 53, 1686–1699. [Google Scholar] [CrossRef] [Green Version]
- Ferro, V.; Liu, L.; Johnstone, K.D.; Wimmer, N.; Karoli, T.; Handley, P.; Rowley, J.; Dredge, K.; Li, C.P.; Hammond, E.; et al. Discovery of PG545: A Highly Potent and Simultaneous Inhibitor of Angiogenesis, Tumor Growth, and Metastasis. J. Med. Chem. 2012, 55, 3804–3813. [Google Scholar] [CrossRef]
- Afosah, D.K.; Al-Horani, R.A.; Sankaranarayanan, N.V.; Desai, U.R. Potent, Selective, Allosteric Inhibition of Human Plasmin by Sulfated Non-Saccharide Glycosaminoglycan Mimetics. J. Med. Chem. 2017, 60, 641–657. [Google Scholar] [CrossRef] [PubMed]
- De Paz, J.L.; Nieto, P.M. Improvement on binding of chondroitin sulfate derivatives to midkine by increasing hydrophobicity. Org. Biomol. Chem. 2016, 14, 3506–3509. [Google Scholar] [CrossRef] [Green Version]
- Maza, S.; Gandia-Aguado, N.; de Paz, J.L.; Nieto, P.M. Fluorous-tag assisted synthesis of a glycosaminoglycan mimetic tetrasaccharide as a high-affinity FGF-2 and midkine ligand. Bioorg. Med. Chem. 2018, 26, 1076–1085. [Google Scholar] [CrossRef]
- Nielsen, M.M.; Pedersen, C.M. Catalytic Glycosylations in Oligosaccharide Synthesis. Chem. Rev. 2018, 118, 8285–8358. [Google Scholar] [CrossRef]
- Zhu, X.M.; Schmidt, R.R. New Principles for Glycoside-Bond Formation. Angew. Chem. Int. Ed. 2009, 48, 1900–1934. [Google Scholar] [CrossRef] [PubMed]
- Jaipuri, F.A.; Pohl, N.L. Toward solution-phase automated iterative synthesis: Fluorous-tag assisted solution-phase synthesis of linear and branched mannose oligomers. Org. Biomol. Chem. 2008, 6, 2686–2691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Curran, D.P. Synthetic applications of fluorous solid-phase extraction (F-SPE). Tetrahedron 2006, 62, 11837–11865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchione, G.; de Paz, J.L.; Nieto, P.M. Synthesis of hyaluronic acid oligosaccharides and exploration of a fluorous-assisted approach. Carbohydr. Res. 2014, 394, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Bhaduri, S.; Pohl, N.L.B. Fluorous-Tag Assisted Syntheses of Sulfated Keratan Sulfate Oligosaccharide Fragments. Org. Lett. 2016, 18, 1414–1417. [Google Scholar] [CrossRef]
- Karst, N.; Jacquinet, J.C. Chemical synthesis of β-d-GlcpA(2SO4)-(1→3)-d-GalpNAc(6SO4), the disaccharide repeating unit of shark cartilage chondroitin sulfate D, and of its methyl β-d-glycoside derivative. J. Chem. Soc., Perkin Trans. 2000, 1, 2709–2717. [Google Scholar] [CrossRef]
- Dinkelaar, J.; Gold, H.; Overkleeft, H.S.; Codee, J.D.C.; van der Marel, G.A. Synthesis of Hyaluronic Acid Oligomers using Chemoselective and One-Pot Strategies. J. Org. Chem. 2009, 74, 4208–4216. [Google Scholar] [CrossRef]
- Mydock, L.K.; Demchenko, A.V. Mechanism of chemical O-glycosylation: From early studies to recent discoveries. Org. Biomol. Chem. 2010, 8, 497–510. [Google Scholar] [CrossRef]
- Yu, Y.; Kononov, A.; Delbianco, M.; Seeberger, P.H. A Capping Step During Automated Glycan Assembly Enables Access to Complex Glycans in High Yield. Chem. Eur. J. 2018, 24, 6075–6078. [Google Scholar] [CrossRef] [PubMed]
- Maza, S.; de Paz, J.L.; Nieto, P.M. Microwave-assisted sulfonation of heparin oligosaccharides. Tetrahedron Lett. 2011, 52, 441–443. [Google Scholar] [CrossRef]
- Maza, S.; Mar Kayser, M.; Macchione, G.; Lopez-Prados, J.; Angulo, J.; de Paz, J.L.; Nieto, P.M. Synthesis of chondroitin/dermatan sulfate-like oligosaccharides and evaluation of their protein affinity by fluorescence polarization. Org. Biomol. Chem. 2013, 11, 3510–3525. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.S.; Jaipuri, F.A.; Pohl, N.L. Fluorous-based carbohydrate microarrays. J. Am. Chem. Soc. 2005, 127, 13162–13163. [Google Scholar] [CrossRef] [PubMed]
- Mamidyala, S.K.; Ko, K.-S.; Jaipuri, F.A.; Park, G.; Pohl, N.L. Noncovalent fluorous interactions for the synthesis of carbohydrate microarrays. J. Fluorine Chem. 2006, 127, 571–579. [Google Scholar] [CrossRef]
- Chen, G.-S.; Pohl, N.L. Synthesis of fluorous tags for incorporation of reducing sugars into a quantitative microarray platform. Org. Lett. 2008, 10, 785–788. [Google Scholar] [CrossRef]
- Tang, S.-L.; Linz, L.B.; Bonning, B.C.; Pohl, N.L.B. Automated Solution-Phase Synthesis of Insect Glycans to Probe the Binding Affinity of Pea Enation Mosaic Virus. J. Org. Chem. 2015, 80, 10482–10489. [Google Scholar] [CrossRef]
- Maza, S.; Macchione, G.; Ojeda, R.; Lopez-Prados, J.; Angulo, J.; de Paz, J.L.; Nieto, P.M. Synthesis of amine-functionalized heparin oligosaccharides for the investigation of carbohydrate-protein interactions in microtiter plates. Org. Biomol. Chem. 2012, 10, 2146–2163. [Google Scholar] [CrossRef]
- Liang, P.H.; Wang, S.K.; Wong, C.H. Quantitative analysis of carbohydrate-protein interactions using glycan microarrays: Determination of surface and solution dissociation constants. J. Am. Chem. Soc. 2007, 129, 11177–11184. [Google Scholar] [CrossRef] [PubMed]
- Medve, L.; Achilli, S.; Serna, S.; Zuccotto, F.; Varga, N.; Thepaut, M.; Civera, M.; Vives, C.; Fieschi, F.; Reichardt, N.; Bernardi, A. On-Chip Screening of a Glycomimetic Library with C-Type Lectins Reveals Structural Features Responsible for Preferential Binding of Dectin-2 over DC-SIGN/R and Langerin. Chem. Eur. J. 2018, 24, 14448–14460. [Google Scholar] [CrossRef] [PubMed]
- Mena-Barragan, T.; de Paz, J.L.; Nieto, P.M. Unexpected loss of stereoselectivity in glycosylation reactions during the synthesis of chondroitin sulfate oligosaccharides. Beilstein J. Org. Chem. 2019, 15, 137–144. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound 17 is available from the authors. |
Scheme 1.
Monosaccharide units employed for the preparation of the target hexasaccharide. Lev = levulinoyl; Piv = pivaloyl; MP = 4-methoxyphenyl; NPhth = N-phthaloyl.
Scheme 1.
Monosaccharide units employed for the preparation of the target hexasaccharide. Lev = levulinoyl; Piv = pivaloyl; MP = 4-methoxyphenyl; NPhth = N-phthaloyl.
Scheme 2.
Reagents and conditions: (a) Lev2O, DMAP, CH2Cl2; TFA, CH2Cl2-H2O, 0 °C → room temperature, 87%; (b) tBu2Si(OTf)2, Py, 0 °C → room temperature, 93%; (c) CAN, CH2Cl2-CH3CN-H2O, 0 °C, 91%; (d) Cl3CCN, DBU, CH2Cl2, 75%.
Scheme 2.
Reagents and conditions: (a) Lev2O, DMAP, CH2Cl2; TFA, CH2Cl2-H2O, 0 °C → room temperature, 87%; (b) tBu2Si(OTf)2, Py, 0 °C → room temperature, 93%; (c) CAN, CH2Cl2-CH3CN-H2O, 0 °C, 91%; (d) Cl3CCN, DBU, CH2Cl2, 75%.

Scheme 3.
Reagents and conditions: (a) TBSOTf, CH2Cl2, 0 °C; (b) NH2NH2·H2O, Py-AcOH, CH2Cl2; (c) 1, TBSOTf, CH2Cl2, room temperature; (d) 2, TBSOTf, CH2Cl2, room temperature; 16: 39% from 3, 9 steps; (e) (HF)n·Py, THF, 0 °C; SO3·Me3N, DMF, 100 °C, MW heating, 56%.
Scheme 3.
Reagents and conditions: (a) TBSOTf, CH2Cl2, 0 °C; (b) NH2NH2·H2O, Py-AcOH, CH2Cl2; (c) 1, TBSOTf, CH2Cl2, room temperature; (d) 2, TBSOTf, CH2Cl2, room temperature; 16: 39% from 3, 9 steps; (e) (HF)n·Py, THF, 0 °C; SO3·Me3N, DMF, 100 °C, MW heating, 56%.
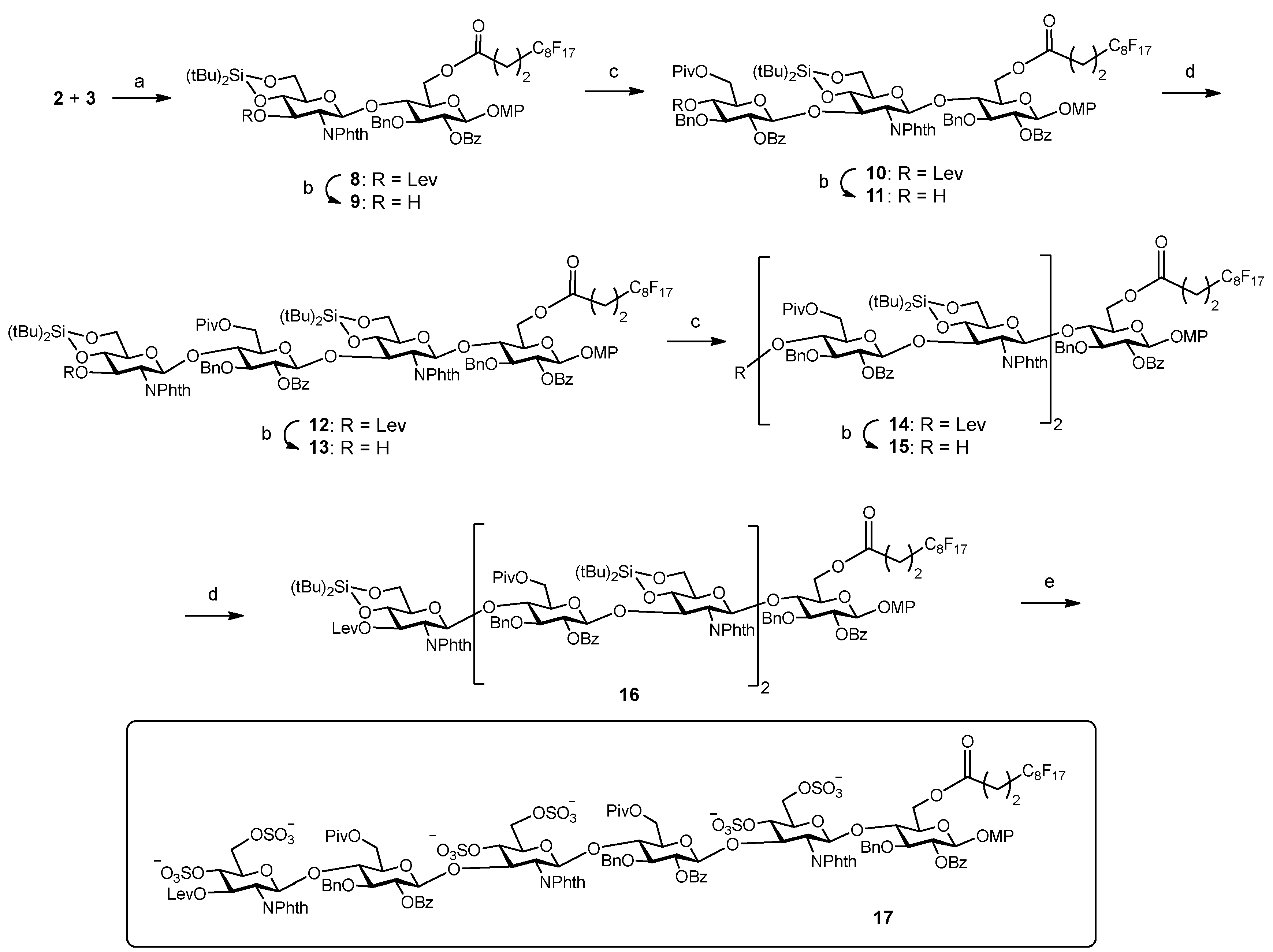
Figure 1.
Schematic representation of the strategy used to study the interactions between fluorinated sugars and proteins on microtiter plates.
Figure 1.
Schematic representation of the strategy used to study the interactions between fluorinated sugars and proteins on microtiter plates.
Figure 2.
Binding curves for the interactions between immobilized hexasaccharide 17 and midkine, FGF-2 and NGF. The displayed fluorescence intensity values are the averages of at least three replicate wells and the error bars represent the standard deviations.
Figure 2.
Binding curves for the interactions between immobilized hexasaccharide 17 and midkine, FGF-2 and NGF. The displayed fluorescence intensity values are the averages of at least three replicate wells and the error bars represent the standard deviations.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
IC50 and KD,surf values for the interaction between fluorinated sugar 17 and midkine, basic fibroblast growth factor (FGF-2) and nerve growth factor (NGF) proteins. The IC50 values were obtained from solution fluorescence polarization (FP) competition experiments while the KD,surf values were calculated from assays using fluorous sugar-coated microplates.
Table 1.
IC50 and KD,surf values for the interaction between fluorinated sugar 17 and midkine, basic fibroblast growth factor (FGF-2) and nerve growth factor (NGF) proteins. The IC50 values were obtained from solution fluorescence polarization (FP) competition experiments while the KD,surf values were calculated from assays using fluorous sugar-coated microplates.
| Midkine | FGF-2 | NGF | |
|---|---|---|---|
| IC50 (nM) | 450 ± 30 | 6300 ± 1600 | not determined |
| KD,surf (nM) | 16 ± 5 | 57 ± 10 | 1600 ± 400 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Maza, S.; de Paz, J.L.; Nieto, P.M. Synthesis of a Fluorous-Tagged Hexasaccharide and Interaction with Growth Factors Using Sugar-Coated Microplates. Molecules 2019, 24, 1591. https://doi.org/10.3390/molecules24081591
AMA Style
Maza S, de Paz JL, Nieto PM. Synthesis of a Fluorous-Tagged Hexasaccharide and Interaction with Growth Factors Using Sugar-Coated Microplates. Molecules. 2019; 24(8):1591. https://doi.org/10.3390/molecules24081591
Chicago/Turabian StyleMaza, Susana, José L. de Paz, and Pedro M. Nieto. 2019. "Synthesis of a Fluorous-Tagged Hexasaccharide and Interaction with Growth Factors Using Sugar-Coated Microplates" Molecules 24, no. 8: 1591. https://doi.org/10.3390/molecules24081591